

Abstract
Objective
Inflammation in the bone microenvironment stimulates osteoclast differentiation, resulting in uncoupling of resorption and formation. Mechanisms contributing to the inhibition of osteoblast function in inflammatory diseases, however, have not been elucidated. Rheumatoid arthritis (RA) is a prototype of an inflammatory arthritis that results in focal loss of articular bone. The paucity of bone repair in inflammatory diseases such as RA raises compelling questions regarding the impact of inflammation on bone formation. The aim of this study was to establish the mechanisms by which inflammation regulates osteoblast activity.
Methods
We characterized an innovative variant of a murine model of arthritis in which inflammation is induced in C57BL/6J mice by transfer of arthritogenic K/B×N serum and allowed to resolve.
Results
In the setting of resolving inflammation, bone resorption ceased and appositional osteoblast-mediated bone formation was induced, resulting in repair of eroded bone. Resolution of inflammation was accompanied by striking changes in the expression of regulators of the Wnt/β-catenin pathway, which is critical for osteoblast differentiation and function. Down-regulation of the Wnt antagonists secreted frizzled-related protein 1 (sFRP1) and sFRP2 during the resolution phase paralleled induction of the anabolic and pro–matrix mineralization factors Wnt10b and DKK2, demonstrating the role of inflammation in regulating Wnt signaling.
Conclusion
Repair of articular bone erosion occurs in the setting of resolving inflammation, accompanied by alterations in the Wnt signaling pathway. These data imply that in inflammatory diseases that result in persistent articular bone loss, strict control of inflammation may not be achieved and may be essential for the generation of an anabolic microenvironment that supports bone formation and repair.
Inflammation-induced pathologic bone loss is characterized by activation of bone resorption and inhibition of bone formation. Although the stimulatory effects of inflammation on osteoclast-mediated bone resorption are well established (1–3), the impact of proinflammatory cytokines on osteoblast function in vivo requires further elucidation (4). Tumor necrosis factor (TNF) has been shown to suppress osteoblast maturation in vitro by inhibiting the expression of runt-related transcription factor 2 (RUNX-2) (5), a transcription factor that is responsible for osteoblast-lineage commitment and gene expression. As a result, expression of the RUNX-2 target genes type I collagen, alkaline phosphatase, and osteocalcin (OCN) is suppressed. Bone matrix deposition and mineralization are thus impaired by TNF (6). Interleukin-1β (IL-1β) elicits similar inhibitory responses in differentiating osteoblasts (7,8). Furthermore, proinflammatory cytokines disrupt the Wnt signaling pathway (9,10), an anabolic pathway that induces the differentiation and maturation of osteoblasts. In this pathway, a family of secreted agonists activate downstream signaling, stabilizing β-catenin, a transcriptional coactivator of osteoblast target genes, including RUNX2 (11,12). Regulation of Wnt signaling is maintained by a number of secreted antagonists, including members of the secreted frizzled-related protein (sFRP) and Dickkopf (DKK) families.
Inflammation-induced focal bone loss is perhaps best exemplified in rheumatoid arthritis (RA), a disease in which osteoclastic resorption leads to the development of articular bone erosions. In the inflamed microenvironment of the RA joint, osteoblast maturation and function are compromised. Studies have shown that although osteoblasts are located in the vicinity of focal articular bone erosions in RA (13) and murine inflammatory arthritis, few mature osteocalcin-expressing osteoblasts are directly associated with eroded bone surfaces (14,15). In the serum transfer model of arthritis, a murine model of inflammatory arthritis, the paucity of alkaline phosphatase and OCN-expressing osteoblasts at erosion sites correlates with limited bone formation (15). Furthermore, up-regulated expression of Wnt signaling antagonists has been implicated in the suppression of osteoblast activity during inflammation-induced bone loss (9,15).
With aggressive treatment of inflammation in RA, bone resorption is suppressed. Although osteoblast-mediated repair of bone erosions occurs, it is infrequent (16,17), and when repair is observed, it correlates with well-controlled clinical disease (18,19). This observation suggests that in patients in whom repair is not detected, subclinical inflammation in the joint may persist, suppressing erosion repair by osteoblasts. We therefore hypothesized that resolution of inflammation would stimulate osteoblast function and ultimately result in the repair of established focal bone erosions. To address this hypothesis, we utilized an innovative variant of the serum transfer model of arthritis, in which inflammation was induced and subsequently allowed to resolve. Using this model, we determined the capacity of osteoblast-lineage cells to recover from inflammation-induced suppression of function and subsequently form bone at erosion sites. For the first time, we show that resolution of inflammation is accompanied by a significant increase in bone formation at previous inflammation–bone interfaces, correlating with altered synovial expression of Wnt signaling components that favor anabolic signaling.
Materials and Methods
K/B×N murine serum transfer model of inflammatory arthritis
All animal procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Massachusetts Medical School. KRN T cell–transgenic mice (provided by Drs. O. Benoist and D. Mathis, Harvard Medical School and the Institut de Genetique et de Biologie Moleculaire et Cellulaire, Illkirch, France) were crossed with NOD/ShiLtJ mice (The Jackson Laboratory) to generate K/B×N mice in which arthritis develops spontaneously (20,21). At 60 days of age, arthritogenic serum was obtained and pooled for studies, as previously described (2,15).
Serum transfer arthritis was induced in 12-week-old male C57BL/6J mice (The Jackson Laboratory) by intraperitoneal injection of 150 μl of arthritogenic serum on days 0, 2, and 7. Nonarthritic mice received 150 μl of sterile phosphate buffered saline at each time point. The method for scoring clinical inflammation was adapted from a previously described scoring system (22). For each paw, a score from 0 to 3 was assigned based on the following criteria: 0 = normal, 1 = slight swelling on the top and bottom of the paw, 2 = obvious swelling that obscures bone landmarks, and 3 = severe swelling that further obscures bone landmarks. Scores of the 4 paws were summed, and the average clinical score for each group was reported.
Histopathologic analyses
For histologic scoring of inflammation and bone erosion, sections were stained with either hematoxylin and eosin or tartrate-resistant acid phosphatase (TRAP). For TRAP staining, sections were incubated in 0.1M Tris HCl, pH 9.0, for 18 hours, followed by 0.1M sodium citrate, pH 5.2, for 3 hours (23). Activated sections were incubated for 30 minutes at 37°C in 0.005% Napthol AS-MX phosphate (Sigma)/0.01% N,N-dimethylformamide/0.03% Fast Red Violet LB salt (Sigma)/50 mM sodium tartrate in 0.1M acetate, pH 5.0 (24,25) and counterstained with hematoxylin. Slides were scored by 2 independent observers (MMM and EMG) using a previously defined histopathologic scoring criteria (2). The data are presented as the average of the scores of both observers. As previously described (15), digoxigenin-labeled antisense and sense riboprobes specific for alkaline phosphatase and OCN messenger RNA (mRNA) were synthesized and used to perform in situ hybridization on serial tissue sections.
Micro–computed tomography (micro-CT)
The hind paws of the mice were imaged at the Musculoskeletal Imaging Core at the University of Massachusetts Medical School, using a Scanco Medical μCT 40 with an isotropic voxel size of 8 μm, at 70 kVp and 114 μA. Images were reconstructed in 1,024 × 1,024–pixel matrices. A threshold of 220–1,000 Hounsfield units was used for segmentation. Axial slices were reformatted to lateral slices and are shown at 3× magnification.
Dynamic histomorphometric analysis
For histomorphometric analysis of in vivo bone formation, mice were administered 50 mg/kg of alizarin and/or 30 mg/kg of calcein by intraperitoneal injection. For quantitative analysis, mice were given alizarin 13 days prior to being killed and calcein 3 days prior to being killed (10-day interlabel period). Mice killed on day 10 received alizarin on day 5. As previously shown (15), the navicular bone of the midfoot is a reproducible site of bone erosion in the serum transfer model of arthritis, and as such, this site was chosen for quantitative measurements. Specifically, cortical and endosteal surfaces within the navicular bone where pannus and inflammatory tissue invaded were analyzed separately from surfaces contacting normal marrow/connective tissue. Following the resolution of active inflammation, the appearance of abnormal marrow, loss of adipocytic structure, and tissue fibrosis were used to identify areas where inflammatory tissue had been present earlier during the course of arthritis. The mineralized surface/bone surface (MS/BS), mineral apposition rate (MAR), and bone formation rate/bone surface (BFR/BS) were determined as previously described (15), using the guidelines established by Parfitt et al (26). For the nonarthritic group, data for mice killed on days 10, 28, 38, and 48 (n = 15 total) were combined.
Synovial gene expression
Synovial and associated soft tissue were isolated from the region of the tibiotalar joint and processed for RNA, as previously described (15). Total RNA (150 ng) was used for iScript complementary DNA (cDNA) synthesis (Bio-Rad). Quantitative reverse transcription–polymerase chain reaction (RT-PCR) analysis was performed on diluted cDNA samples using iScript SYBR Green RT-PCR mix (Bio-Rad) and an Eppendorf realplex quantitative PCR system. All primers were obtained from Qiagen, except for sFRP1 primers, which were obtained from Eurofins MWG Operon. Gene expression was normalized to the expression of HMBS, a housekeeping gene, and data are expressed as the fold increase in gene expression in arthritic mice compared with that in nonarthritic mice averaged over the course of the experimental time points (n = 7–8 mice in each group per time point), using the 2−ΔΔCt method (27). Gene expression in the nonarthritic controls was stable over time in comparison with HMBS, and similar variability in gene expression among individual mice was observed at each time point.
Serum measurements
TRAP-5b levels were determined by activity assay (Immunodiagnostic Systems) according to the manufacturer's instructions.
Statistical analysis
Statistical analysis of the quantitative RT-PCR data was performed using a type III F test of homogeneity from analysis of variance (ANOVA) modeling to determine if differences existed among the days. For genes with a P value of ≤ 0.05, the relative expression on day 10 was tested against the relative expression on subsequent days, using a standard 2-group t-test assuming independent observations. An informal Bonferroni correction for significance (0.05/5 = 0.01) was used. Outliers (defined as the mean ± 3SD) were removed prior to statistical analysis.
ANOVA with a Dunnett's post-test was used to determine statistical significance for the histomorphometry data, where samples from the nonarthritic controls served as the comparator control. Student's unpaired t-tests were used to calculate statistical significance for histology scores and TRAP-5b data.
Results
Association between resolution of inflammation, proinflammatory cytokine expression, and osteoclastogenesis
To evaluate the effects of abating inflammation on osteoblast activity, we characterized a variant of the serum transfer model of arthritis (20), in which inflammation was induced with 3 injections of arthritogenic serum and subsequently allowed to decline. After administration of arthritogenic serum on days 0, 2, and 7, clinical signs of inflammation developed rapidly (Figure 1A), peaking on day 10. Synovial tissue isolated from the tibiotalar joint of arthritic mice showed an up-regulation of IL-1β and TNF mRNA at the peak of clinical inflammation (Figures 1B and C). In arthritic mice, the expression of IL-1β mRNA was up-regulated ∼20-fold, and expression of TNF mRNA was up-regulated ∼1.5-fold compared with nonarthritic controls, consistent with the dominance of IL-1β as the critical cytokine in this model (28). Up-regulation of proinflammatory cytokines was accompanied by significant infiltration of inflammatory cells into the joint, synovial hyperplasia, and invasion of pannus into the marrow cavities of the navicular bone (Figure 2A).
Figure 1.
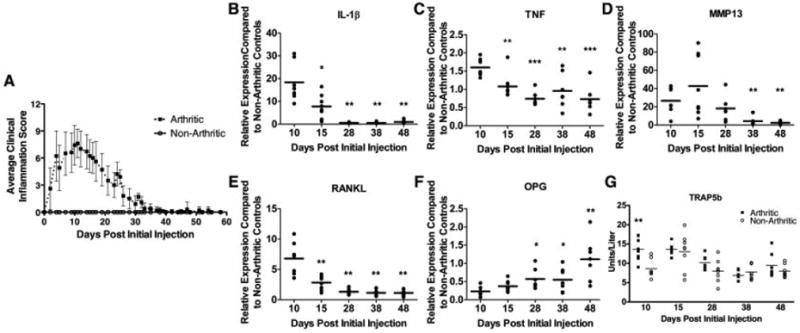
Discontinuation of arthritogenic serum injections promotes resolution of proinflammatory cytokine expression and osteoclastogenesis. A, Clinical inflammation scores of arthritic and nonarthritic mice. Values are the mean ± SD of 3 independent experiments (n = 7–8 mice per time point). B–F, Expression of interleukin-1β (IL-1β) (B), tumor necrosis factor (TNF) (C), matrix metalloproteinase 13 (MMP-13) (D), RANKL (E), and osteoprotegerin (OPG) (F) in synovial tissue as determined by quantitative reverse transcription–polymerase chain reaction (n = 7–8 mice per time point). * = P < 0.05; ** = P < 0.01; *** = P < 0.001 versus day 10. G, Serum tartrate-resistant acid phosphatase 5b (TRAP-5b) levels in arthritic and nonarthritic mice combined from 2 independent experiments (n = 7–8). ** = P < 0.01 versus nonarthritic controls on day 10. Each symbol represents the mean of duplicate samples for a single mouse. Horizontal lines represent the group means.
Figure 2.
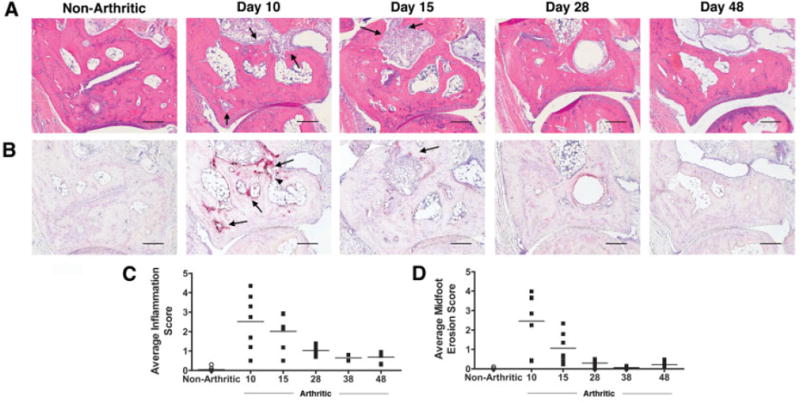
Histologic evaluation demonstrates resolution of inflammation and osteoclast-mediated bone resorption. A and B, Representative serial hematoxylin and eosin–stained sections (A) and tartrate-resistant acid phosphatase–stained sections (B) of the navicular bone of nonarthritic mice (left panel) and arthritic mice (right panels) (n = 3 independent experiments). Arrows denote pannus tissue and inflammation in A and osteoclasts in B. The arrowhead indicates a full-thickness cortical defect. Bars = 200 μm. C and D, Histologic quantification of inflammation (C) and midfoot erosion (D) in arthritic mice and nonarthritic mice. Each symbol represents the mean histologic score per mouse (n = 7–8 mice per time point). Horizontal lines represent the group means.
Synovitis promotes osteoclast-mediated articular bone erosion through the up-regulation of RANKL (2) and reproducibly generates cortical and endosteal bone erosions in the navicular bone (15). As expected, RANKL mRNA was induced in the synovium of arthritic mice by day 10 (Figure 1E). In contrast, mRNA expression of osteoprotegerin (OPG), a soluble decoy receptor that binds RANKL and inhibits osteoclastogenesis, was down-regulated by the inflammatory process (Figure 1F). As a result, the RANKL:OPG ratio was markedly elevated on day 10, leading to a significant increase in osteoclast numbers, as measured by serum TRAP-5b levels (Figure 1G) and osteoclast-mediated bone erosion (Figures 2B and D). By day 10, the presence of TRAP-positive osteoclasts on the endosteal and cortical surfaces of the navicular bone was associated with full-thickness cortical defects (Figure 2B). The expression of matrix metalloproteinase 13 (MMP-13), which is implicated in cartilage destruction and plays a role in preparation of bone surfaces for osteoclast-mediated resorption (29), was also induced by the inflammatory process (Figure 1D).
With discontinuation of arthritogenic serum injections, clinical inflammation progressively decreased from day 10 onward (Figure 1A). The expression of IL-1β and TNF mRNA in synovial tissue decreased rapidly and returned to the levels in nonarthritic controls (Figures 1B and C), resulting in little cellular infiltration in the synovium and decreased synovial thickening by day 28 (Figure 2). A decline in proinflammatory cytokine expression was accompanied by a precipitous down-regulation of RANKL mRNA and up-regulation of OPG mRNA (Figures 1E and F). The resulting reversal in the RANKL:OPG ratio correlated with a reduction in the osteoclast number and a marked decrease in active resorption of bone by day 15 (Figure 2B). During the repair and remodeling phase from day 28 onward, the absence of IL-1β, TNF, and RANKL mRNA expression paralleled the absence of cellular infiltrates and osteoclast activity, with a slightly thickened synovium remaining (Figures 2A and B). Furthermore, bone marrow cavities previously penetrated by inflammatory tissue were now devoid of normal marrow components and adipocytes (Figure 2A).
The impact of resolving inflammation on bone architecture was evaluated by micro-CT. By day 15, endosteal and cortical erosion of the ankle and midfoot bones was clearly evident, resulting in roughened, irregular bone surfaces. Consistent with the histologic data, full-thickness defects in the midfoot bones were apparent (Figure 3, arrows). By day 38, with the complete resolution of inflammation, the bone architecture of arthritic mice was similar to that of nonarthritic control mice.
Figure 3.
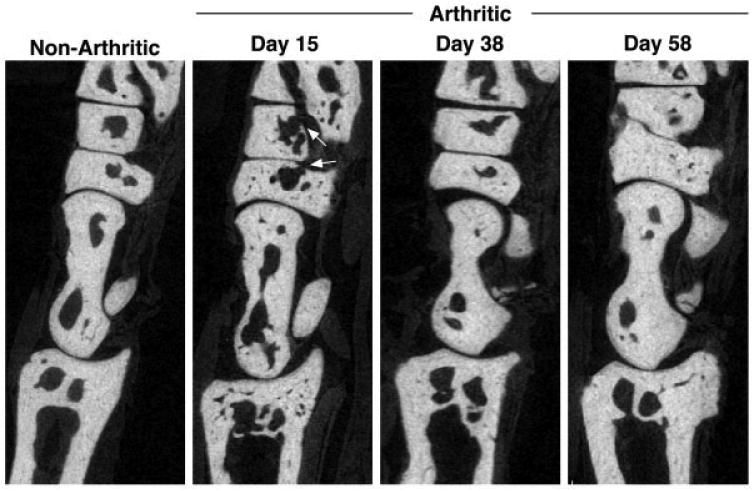
Restoration of cortical and trabecular bone architecture correlates with resolution of inflammation. Sagittal images of the ankle and midfoot bones are shown. Arrows indicate full cortical breaks in the midfoot bones due to osteoclast-mediated erosion. At each time point, 3–4 arthritic mice and 4 nonarthritic mice were examined. Original magnification × 3.
Thus, using quantitative molecular and morphologic parameters, we characterized 3 reproducible phases in this model of inflammation resolution, including: 1) an inflammatory phase (days 0–10) exemplified by expression of proinflammatory cytokines and RANKL that induce bone erosion, 2) a resolution phase (days 10–28) of abating proinflammatory cytokine and catabolic factor expression leading to declining osteoclastogenesis, and 3) a remodeling and repair phase (day 28 onward).
Effect of resolution of inflammation on osteoblast-mediated bone formation and erosion repair
Given the apparently normal bone architecture observed on days 38 and 58 by micro-CT, we hypothesized that the reduction of inflammation promoted osteoblast-mediated bone formation. To quantify the extent of osteoblast activity, in vivo fluorochrome incorporation at inflammation–bone interfaces was assessed using dynamic histomorphometry. Little incorporation of alizarin was evident at inflammation–bone interfaces in the navicular bone on day 10, when peak inflammation was observed (Figure 4A). Quantification on day 10 revealed a slight decrease in MS/BS in arthritic mice compared with nonarthritic controls (Figure 4B). In contrast, with resolution of inflammation, significant alizarin and calcein incorporation was observed at inflammation–bone interfaces on days 28, 38, and 48 (Figure 4A). At these time points, a statistically significant increase in MS/BS in arthritic mice compared with nonarthritic controls indicated an increase in the mineralizing activity of osteoblasts at erosion sites (Figure 4B). In addition, there was a statistically significant increase in the MAR, a measure of the activity of individual osteoblasts (Figure 4C), and BFR/BS, a value calculated from the MAR and MS/BS (Figure 4D). The MAR and BFR could not be calculated on day 10, because only a single label (alizarin) can be administered within this time period.
Figure 4.
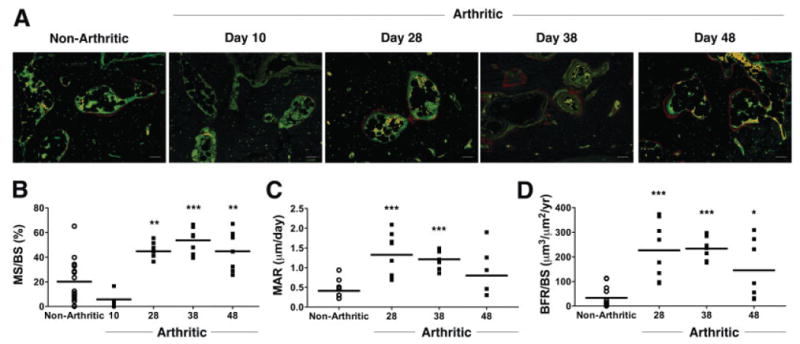
Quantification of bone histomorphometric parameters demonstrates increased bone formation with inflammation resolution. A, Representative images of alizarin (red) and calcein (green) incorporation in 2 independent experiments are shown. Mice killed on day 10 received a single injection of alizarin on day 5. For quantitative analysis, mice were given alizarin 13 days prior to being killed and calcein 3 days prior to being killed. Bars = 100 μm. B–D, Fluorochrome incorporation at navicular erosion sites was used to quantify the bone formation parameters mineralized surface/total bone surface (MS/BS) (B), mineral apposition rate (MAR) (C), and bone formation rate/total bone surface (BFR/BS) (D). Each symbol represents a single nonarthritic mouse (n = 15) or a single arthritic mouse (n = 4 on day 10, n = 7–8 on days 28, 38, and 48). Horizontal lines represent the means. * = P < 0.05; ** = P < 0.01; *** = P < 0.001 versus nonarthritic controls.
To observe bone formation over time, 2 fluorochromes were administered at alternating intervals every 10 days throughout the course of arthritis, resulting in a total of 6 fluorochrome injections over the course of 58 days (3 alizarin, 3 calcein). Strikingly, multiple layers of fluorochrome incorporation were observed over the 58-day period following the induction of arthritis, indicating that bone formation was a dynamic process that occurred throughout the resolution and remodeling phases (results not shown).
The presence of mature osteoblasts at inflammation–bone interfaces paralleled the increase in bone formation that occurred with resolution of inflammation. At the time of peak inflammation (day 10), there was limited to no expression of alkaline phosphatase, a mid-stage osteoblast marker, or OCN, a late-stage osteoblast marker, by cells along the inflammation–bone interfaces (Figures 5A and B). However, as inflammation abated during the resolution phase, there was a progressive increase in the number of osteoblasts expressing alkaline phosphatase and OCN, as well as an increase in the number of these cells lining the eroded endosteal bone surfaces. Alkaline phosphatase–expressing cells were observed beginning on day 15, followed by an abundance of osteoblasts expressing OCN on endosteal bone surfaces as well as some eroded cortical surfaces by day 21. From day 28 onward, there were fewer alkaline phosphatase– and OCN-expressing osteoblasts and surfaces lined by these cells. Taken together, these results demonstrate that osteoblasts populate bone surfaces and synthesize bone in parallel with declining inflammation.
Figure 5.
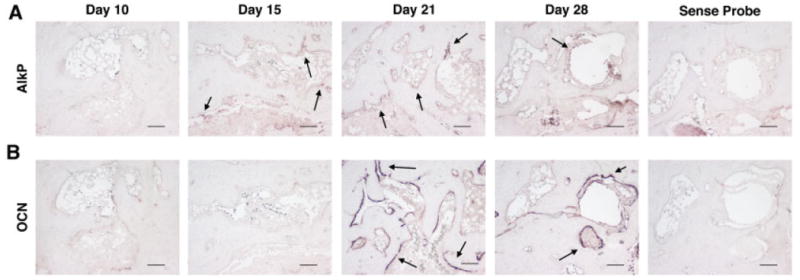
Mature osteoblasts are identified at eroded surfaces during the resolution phase of inflammation. Histologic sections show in situ hybridization for alkaline phosphatase (AlkP) (A) and osteocalcin (OCN) (B) mRNA expression at eroded bone surfaces in the navicular bone of arthritic mice (n = 3–4 mice per time point in 2 independent experiments). Positive cells are identified by purple staining. Arrows denote areas of mRNA expression. Hybridization with sense probes was used as the negative control. Bars = 100 μm.
Alterations in expression of Wnt signalling factors with resolution of inflammation
To address mechanisms related to osteoblast maturation and bone formation at eroded bone surfaces, we investigated the impact of inflammation on the expression of antagonists and agonists of the Wnt signaling pathway. Within the synovial tissue of arthritic mice, induction of sFRP1 and sFRP2 mRNA expression was observed in relation to nonarthritic controls on day 10 (Figures 6A and B). The timing of this expression corresponded to the inflammatory phase of the model, when inflammation peaks and bone formation is suppressed. The expression of sFRP1 and sFRP2 mRNA subsequently declined as inflammation resolved, until day 28, when the expression levels in arthritic mice were similar to those in nonarthritic controls. In contrast, DKK1 mRNA was detected later in the inflammatory process, with peak ∼2-fold expression occurring on days 28 and 38 (Figure 6C).
Figure 6.
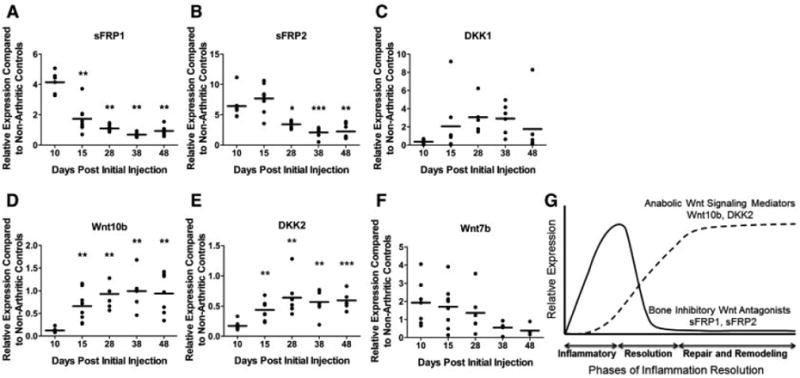
Resolution of inflammation and erosion repair parallel alterations in Wnt antagonist and agonist mRNA expression. A–F, Synovial expression of secreted frizzled-related protein 1 (sFRP1) (A), sFRP2 (B), DKK1 (C), Wnt10b (D), DKK2 (E), and Wnt7b (F) mRNA. Gene expression was calculated relative to the average gene expression in nonarthritic controls (n = 7–8 mice per time point). Each symbol represents the mean of duplicate samples for a single mouse. Horizontal lines represent the group means. * = P < 0.05; ** = P < 0.01; *** = P < 0.001 versus day 10. G, Expression patterns of Wnt antagonists and agonists over the course of inflammation, resolution, and remodeling.
We further investigated the ability of declining inflammation to promote the expression of anabolic Wnt ligands. As inflammation declined, the mRNA expression of Wnt10b, a Wnt agonist that promotes osteoblastogenesis and in vivo bone formation (30), was up-regulated in arthritic mice compared with that in nonarthritic controls (Figure 6D). Although mRNA levels were low, we also examined the trends of DKK2 and Wnt7b expression in this model. Synovial mRNA expression of DKK2, a Wnt antagonist that promotes matrix mineralization, was suppressed at the peak of inflammation and bone erosion (day 10). In contrast, expression increased during the resolution phase of inflammation (Figure 6E). Up-regulation of DKK2 mRNA correlated with a reciprocal down-regulation of the canonical Wnt signaling agonist, Wnt7b (Figure 6F). A similar reciprocal relationship between DKK2 and Wnt7b expression in osteoblasts has been reported previously (31). These results demonstrate that although inflammation induces expression of Wnt antagonists that inhibit osteoblast differentiation, the reduction in inflammation up-regulates anabolic and pro–matrix mineralization Wnt signaling factors (Figure 6G).
Discussion
Pathologic bone loss in inflammatory diseases is characterized by an imbalance of osteoclast-mediated bone resorption relative to osteoblast-mediated bone formation, resulting in a net loss of bone. We previously showed in a murine model of inflammatory arthritis that the presence of inflammation at sites of articular bone erosion correlated with limited osteoblast activity (15). This observation demonstrated that inflammation impairs the ability of osteoblasts to form bone and raised the question as to whether resolution of inflammation would promote osteoblast-mediated bone formation. We adapted the serum transfer model of arthritis, based on the original observations by Korganow et al (20), in order to establish the phases of inflammation-induced bone erosion repair and demonstrated that in the setting of declining inflammation, bone formation occurs, leading to repair of focal articular bone loss.
By elucidating the temporal sequence of bone formation relative to the inflammatory process in this model, an understanding of the key events leading to inflammation-induced bone loss and repair was established. The inflammatory phase was characterized by decreased mineralized bone surface and by a paucity of alkaline phosphatase− and OCN-expressing osteoblasts at eroded surfaces, consistent with our previous studies (15). During the resolution phase in the present study, osteoblasts prominently expressing OCN populated previous inflammation–bone interfaces, in particular endosteal surfaces, coincident with a significant increase in MS/BS, MAR, and BFR. This increase in MS/BS, MAR, and BFR persisted through the repair and remodeling phase, demonstrating that resolving inflammation stimulates repair of bone loss.
Repair of inflammation-induced bone erosion in this model is a result of both an increase in osteoblast activity and a reciprocal decrease in osteoclast-mediated bone resorption. The removal of arthritogenic stimuli results in a decrease in expression of proinflammatory cytokines and RANKL, leading to a decrease in osteoclast formation and bone resorption. Activation of the canonical Wnt/β-catenin pathway has been shown to up-regulate OPG expression to inhibit osteoclast differentiation (32,33). Our data demonstrate an increase in synovial OPG expression concurrent with the decline in osteoclast formation with resolving inflammation, consistent with activation of this pathway.
Previously, synovial fibroblasts in RA have been shown to express inhibitors of the Wnt signaling pathway, including DKK1, sFRP1, sFRP3, and sFRP4 (9,34,35). There is also increasing evidence implicating the Wnt antagonists, DKK1 (9) and sFRP1 (15,36), as factors that suppress osteoblast activity in models of inflammatory bone loss. This study establishes that expression of both sFRP1 and sFRP2 is induced with inflammation and declines with decreasing inflammation.
Restoration of osteoblast activity following the decline of inflammation in this study clearly demonstrates that inflammation perturbs the balance of anabolic bone signaling, with profound effects on the Wnt signaling pathway. Elevated DKK1 expression has been associated with inflammatory bone loss and suppression of osteoblast activity in RA (9) as well as in multiple myeloma (37), while prophylactic DKK1 blockade stimulated periosteal osteophyte formation in the human TNFα–transgenic (hTNF-Tg) murine model of RA (9). DKK1 is expressed in the synovium of arthritic hTNF-Tg mice and is up-regulated in cultured human RA and murine synovial fibroblasts. In the current study, we observed that synovial DKK1 mRNA expression was not significantly elevated at the time point examined during the inflammatory phase of serum transfer–induced arthritis (day 10) but trended upward later in the course of arthritis (days 28 and 38), during the phase of resolving inflammation. However, as previously demonstrated, when more robust inflammation was induced in this model following 4 injections of arthritogenic serum, an up-regulation of DKK1 mRNA was seen during the period of peak inflammation (15). In the model of serum transfer–induced arthritis, IL-1β is a predominant proinflammatory cytokine (28), and as such, a pattern of Wnt antagonist expression different from that seen in the hTNF-Tg model may ensue. In addition, in the serum transfer model of arthritis, expression of DKK1 mRNA is low; therefore, it is difficult to draw conclusions about the impact of DKK1 on osteoblast function following the resolution of inflammation in this study.
Regulated Wnt agonist expression directs vertebrate limb and synovial joint development (38,39) and as such may promote bone formation. In this study, bone formation correlated with increasing synovial expression of Wnt10b, a canonical Wnt agonist. Overexpression of Wnt10b was shown to increase bone mass and the trabecular number by increasing osteoblast number and osteoblast activity, while deletion of Wnt10b led to a decrease in bone formation (30). In this study, the up-regulation of Wnt10b in the synovium following resolution of inflammation is a likely mechanism contributing to bone formation.
The expression of Wnt7b is down-regulated with declining inflammation. Previous studies have demonstrated that Wnt7b mRNA is expressed by trabecular osteoblasts in vivo (40), and that expression peaks early during osteoblast differentiation in vitro (31,40), suggesting that Wnt7b may promote bone formation. Importantly, down-regulation of Wnt7b expression in osteoblasts correlates with induction of expression of DKK2, a Wnt antagonist that promotes osteoblast maturation and mineralization of newly synthesized bone matrix (31). Indeed, in this study, we observed a similar inverse relationship between Wnt7b and DKK2 expression in synovial tissue, which was regulated by the presence of inflammation. With resolution of inflammation, decreasing Wnt7b expression correlates with the expression of DKK2 by osteoblasts recruited to the sites of bone erosion, which could subsequently promote osteoblast maturation and direct mineralization of the newly formed bone at erosion sites.
These findings provide insight into an important conundrum in clinical disease, the relative lack of repair of articular erosions in RA patients in whom clinical parameters suggest excellent disease control or even remission. Therapy with potent antiinflammatory drugs such as anti-TNF agents has been shown to slow or halt the progression of articular erosion, but repair of erosions is still limited, even when sensitive radiographic techniques such as high-resolution computed tomography are employed to identify repair (41). Our findings in this murine model of RA demonstrate that if inflammation is controlled, bone formation and repair of erosions occur and suggest that in clinical disease in which repair is not seen, inflammation in synovial tissues likely persists. Recent clinical data are consistent with this notion. When studying patients with RA who were receiving disease-modifying antirheumatic drug therapy and in whom disease was deemed to be in clinical remission, Brown et al demonstrated that subclinical inflammation could be identified by the sensitive techniques of magnetic resonance imaging (MRI) and musculoskeletal ultrasonography. Furthermore, a direct association between ongoing synovitis and structural progression was shown (42,43). In addition, in studies of patients with disease judged to be in clinical remission, MRI, as well as synovial biopsies combined with ultrasonography or MRI, demonstrated the persistence of synovitis (44,45). Our findings, however, cannot exclude the possibility that other environmental, cellular, and/or epigenetic changes could have occurred that alter Wnt antagonist and agonist expression independent of the requirement of ongoing inflammation.
In conclusion, these studies in a murine model of resolving inflammation provide a direct demonstration that reduced inflammation within the bone microenvironment favors the restoration of osteoblast function and the repair of focal bone erosions. Furthermore, by altering the expression of Wnt antagonists and agonists in the synovium, inflammation significantly contributes to the regulation of Wnt signaling, a pathway that ultimately determines the fate of bone. Therefore, aggressive control of synovial inflammation and close monitoring of synovitis with sensitive radiologic techniques may be required in order to stimulate bone formation and restore bone structural integrity in diseases involving inflammation-induced bone loss.
Acknowledgments
We would like to thank Dr. Sougata Karmakar and Teresa Bowman for technical assistance, Dr. Bruce Barton for conducting the statistical analysis, and Dr. Tripti Gaur for critical discussions.
Ms Matzelle's work was supported by an Abbott Bioresearch Fellowship in Translation Science. Dr. Gravallese's work was supported by the American College of Rheumatology Research and Education Foundation (Within Our Reach Program grant).
Footnotes
Author Contributions: All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Gravallese had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Matzelle, Walsh, Burr, Gravallese.
Acquisition of data. Matzelle, Gallant, Condon, Manning, Burr, Gravallese.
Analysis and interpretation of data. Matzelle, Gallant, Walsh, Stein, Lian, Burr, Gravallese.
References
- 1.Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, et al. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000;43:250–8. doi: 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 2.Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, et al. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001;159:1689–99. doi: 10.1016/S0002-9440(10)63016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Redlich K, Hayer S, Ricci R, David JP, Tohidast-Akrad M, Kollias G, et al. Osteoclasts are essential for TNF-α-mediated joint destruction. J Clin Invest. 2002;110:1419–27. doi: 10.1172/JCI15582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walsh NC, Gravallese EM. Bone remodeling in rheumatic disease: a question of balance. Immunol Rev. 2010;233:301–12. doi: 10.1111/j.0105-2896.2009.00857.x. [DOI] [PubMed] [Google Scholar]
- 5.Gilbert L, He X, Farmer P, Rubin J, Drissi H, van Wijnen AJ, et al. Expression of the osteoblast differentiation factor RUNX2 (Cbfa1/AML3/Pebp2α A) is inhibited by tumor necrosis factor-α. J Biol Chem. 2002;277:2695–701. doi: 10.1074/jbc.M106339200. [DOI] [PubMed] [Google Scholar]
- 6.Nanes MS. Tumor necrosis factor-α: molecular and cellular mechanisms in skeletal pathology. Gene. 2003;321:1–15. doi: 10.1016/s0378-1119(03)00841-2. [DOI] [PubMed] [Google Scholar]
- 7.Stashenko P, Dewhirst FE, Rooney ML, Desjardins LA, Heeley JD. Interleukin-1 β is a potent inhibitor of bone formation in vitro. J Bone Miner Res. 1987;2:559–65. doi: 10.1002/jbmr.5650020612. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen L, Dewhirst FE, Hauschka PV, Stashenko P. Interleukin-1 β stimulates bone resorption and inhibits bone formation in vivo. Lymphokine Cytokine Res. 1991;10:15–21. [PubMed] [Google Scholar]
- 9.Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, et al. Dickkopf-1 is a master regulator of joint remodeling. Nat Med. 2007;13:156–63. doi: 10.1038/nm1538. [DOI] [PubMed] [Google Scholar]
- 10.Vincent C, Findlay DM, Welldon KJ, Wijenayaka AR, Zheng TS, Haynes DR, et al. Pro-inflammatory cytokines TNF-related weak inducer of apoptosis (TWEAK) and TNFα induce the mitogen-activated protein kinase (MAPK)-dependent expression of sclerostin in human osteoblasts. J Bone Miner Res. 2009;24:1434–49. doi: 10.1359/jbmr.090305. [DOI] [PubMed] [Google Scholar]
- 11.Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS, et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem. 2005;280:33132–40. doi: 10.1074/jbc.M500608200. [DOI] [PubMed] [Google Scholar]
- 12.Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202–9. doi: 10.1172/JCI28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gravallese EM, Harada Y, Wang JT, Gorn AH, Thornhill TS, Goldring SR. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am J Pathol. 1998;152:943–51. [PMC free article] [PubMed] [Google Scholar]
- 14.Zwerina J, Tuerk B, Redlich K, Smolen JS, Schett G. Imbalance of local bone metabolism in inflammatory arthritis and its reversal upon tumor necrosis factor blockade: direct analysis of bone turnover in murine arthritis. Arthritis Res Ther. 2006;8:R22. doi: 10.1186/ar1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walsh NC, Reinwald S, Manning CA, Condon KW, Iwata K, Burr DB, et al. Osteoblast function is compromised at sites of focal bone erosion in inflammatory arthritis. J Bone Miner Res. 2009;24:1572–85. doi: 10.1359/jbmr.090320. [DOI] [PubMed] [Google Scholar]
- 16.Rau R, Wassenberg S, Herborn G, Perschel WT, Freitag G. Identification of radiologic healing phenomena in patients with rheumatoid arthritis. J Rheumatol. 2001;28:2608–15. [PubMed] [Google Scholar]
- 17.Sharp JT, van der Heijde D, Boers M, Boonen A, Bruynesteyn K, Emery P, et al. Repair of erosions in rheumatoid arthritis does occur: results from 2 studies by the OMERACT Subcommittee on Healing of Erosions. J Rheumatol. 2003;30:1102–7. [PubMed] [Google Scholar]
- 18.Sokka T, Hannonen P. Healing of erosions in rheumatoid arthritis. Ann Rheum Dis. 2000;59:647–9. doi: 10.1136/ard.59.8.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ideguchi H, Ohno S, Hattori H, Senuma A, Ishigatsubo Y. Bone erosions in rheumatoid arthritis can be repaired through reduction in disease activity with conventional disease-modifying antirheumatic drugs. Arthritis Res Ther. 2006;8:R76. doi: 10.1186/ar1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–61. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- 21.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–22. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 22.Ohmura K, Nguyen LT, Locksley RM, Mathis D, Benoist C. Interleukin-4 can be a key positive regulator of inflammatory arthritis. Arthritis Rheum. 2005;52:1866–75. doi: 10.1002/art.21104. [DOI] [PubMed] [Google Scholar]
- 23.Sankaramanivel S, Jeyapriya R, Hemalatha D, Djody S, Arunakaran J, Srinivasan N. Effect of chromium on vertebrae, femur and calvaria of adult male rats. Hum Exp Toxicol. 2006;25:311–8. doi: 10.1191/0960327105ht627oa. [DOI] [PubMed] [Google Scholar]
- 24.Lee SK, Goldring SR, Lorenzo JA. Expression of the calcitonin receptor in bone marrow cell cultures and in bone: a specific marker of the differentiated osteoclast that is regulated by calcitonin. Endocrinology. 1995;136:4572–81. doi: 10.1210/endo.136.10.7664679. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi N, Udagawa N, Tanaka S, Suda T. Generating murine osteoclasts from bone marrow. Methods Mol Med. 2003;80:129–44. doi: 10.1385/1-59259-366-6:129. [DOI] [PubMed] [Google Scholar]
- 26.Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Ji H, Pettit A, Ohmura K, Ortiz-Lopez A, Duchatelle V, Degott C, et al. Critical roles for interleukin 1 and tumor necrosis factor α in antibody-induced arthritis. J Exp Med. 2002;196:77–85. doi: 10.1084/jem.20020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holliday LS, Welgus HG, Fliszar CJ, Veith GM, Jeffrey JJ, Gluck SL. Initiation of osteoclast bone resorption by interstitial collagenase. J Biol Chem. 1997;272:22053–8. doi: 10.1074/jbc.272.35.22053. [DOI] [PubMed] [Google Scholar]
- 30.Bennett CN, Ouyang H, Ma YL, Zeng Q, Gerin I, Sousa KM, et al. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J Bone Miner Res. 2007;22:1924–32. doi: 10.1359/jbmr.070810. [DOI] [PubMed] [Google Scholar]
- 31.Li X, Liu P, Liu W, Maye P, Zhang J, Zhang Y, et al. Dkk2 has a role in terminal osteoblast differentiation and mineralized matrix formation. Nat Genet. 2005;37:945–52. doi: 10.1038/ng1614. [DOI] [PubMed] [Google Scholar]
- 32.Glass DA, 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–64. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 33.Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere MC, Bouxsein ML, et al. Essential role of β-catenin in postnatal bone acquisition. J Biol Chem. 2005;280:21162–8. doi: 10.1074/jbc.M501900200. [DOI] [PubMed] [Google Scholar]
- 34.Imai K, Morikawa M, D'Armiento J, Matsumoto H, Komiya K, Okada Y. Differential expression of WNTs and FRPs in the synovium of rheumatoid arthritis and osteoarthritis. Biochem Biophys Res Commun. 2006;345:1615–20. doi: 10.1016/j.bbrc.2006.05.075. [DOI] [PubMed] [Google Scholar]
- 35.Ijiri K, Nagayoshi R, Matsushita N, Tsuruga H, Taniguchi N, Gushi A, et al. Differential expression patterns of secreted frizzled related protein genes in synovial cells from patients with arthritis. J Rheumatol. 2002;29:2266–70. [PubMed] [Google Scholar]
- 36.Li CH, Amar S. Inhibition of SFRP1 reduces severity of periodontitis. J Dent Res. 2007;86:873–7. doi: 10.1177/154405910708600913. [DOI] [PubMed] [Google Scholar]
- 37.Heath DJ, Chantry AD, Buckle CH, Coulton L, Shaughnessy JD, Jr, Evans HR, et al. Inhibiting Dickkopf-1 (Dkk1) removes suppression of bone formation and prevents the development of osteolytic bone disease in multiple myeloma. J Bone Miner Res. 2009;24:425–36. doi: 10.1359/jbmr.081104. [DOI] [PubMed] [Google Scholar]
- 38.Guo X, Day TF, Jiang X, Garrett-Beal L, Topol L, Yang Y. Wnt/β-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev. 2004;18:2404–17. doi: 10.1101/gad.1230704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamagami T, Molotkov A, Zhou CJ. Canonical Wnt signaling activity during synovial joint development. J Mol Histol. 2009;40:311–6. doi: 10.1007/s10735-009-9242-1. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–7. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 41.Finzel S, Rech J, Schmidt S, Engelke K, Englbrecht M, Stach C, et al. Repair of bone erosions in rheumatoid arthritis treated with tumour necrosis factor inhibitors is based on bone apposition at the base of the erosion. Ann Rheum Dis. 2011;70:1587–93. doi: 10.1136/ard.2010.148395. [DOI] [PubMed] [Google Scholar]
- 42.Brown AK, Quinn MA, Karim Z, Conaghan PG, Peterfy CG, Hensor E, et al. Presence of significant synovitis in rheumatoid arthritis patients with disease-modifying antirheumatic drug–induced clinical remission: evidence from an imaging study may explain structural progression. Arthritis Rheum. 2006;54:3761–73. doi: 10.1002/art.22190. [DOI] [PubMed] [Google Scholar]
- 43.Brown AK, Conaghan PG, Karim Z, Quinn MA, Ikeda K, Peterfy CG, et al. An explanation for the apparent dissociation between clinical remission and continued structural deterioration in rheumatoid arthritis. Arthritis Rheum. 2008;58:2958–67. doi: 10.1002/art.23945. [DOI] [PubMed] [Google Scholar]
- 44.McQueen FM. The MRI view of synovitis and tenosynovitis in inflammatory arthritis: implications for diagnosis and management. Ann N Y Acad Sci. 2009;1154:21–34. doi: 10.1111/j.1749-6632.2009.04382.x. [DOI] [PubMed] [Google Scholar]
- 45.Anandarajah A, Thiele R, Monu J, Seo G, Bokhari A, Ritchlin C. Rheumatoid arthritis patients in clinical remission manifest persistent joint inflammation on histology and imaging. Arthritis Rheum. 2010;62:S438. doi: 10.3899/jrheum.140411. abstract. Suppl. [DOI] [PubMed] [Google Scholar]