

Abstract
Despite viral vectors being potent inducers of antigen-specific T cells, strategies to further improve their immunogenicity are actively pursued. Of the numerous approaches investigated, fusion of the encoded antigen to major histocompatibility complex class II–associated invariant chain (Ii) has been reported to enhance CD8+ T-cell responses. We have previously shown that adenovirus vaccine encoding nonstructural (NS) hepatitis C virus (HCV) proteins induces potent T-cell responses in humans. However, even higher T-cell responses might be required to achieve efficacy against different HCV genotypes or therapeutic effect in chronically infected HCV patients. In this study, we assessed fusion of the HCV NS antigen to murine and human Ii expressed by the chimpanzee adenovirus vector ChAd3 or recombinant modified vaccinia Ankara in mice and nonhuman primates (NHPs). A dramatic increase was observed in outbred mice in which vaccination with ChAd3 expressing the fusion antigen resulted in a 10-fold increase in interferon-γ+ CD8+ T cells. In NHPs, CD8+ T-cell responses were enhanced and accelerated with vectors encoding the Ii-fused antigen. These data show for the first time that the enhancement induced by vector vaccines encoding li-fused antigen was not species specific and can be translated from mice to NHPs, opening the way for testing in humans.
Introduction
Replication-defective adenoviruses (Ads) have emerged as the most promising vectors for genetic vaccination, given their capability to elicit a robust cellular immune response, particularly of the CD8+ T-cell subset that might be crucial for protection against pathogens, such as human immunodeficiency virus (HIV), hepatitis C virus (HCV), Plasmodium species, and Mycobacterium species, which establish chronic infection.1 Ads can be considered self-adjuvanting vaccines because they have a natural aptitude to trigger innate immunity, largely due to the activity of their structural viral proteins. Several innate signaling pathways have been implicated in the recognition of components of Ads, including toll-like receptors (TLRs).2 Engagement of TLRs initiates a signaling cascade involving the intracellular adaptor molecules MyD88 and/or TRI-domain-containing adapter-inducing interferon-β, which in turn mediates the release of a number of inflammatory cytokines, thus influencing the induction of adaptive immune responses to the Ad-encoded transgene. Although immunogenicity studies in knockout mice clearly indicate that MyD88 plays a crucial role in enhancing Ad-mediated immunogenicity,3,4 a systematic assessment of Ad-based vaccines in mice deficient in individual upstream TLRs has shown a minimal impact on Ad potency, suggesting that there is no single TLR accounting for MyD88 dependence.4 Consistent with this redundancy, conflicting results were obtained when various innate receptor agonists were tested as adjuvants in Ad vaccine formulations. As examples, TLR3 ligand polyinosinic–polycytidylic acid (poly(I:C)) markedly inhibited Ad-induced antigen-specific response,5 and reduced CD8+ T-cell responses have also been observed on adding a TLR9 agonist to Ad5-based tumor vaccine,6 despite the antitumor protection elicited. By contrast, the activation of TLR4 by lipopolysaccharides or monophosphoryl lipid A potentiated the magnitude and functionality of Ad-mediated immune response.5
An alternative approach to augment genetic vaccine–induced T-cell responses is the fusion of enhancing elements directly to the encoded antigen. To date, the encoded adjuvant most consistently enhancing cell-mediated immunity is the major histocompatibility complex (MHC) class II–associated invariant chain (CD74 or Ii). Ii is a type II transmembrane glycoprotein that associates with the MHC class II α and β chains and directs the transport of the αβIi complexes to endosomes and lysosomes, exerting its main function in antigen presentation.7 A lentiviral vector expressing ovalbumin fused to Ii was shown to be the most potent inducer of CD4 and CD8– T cells and the only vector to protect mice from challenge with ovalbumin-expressing tumor cells.8 The linkage of murine Ii (mIi) to human Ad5- or naked DNA-encoded antigens has been shown to enhance antigen presentation in MHC class I, resulting in augmented and broadened CD8+ T-cell response9,10 and increased protection in a murine cancer model.11 Ii linkage was also shown to enhance protection against infections caused by viruses or intracellular bacteria by using either lymphocytic choriomeningitis virus10 or Listeria monocytogenes as models .12 This effect was shown to be independent of CD4 help because it was preserved in MHC class II–deficient mice,13 and the result was confirmed in several inbred mice strains, independently, by the fused antigen.11,13,14
Currently, there is no vaccine for the prevention or treatment of HCV infection. Because effective cellular immunity is known to play a crucial role in spontaneous viral eradication, HCV infection may be particularly suitable for T-cell vaccination strategies.15,16 We have previously shown that a vaccine based on simian Ad 3 (ChAd3) encoding nonstructural (NS) HCV proteins induces potent T-cell responses in humans.17 Heterologous boost with recombinant modified vaccinia Ankara (MVA) can further augment ChAd-primed T cells (refs. 18,19,20 and Swadling et al., submitted); nonetheless, even higher T-cell responses might be required both to achieve efficacy against the different HCV genotypes or in “at-risk populations” such as HIV-infected individuals with compromised CD4 function and to exert a therapeutic effect in chronically infected HCV patients.
In this study, we assessed the immunogenicity of the HCV NS antigen fused to murine and human Ii expressed by ChAd3 and MVA in outbred mice and nonhuman primates (NHPs). CD8 T-cell responses were enhanced, accelerated, and sustained with vectors encoding the Ii-fused antigen, demonstrating that the enhancement effect is not species specific and translates from mice to NHPs, setting the ground for the development of improved genetic vaccines against major pathogens that require cellular immunity with potent and rapid effector functions.
Results
Fusion to murine Ii enhances CD8 T-cell response to vaccine-encoded homologous and heterologous HCV NS antigens in outbred mice
It has been recently demonstrated14 that an Ad5 encoding mIi fused to HCV NS3 markedly accelerated and enhanced the CD8+ T-cell response in inbred mice. We extended this observation to our clinically validated HCV vaccine candidate, ChAd3 encoding the entire NS3-5B HCV genotype 1b region, using outbred CD1 mice. Magnitude and breadth of T-cell responses were assessed 2 weeks after ChAd3NS or ChAd3mIiNS vaccination by interferon-γ (IFN-γ) enzyme-linked immunosorbent spot (ELISpot) assay using six pools of 15-mer peptides encompassing either genotype 1b HCV NS3-NS5B sequence homologous to the vaccine or the heterologous genotype 3a, the most divergent from genotype 1b. ChAd3mIiNS induced T-cell responses to genotype 1b antigens to a significantly higher level than ChAd3NS, with a geometric mean (GM) of 4,888 spot-forming cells (SFCs) versus 646 SFCs per million splenocytes (P = 0.01; Figure 1a). Moreover, ChAd3mIiNS vaccination resulted in homogeneously strong T-cell responses of more than 1,000 SFCs, whereas a wide variability was seen in control animals vaccinated with ChAd3NS. Importantly, the reactivity to the heterologous 3a antigens was also enhanced by mIi fusion, with a GM of 672 SFCs in the ChAd3mIiNS-vaccinated group compared with 159 SFCs in the control group (P = 0.0002; Figure 1a). The magnitude of overall T-cell responses to 1b antigens correlates (P = 0.016; Figure 1b) with the responses to 3a antigens, as we already reported for vaccinated human volunteers.17 All HCV NS proteins encoded in the vaccine were recognized with variable magnitude and hierarchy in individual mice (Figure 1c). ChAd3mIiNS vaccination clearly improved the breadth of T-cell responses evoked in individual animals compared with the result of ChAd3NS vaccination. Indeed, a median of six peptide pools (range: 4–6) was recognized in ChAd3mIiNS-vaccinated mice compared with a median of three pools (range: 1–6) in ChAd3NS-vaccinated mice (P = 0.004; Figure 1c,d). In order to characterize the T-lymphocyte subset that was most affected by the enhancing effect of the mIi fusion, intracellular cytokine staining (ICS) and fluorescence-activated cell sorting (FACS) analyses were performed on splenocytes, revealing a higher frequency of both CD4+ and CD8+ IFN-γ–producing cells in presence of mIi. However, the predominant effect was seen on the CD8+ T-cell subset, with a remarkable, almost 20-fold increase (GM of 7.7% IFN-γ+ CD8+ in the group receiving the mIi fusion versus GM of 0.4% in control mice, P = 0.01; Figure 1e).
Figure 1.
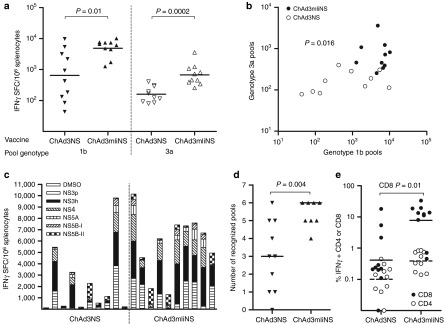
Fusion of nonstructural (NS) antigen to murine invariant chain (mIi) enhances magnitude and breadth of T-cell response in CD1 mice after priming with ChAd3. CD1 mice were vaccinated with ChAd3NS or ChAd3mIiNS with 108 viral particles (vp) dose, and T-cell response was tested on splenocytes 2 weeks later. (a) Cross-reactivity of T-cell responses, measured by interferon-γ (IFN-γ) enzyme-linked immunosorbent spot (ELISpot) using two comparable sets of six 15-mer peptide pools spanning NS3-5B polyprotein of either hepatitis C virus (HCV) genotype 1b (corresponding to the vaccine sequence) or 3a. Vaccines received and pools' genotypes are indicated at the bottom of the graph. Each symbol represents the total NS response in individual mice, defined as the sum of responses to the six peptide pools less the dimethyl sulfoxide (DMSO) background and expressed as spot-forming cells (SFCs) per million splenocytes. Lines represent geometric mean (GM). (b) Correlation of total NS ELISpot response to genotype 1b or 3a peptide pools in individual mice. Open and filled symbols correspond to animals vaccinated with ChAd3NS and ChAd3mIiNS, respectively. (c) Magnitude and breadth of ELISpot response. Each bar shows individual mouse response to DMSO and to each of the six 15-mer peptide pools, irrespective of whether it is positive or negative, spanning the entire NS3-5B polyprotein length (genotype 1b), as shown in the graph's legend. Data are expressed as IFN-γ SFCs/million splenocytes. (d) Breadth analysis in ChAd3NS- and ChAd3mIiNS-vaccinated mice. Each symbol corresponds to the number of positive pools, according to ELISpot positivity criteria defined in Materials and Methods, recognized in individual mice. Lines represent the group median. (e) Frequencies of IFN-γ–secreting cells analyzed by intracellular staining, expressed as the percentage of NS-specific CD4+ (open symbols) or CD8+ (filled symbols) response over total CD4+ or CD8+. Lines represent GM (filled for CD8 and dashed for CD4).
Fusion of mIi to antigen also enhances T-cell responses after booster immunization and allows multiple readministrations of the same chimpanzee adenoviral vector
We next extended our study to prime/boost regimens to assess whether incorporating mIi in Ad-based vaccine could also confer an advantage in recall vaccination. With this aim, CD1 mice were primed with ChAd3NS and then boosted 11 weeks later, when T-cell responses contracted, with ChAd3mIiNS or ChAd3NS. As benchmark of efficient boosting, we included additional groups receiving MVA encoding the NS antigen (MVA-NS). MVA has been shown in humans18,19,20 to efficiently boost the T-cell response primed by an Ad vector. In parallel, groups of animals were primed with ChAd3mIiNS and boosted either with the same vector or with MVA-NS. T-cell response was measured at week 11 to establish levels before and shortly after boost to evaluate peak response. As expected, in ChAd3NS-primed animals, homologous vector readministration modestly increased CD8+ responses compared with preboost levels (GM CD8+ frequency: 0.85 versus 0.35%; Figure 2a), possibly due to antivector immunity raised by first injection. However, a potent 30-fold increase in GM CD8+ frequency was observed when mice were boosted with ChAd3mIiNS (GM of 10.8%; P = 0.0002; Figure 2a), and peak CD8 response after the boost in these mice was significantly higher than that in mice boosted with the vector without mIi (ChAd3NS; P = 0.0039 Figure 2a). A similar pattern, although less impressive in terms of overall magnitude, was observed for CD4 responses (Figure 2b). On comparison with MVA-NS, the boost with ChAd3mIiNS was similarly efficient, with CD8 levels higher for ChAd3mIiNS and CD4 responses more homogeneously boosted by MVA, reflecting well-known intrinsic properties of the two viral vector platforms.
Figure 2.

Enhancing effect of murine invariant chain (mIi) fusion to nonstructural (NS) antigen at both priming and boosting in CD1 mice. CD1 mice were primed by ChAd3NS (filled symbols) or ChAd3mIiNS (open symbols) at 108 viral particle (vp) dose and boosted at week 11 with either ChAd3mIiNS or ChAd3NS at the same dosage, or with MVA-NS at 106 plaque-forming units (pfu) dose, as specified at the bottom of each graph. Analysis of T-cell response at boost (week 11) and at peak postboost (week 12 for MVA or week 13 for ChAd) was performed by interferon-γ (IFN-γ) intracellular cytokine staining on splenocytes. Each symbol shows frequency of NS-specific, IFN-γ–secreting (a) CD8+ or (b) CD4+ in individual animals. Lines represent geometric mean. MVA, modified vaccinia Ankara; ns, nonsignificant.
Residual CD8 T-cell levels at the time of boosting (week 11) were higher in ChAd3mIiNS-primed animals than in those vaccinated with unfused antigen (GM CD8: 1.03 versus 0.36%; Figure 2a), suggesting that the enhancing effect of mIi is not confined only to peak responses but also to the subsequent contraction phase. Nonetheless, a significant boost in both CD8 and CD4 responses was observed when the same Ii-fused vector was readministered (GM CD8: 8.5% and GM CD4: 0.42%; Figure 2a,b), with boosting efficiency similar to that of MVA, again confirming that incorporating mIi into genetic vaccine vectors leads to improved CD8, and to a lesser extent CD4, T-cell responses both at priming and at boosting.
Both murine and human Ii enhance and accelerate CD8 T-cell responses after priming with ChAd3 in mice and NHPs
Prompted by the results obtained with mIi, we extended our studies investigating the effect of NS transgene linkage to the human Ii isoform 2, p35 (hIi). Indeed, although Ii is structurally and functionally conserved in mammalians, so far, the human isoform has never been tested in the context of Ad vaccine. In addition, both mouse and human variants have not been tested yet in NHPs. Comparing the immunogenicity of ChAd3hIiNS with that of ChAd3mIiNS in CD1 mice revealed that both vectors were equally potent and were significantly stronger than the unfused vaccine, inducing IFN-γ-T-cell responses with GM, respectively, of 4,676 and 4,336 SFCs 2 weeks after vaccination (Figure 3a). Similarly, in NHPs, both Ii fusion vectors were effective in enhancing priming of T-cell responses compared with ChAd3NS, with possibly a better effect of the ChAd3hIiNS (Figure 3b). Of note, T-cell response development was accelerated in NHPs receiving either Ii fusions because all animals were already positive 2 weeks after vaccination, confirming in a more relevant animal model what has been previously described in mice. A broad response targeted to all the NS peptide pools tested was seen in animals primed with ChAd3hIiNS, with an increased median of five positive peptide pools (range: 3–6) versus a median of three pools (range: 1–6) in control animals receiving ChAd3NS, thus confirming CD1 data (Figure 4a). Similar broad responses were seen in ChAd3mIiNS-vaccinated animals (not shown). IFN-γ ICS analysis confirmed that, similar to what was found in mouse models, a more vigorous CD8+ T-cell response was evoked in animals vaccinated with ChAd3hIiNS, quantified as fourfold stronger than the one induced by unlinked control vector vaccine (GM CD8: 0.53 versus 0.16%, respectively; Figure 4b).
Figure 3.

Fusion to murine invariant chain (mIi) or human invariant chain (hIi) enhances and accelerates T-cell responses in mice and nonhuman primates (NHPs) in a non–species-specific manner. Total NS response on genotype 1b peptide pools, measured by interferon-γ (IFN-γ) enzyme-linked immunosorbent spot (ELISpot) after vaccination with ChAd3NS, ChAd3mIiNS, or ChAd3hIiNS, as indicated at the bottom of each graph. Symbols represent response in individual animals, and lines represent the groups' geometric mean. (a) CD1 mice vaccinated with 108 viral particles (vp) dose of each vector. ELISpot performed on splenocytes 2 weeks after vaccination. (b) Cynomolgus macaques vaccinated with 1010 vp of each vector. ELISpot performed on peripheral blood mononuclear cells (PBMCs) 2 weeks after vaccination. Frequency of responding animals at each time point is indicated at the bottom of the graph. SFC, spot-forming cell.
Figure 4.

Fusion to human invariant chain (hIi) improves magnitude and breadth of nonstructural (NS)-specific T-cell response and CD8+ T cells frequency in nonhuman primates (NHPs). Cynomolgus macaques were immunized with ChAd3NS or ChAd3hIiNS at 1010 vp dose. T-cell response was measured in peripheral blood mononuclear cells (PBMCs) 4 weeks after vaccination by means of interferon-γ (IFN-γ) enzyme-linked immunosorbent spot (ELISpot) and intracellular cytokine staining (ICS). (a) Magnitude and breadth of ELISpot response. Each bar shows individual NHP response to dimethyl sulfoxide (DMSO) and to each of the six genotype 1b peptide pools irrespective of whether it is positive or negative, as shown in the graph's legend. Below each bar, the number of positive pools, according to ELISpot positivity criteria defined in Materials and Methods (≥50 spot-forming cells (SFCs)/million PBMCs to at least one peptide pool; ≥ three times of DMSO background), recognized in individual NHPs are shown. Data are expressed as IFN-γ SFCs per million PBMCs. (b) Frequencies of IFN-γ–secreting cells analyzed by ICS, expressed as the percentage of NS-specific CD4+ (open symbols) or CD8+ (filled symbols) response over total CD4+ or CD8+ in individual animals. Lines represent geometric mean (filled for CD8 and dashed for CD4).
Fusion of antigen to Ii improves T-cell memory levels and peak postboost responses if encoded by the boosting vector in NHPs
We next explored if the Ii technology might provide further advantage in terms of magnitude, quality, and longevity of immune responses in the context of ChAd and MVA prime/boost regimens in NHPs. Two groups of four macaques each were primed with either ChAd3mIiNS or ChAd3NS and boosted 8 weeks later with MVA-NS. IFN-γ ELISpot analysis at key time points (peak postprime/postboost and 3 months after boost; Figure 5a) revealed that a stronger response was seen at peak postprime in animals vaccinated with ChAd3mIiNS. However, the heterologous MVA-NS boost was equally effective in both groups, inducing potent recall T-cell response with similar GM around 7,000 SFCs (Figure 5a). ICS analysis at peak post-MVA boost confirmed this finding, with remarkably high levels of IFN-γ-secreting, NS-specific CD8 and CD4 in both groups (Figure 5b). However, after natural T-cell contraction, somewhat higher levels of memory T cells were detectable ex vivo in the group primed with ChAd3mIiNS.
Figure 5.

Fusions to murine invariant chain (mIi) or human invariant chain (hIi) in the priming or boosting vector increases T-cell magnitude, CD8 frequency and persistence in nonhuman primates (NHPs) receiving different prime/boost combinations. (a,b) Cynomolgus macaques received ChAd3NS (filled symbols) or ChAd3mIiNS (open symbols) vaccination at 1010 viral particles (vp) dose and were boosted 8 weeks later with MVA-NS at 2 × 108 plaque-forming units (pfu) dose. (c,d) Additional animals received ChAd3mIiNS (filled symbols) or ChAd3hIiNS (open symbols) vaccination at 1010 vp dose and were boosted 50 weeks later with MVA-NS or MVA-hIiNS, respectively, at 2 × 108 pfu dose. (a,c) Interferon-γ (IFN-γ) enzyme-linked immunosorbent spot responses measured 4 weeks after prime, 1 week after boost, and 3 months after boost (memory), as indicated on x-axis labels. Each symbol corresponds to total anti-NS response in individual animals, expressed as IFN-γ spot-forming cells (SFCs) per million peripheral blood mononuclear cells (PBMCs). Lines represent geometric mean (GM). (b,d) Frequencies of IFN-γ–secreting cells analyzed 1 week after MVA boost by intracellular cytokine staining, expressed as the percentage of NS-specific CD4+ (open symbols) or CD8+ (filled symbols) response over total CD4+ or CD8+ in individual animals. Lines represent GM (filled for CD8 and dashed for CD4). MVA, modified vaccinia Ankara; NS, nonstructural.
To further improve the vaccine-induced T-cell responses, we generated an MVA vector encoding hIi fused to NS antigen and boosted two additional groups of macaques that were primed 50 weeks before with either ChAd3mIiNS or ChAd3hIiNS, with barely detectable ex vivo T-cell responses at the time of long-term boost (not shown). The animals primed with ChAd3hIiNS were boosted with the novel MVA-hIiNS and the ChAd3mIiNS-primed animals with MVA-NS as control. Vigorous recall responses were elicited in both groups, with twofold higher GM ELISpot response in the group receiving MVA-hIiNS (9,260 versus 5,031 SFCs; Figure 5c) and an almost threefold higher GM CD8 frequency (4.16 versus 1.47%, Figure 5d) by IFN-γ ICS, whereas CD4 levels were comparable in the two groups. This difference was maintained 3 months postboost because stronger and more homogeneous ex vivo responses were still detectable in the group boosted with MVA-hIiNS (Figure 5c). Thus, transgene fusion to Ii encoded in the MVA boosting vector provides stronger peak postboost responses, which persist at higher frequencies over time.
Vaccination with viral vectors encoding hIi-antigen fusion does not break tolerance in mice and NHPs
The inclusion of a self-protein to vaccine-encoded antigen raises safety concerns because autoimmunity may be triggered. Potential induction of T-cell and antibody responses to the autologous Ii in mice was measured in animals receiving prime only or prime/boost schedules including mIi-encoding vectors. We found that there was no induction of such autoreactive T or B cells in any of the tested animals (data not shown).
Macaque Ii has very high amino acid sequence homology with human p35 (92.4% identity), suggesting that it should be regarded as a self-antigen in NHPs. We thus measured T-cell responses to mIi and hIi by means of specific 15-mer peptide pools at key time points during our macaque studies. At peak postprime, a low-level T-cell response to mIi was detectable in ChAd3mIiNS-primed animals (Figure 6a), not surprisingly because macaque and murine Ii have a much lower homology (66.4% identity). However, no reactivity to human peptide pool was seen in the same animals, demonstrating that T-cell responses were raised against regions of significant diversity between the murine and macaque Ii. Most importantly, animals primed with ChAd3hIiNS did not show any induction of anti hIi-T-cell response on priming and no self-reactive T cells to Ii were detected even after a second exposure to hIi by MVA-hIiNS boost (Figure 6a). Sera collected after ChAd3hIiNS prime and MVA-hIiNS boost in these four macaques were tested by enzyme-linked immunosorbent assay on recombinant full-length hIi to check for induction of autoantibodies. No reactivity could be detected in this assay even at very low serum dilution (1:20; Figure 6b), demonstrating preservation of T- and B-cell tolerance to self Ii.
Figure 6.

Murine invariant chain (mIi)- or human invariant chain (hIi)-encoding vector vaccinations do not induce T-cell or antibody responses to hIi in nonhuman primates (NHPs). (a) T-cell response to two 15-mer peptide pools spanning the entire length of hIi or mIi proteins was tested by interferon-γ (IFN-γ) enzyme-linked immunosorbent spot in peripheral blood mononuclear cells (PBMCs) collected 4 weeks after prime from Cynomolgus macaques vaccinated with ChAd3mIiNS (open symbols), or collected 4 weeks after prime and 1 week after boost in animals vaccinated with ChAd3hIiNS and boosted 50 weeks later with MVA-hIiNS (filled symbols). Responses corrected for dimethyl sulfoxide background are shown, expressed as IFN-γ spot-forming cells (SFCs) per million PBMCs, and lines (dashed for mIi primed animals and filled for animals receiving hIi prime/boost) represent geometric mean. A dotted line set at 50 SFCs indicates threshold for a positive response. (b) Anti-hIi antibody detection by enzyme-linked immunosorbent assay in the four Cynomolgus macaques (indicated at the bottom of the graph) receiving ChAd3hIiNS/MVA-hIiNS vaccinations at weeks 0 and 50. Sera collected before first vaccination (preimmune), 8 weeks after prime and 5 weeks after boost were tested at dilution of 1:20 for binding to purified hIi protein. A commercial anti-human Ii antibody was used as positive control. Data are shown as optical density (OD) readings at 405 nm. Each bar represents the mean + SD of six replica wells. A dotted line is set at an OD of 0.32, corresponding to the mean of all preimmune wells + 3 × SD, indicating a reasonable threshold for positivity. MVA, modified vaccinia Ankara; NS, nonstructural.
Discussion
Various attempts to further improve the immunogenicity of recombinant adenoviral vectors, most of them based on the use of TLR agonists as adjuvants in Ad vaccine formulations, have been made without much success. Given the large “capacity” of Ad and MVA vaccine vectors, it is possible to incorporate DNA sequences capable of enhancing the immune response to the encoded antigen. A number of studies have shown that fusion of mouse MHC class II–associated Ii to proteins delivered by Ad5 increased, accelerated, broadened, and prolonged antigen-specific T-cell–mediated immunity. Proteins that have been fused to Ii include lymphocytic choriomeningitis virus glycoprotein,10 HCV NS3 protein,14 and listeriolysin O and p60 from L. monocytogenes.12
This study extends those findings to different classes of viral vectors because the chimpanzee Ad and MVA have recently been demonstrated to be safe in humans, particularly being immunogenic when used in heterologous prime and boost regimen (reviewed in ref. 21). ChAds are emerging as a promising new class of genetic vaccine carriers due to the low frequency of neutralizing antibodies in human population, which, instead, represents a major issue for human-derived Ads of which Ad5 is the prototype.22,23,24 Safety concerns regarding the use of Ad5 have also recently emerged from efficacy trials of Ad5-based HIV vaccines, particularly in volunteers with high level of neutralizing antibodies to this vector at baseline.25,26 ChAd3 encoding the HCV NS antigen fused to either murine Ii or human p35 Ii was evaluated in outbred mice and NHPs. In outbred mice, ChAd3mIiNS significantly improved the magnitude and breadth of T-cell response against the encoded antigen (1b HCV genotype), and this effect was even more striking when peptides corresponding to the divergent HCV 3a genotype were used in the assay. These data are of great relevance considering that HCV exists in six different genotypes and circulates as a quasispecies (a swarm of related but divergent sequences) within each infected host, due to its high replication rate and the lack of proofreading function in the viral polymerase NS5B. These features allow the virus to rapidly accumulate mutations, leading to viral escape from immune pressure.27,28 Thus, an ideal HCV vaccine should elicit strong CD8 T-cell responses targeting multiple epitopes from divergent HCV virus genotypes to successfully counteract the viral evasion strategies (reviewed in ref. 16).
One main drawback of vaccination strategies based on Ad is the induction of antivector immunity on priming, which can prevent efficient boost by the same vector. Here, we showed that ChAd3mIiNS more efficiently boosts CD8 response when readministered, with respect to ChAd3NS, in line with the recent report by Steffensen et al.29 with Ad5-Ii-GP. Consistently with previous data, a potent boost was observed with MVA. The effectiveness of readministration of the vector encoding the Ii fusion may thus overcome the need to generate a different vaccine (i.e., heterologous Ad or MVA) encoding the same transgene when a prime/boost vaccination schedule is needed.
The human Ii was tested here for the first time as an encoded adjuvant in the context of Ad-based vaccines, and its evaluation in NHPs is remarkably important for applications in humans. In both CD1 mice and NHPs, ChAd3mIiNS and ChAd3hIiNS vaccines elicited significantly higher CD8 T-cell response in comparison with the vector encoding NS alone, thus validating the function of Ii as an enhancer of antigen-specific T-cell responses in relevant models. Magnitude, quality, breadth, and interindividual variability of responses were similar between CD1 and NHPs, suggesting that outbred mice constitute a valuable model to predict vaccine responses in superior species, with the further advantage of enabling use of experimental groups numerically large enough to perform statistical evaluation, which is ethically and practically unfeasible in NHPs.
To evaluate whether in the context of heterologous prime and boost regimens, Ii could enhance T-cell responses when encoded by MVA, two groups of macaques were primed with ChAd3 encoding Ii-NS and boosted with MVA encoding either NS or Ii-NS. Increased T-cell responses were induced right after boost and were maintained over time, suggesting that an even more immunogenic HCV vaccine could be developed based on prime and boost with ChAd3 and MVA vectors encoding the HCV-NS antigen fused to the human Ii. Such an improved vaccine may be more effective in a prophylactic setting and, more importantly, it may overcome viral suppression and rescue or may generate de novo T-cell responses in chronically infected patients known to mount weak, narrow, and impaired immune responses.30,31,32,33,34
Ii-mediated CD8 T-cell enhancement effect has been shown to be independent of CD4+ T-cell help because it has been confirmed in MHC class II–deficient mice.13 This finding extends the applicability of Ii-NS viral vector vaccine in settings where the CD4 T-cell number and function are impaired, as in HIV-infected patients. Although injection drug use accounts for 40% of all cases of acute HCV infection, in the past 15 years, there has also been a more than 10-fold increase in the incidence of acute HCV infection in HIV-1–seropositive men who have sex with men (data from European Monitoring Centre for Drugs and Drug Addiction). Because HIV–HCV coinfection is associated with an accelerated course of HCV infection and worse prognosis, HIV-positive individuals represent a key target population in whom an Ii-based HCV vaccine may be beneficial, its immunogenicity being potentially unaffected by impairment of the CD4 T-cell compartment.
Because Ii is a self-antigen expressed throughout the immune system by B cells, activated T cells, dendritic cells, monocytes, and macrophages and is widely expressed in the thymus, it should be highly tolerated. Two recent publications35,36 have shown that autoantibodies targeting the class II-associated invariant chain peptide of Ii constitute a highly specific biomarker for inflammatory spondyloarthritis. Although a direct link of such autoantibodies with pathogenesis has not been established, these studies highlight potential safety concerns. Our analysis of T-cell and antibody response against human Ii in NHPs receiving ChAd3hIiNS priming and subsequent MVA-hIiNS boost showed no reactivity against the human Ii. Low-level T-cell responses to Ii were only detected in NHPs vaccinated with the vector encoding the murine Ii version, suggesting that those T cells targeted regions of diversity between murine and macaque li sequences. Immune responses targeting autologous Ii were also not induced in mice even after repeated administrations of ChAd3mIiNS.
This study extended the benefit of the Ii–Ad technology by applying it to vaccine vectors and antigens already in clinical trials and showed in relevant animal models that an improved HCV vaccine could be developed to better target different HCV genotypes or special patient populations, including HIV+ subjects at risk for HCV coinfection and chronic HCV patients. Deeper knowledge and comprehension of the mechanism responsible for the Ii-mediated enhancement of the CD8+ T-cell response will help to address potential safety concerns and open the way to clinical testing of new vaccines with enhanced antigen-specific T-cell responses.
Materials and Methods
Adenoviral and MVA vectors. The ChAd3 vector expressing the entire HCV NS3-5B (NS) region from genotype 1b, strain bk, has been described previously.37,38 Murine Ii insert (accession number NM_1042605.1) was obtained from plasmid Murine Ii opt. PJH#66, kindly provided by Peter Holst (University of Copenhagen), and the human Ii (p35, NCBI reference sequence: NM_004355.2) insert was synthesized by GeneArt (Life Technologies, Paisley, UK). Both sequences were cloned at the N-terminus of the NS transgene under human cytomegalovirus and bovine growth hormone polyadenylation signal control and then transferred into the ChAd3 vector by homologous recombination in BJ5183 cells. Recombinant viruses ChAd3mIiNS and ChAd3hIiNS were purified as described previously.38
To generate MVA-hIiNS, transfer plasmid green (TPG) shuttle vector, kindly provided by Antonio Siccardi, Istituto San Raffaele, Milan, Italy, was used to clone hIiNS gene under the control of P7.5 MVA early promoter and flanked by FlankIII-2 and FlankIII-1 regions, necessary to allow insertion in the del III of MVA by in vivo homologous recombination39,40; the TPG-hIiNS shuttle vector also contains the expression cassette for green fluorescent protein (GFP) under control of the synthetic promoter sP. Briefly, primary chick embryo fibroblasts were infected with a recombinant MVA carrying hcRed gene and then transfected with TPG-hIiNS plasmid. The resulting virus lysate was used to infect fresh chick embryo fibroblasts in the presence of 1 µmol/l cytochalasin D (Sigma-Aldrich, St. Louis, MO). Twenty-four hours after infection, the cells were collected by trypsinization, and green cells were either bulk sorted or single cell sorted by a Becton Dickinson FACS Vantage SE flow cytometer (Becton Dickinson, San Jose, CA). Sorted cells were seeded onto chick embryo fibroblast monolayers in microplate cultures to produce virus lysates. Finally, markerless recombinant viruses (MVA-hIiNS) were cloned by terminal dilution, screened by Typhoon fluorography, cloned again by terminal dilution, expanded in chick embryo fibroblasts, purified over sucrose cushions, and titrated by plaque assay using conventional methods. Polymerase chain reaction was performed to check for identity and purity of MVA isolates.
A similar method was used to generate MVA-NS; the NS expression cassette was subcloned into the MVA shuttle vector pMVA-GFP-TD, kindly provided by Sarah Gilbert, University of Oxford, UK, flanked by TKL (thymidine kinase gene left region) and TKR (thymidine kinase gene right region), under the control of P7.5 MVA early promoter generating the transfer vector pMVA-GFP-TD-NS, which also expresses GFP under the fowlpox late promoter, FP4b control. The production of the recombinant MVA-NS virus was based on in vivo recombination between the MVA-Red genome and the homologous sequence (TKL and TKR) within the transfer vector pMVA-GFP-TD-NS.
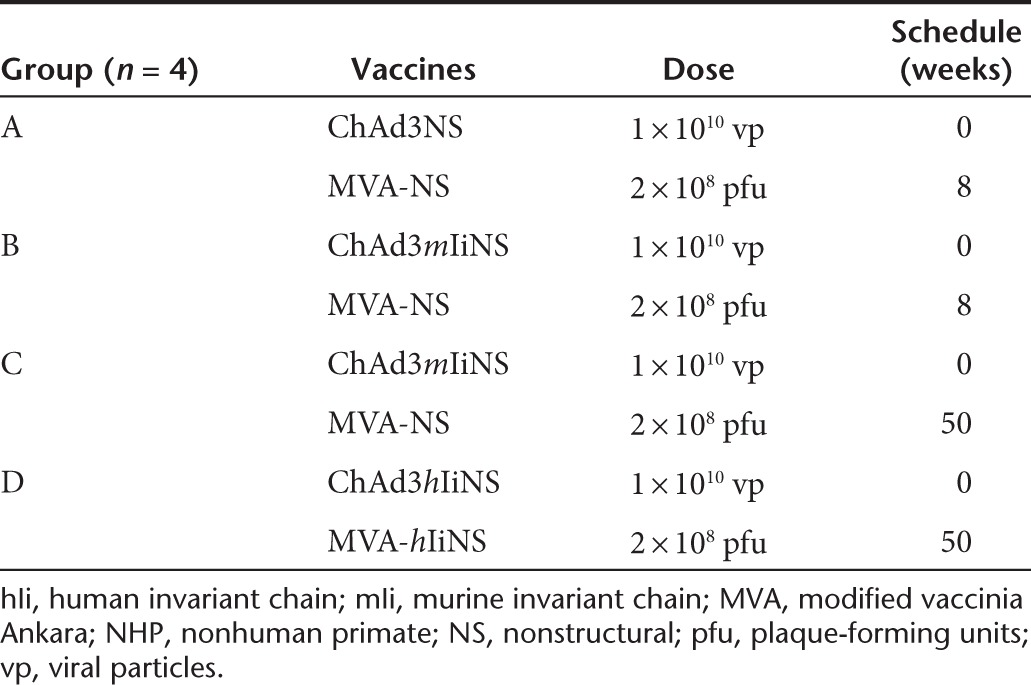
Animals and vaccinations. All experimental procedures were approved by the local animal ethics council and were performed in accordance with national and international laws and policies (EEC Council Directive 86/609; Italian Legislative Decree 116/92). The ethical committee of the Italian Ministry of Health approved this research. Animal handling procedures were performed under anesthesia, and all efforts were made to reduce animal numbers and minimize suffering. Six-week-old female CD1 mice were purchased from Charles River (Como, Italy), and experimental groups of 10 mice each were set. Viral vectors were administered i.m. in the quadriceps by delivering a volume of 50 µl per side (100 µl final volume). The injected dose for all mouse experiments was 1 × 108 viral particles (vp) for Ad vectors and 2 × 106 plaque-forming units (pfu) for MVA vectors. Cynomolgus macaques (Macaca fascicularis) from a purpose-bred colony housed at the Institute of Cell Biology and Neurobiology primate facility (ENEA-Casaccia, Rome, Italy), distributed in groups of four animals each, were immunized by the i.m. route in the deltoid muscle by injecting 0.5 ml of virus diluted in stabilizing buffer. The injected dose was 1 × 1010 vp for Ad vectors and 2 x 108 pfu for MVA vectors. For immunization schedule, see Table 1. During handling, the animals were anesthetized by i.m. injection of 10 mg/kg ketamine hydrochloride.
Table 1. NHP vaccination schedule.
Peptides. A set of 494 (495 for genotype 3a) peptides, 15 amino acids in length, overlapping by 11 amino acids, and spanning the open reading frame from NS3 to NS5B (1985 amino acids) of HCV genotype 1b strain BK or genotype 3a (GenBank accession D28917), were obtained from BEI Resources (Manassas, VA). Two further sets of 50 peptides, 15 amino acids in length, overlapping by 11 amino acids, and spanning the entire sequence of hIi (accession number NM_004355.2) or mIi (accession number NM_1042605.1), were obtained from Mimotopes (Clayton, Victoria, Australia).
Ex vivo IFN-γ ELISpot. MSIP S4510 plates (Millipore, Billerica, MA) were coated with 10 µg/ml of anti-mouse or anti-monkey IFN-γ antibody (both from U-CyTech Utrecht, The Netherlands) and incubated overnight at 4 °C. After washing and blocking, mouse splenocytes or macaque peripheral blood mononuclear cells (PBMCs) were plated in duplicate at two different densities (2 × 105 and 4 × 105 cells per well) and stimulated overnight with overlapping 15-mer peptide pools at a final concentration of 4 µg/ml of each single peptide. The peptide diluent dimethyl sulfoxide (Sigma-Aldrich, Milan, Italy) and concanavalin A (Sigma-Aldrich, Milan, Italy) were used, respectively, as negative and positive controls. Plates were developed by subsequent incubations with biotinylated anti-mouse or anti-monkey IFN-γ antibody (both from U-CyTech Utrecht, The Netherlands), conjugated streptavidin–alkaline phosphatase (BD Biosciences, San Jose, CA) and finally with 5-bromo-4-chloro-3-indoyl-phosphate/nitro blue tetrazolium 1-Step solution (Thermo Fisher Scientific, Rockford, IL). Plates were analyzed by an automated ELISA-spot assay video analysis system automated plate reader. The ELISpot response was considered positive when all of the following conditions were met: IFN-γ production present in Con-A stimulated wells; at least 50 specific spots/million splenocytes or PBMCs to at least one peptide pool; the number of spots seen in positive wells was three times the number detected in the mock control wells (dimethyl sulfoxide); and responses decreased with cell dilutions. ELISpot data were expressed as IFN-γ SFCs per million splenocytes or PBMCs.
ICS and FACS analysis. Briefly, 4 × 106 mice splenocytes or 2 × 106 monkey PBMCs were stimulated at 37 °C in 5% CO2 for 15–20 hours using peptide pools as antigen at final concentration of 2 µg/ml for each peptide in presence of either Golgi plug (BD Biosciences) for mouse experiments or anti-human CD28/CD49d costimulatory antibodies (BD Biosciences) and Brefeldin A (Sigma-Aldrich, Milan, Italy) for macaque assays. Dimethyl sulfoxide (Sigma-Aldrich, Milan, Italy) was used as negative control, and phorbol myristate acetate/ionomycin (Sigma-Aldrich, Milan, Italy) or Staphylococcal enterotoxin B (Sigma-Aldrich, Milan, Italy) were used as positive controls in mice and macaque experiments, respectively. After overnight stimulation, mouse splenocytes were incubated with purified anti-mouse CD16/CD32 clone 2.4G2 (Fc block: BD Biosciences) and then stained in FACS buffer (phosphate-buffered saline, 1% fetal calf serum) with the following surface antibodies: allophycocyanin anti-mouse CD3e, clone 145-2C11; phycoerythrin anti-mouse CD4, clone L3T4; and PerCP anti-mouse CD8a, clone 53–6.7 (all from BD Biosciences). Macaque PBMCs were stained with the following surface antibodies: allophycocyanin anti-monkey CD3, clone SP34-2; PerCp-Cy5.5 anti-monkey CD4, clone L200; and phycoerythrin anti-human CD8, clone RPA-T8 (all from BD Biosciences). Intracellular staining was performed after treatment with Cytofix/Cytoperm and in the presence of PermWash (BD Biosciences) using fluorescein isothiocyanate anti-mouse IFN-γ, clone XMG1.2 (BD Biosciences) or fluorescein isothiocyanate anti-human IFN-γ, clone MD-1 (U-CyTech Utrecht, The Netherlands). Stained cells were acquired on a FACS Canto flow cytometer and analyzed using DIVA software (BD Biosciences). At least 30,000 CD8+, CD3+ gated events were acquired for each sample.
Enzyme-linked immunosorbent assay with purified human invariant chain protein. Polystyrene 96-well plates (Immunoplate Maxisorp; Nunc, Langenselbold, Germany) were coated with purified hIi (human CD74 transcript 1, p43, GenBank accession number NM_001025159 OriGene, Rockville, MD) protein at 500 ng per well in 50 mmol/l NaHCO3 buffer overnight at 4 °C. Plates were washed with wash buffer (phosphate-buffered saline/0.05% Tween 20) and incubated for 1 hour at 37 °C with 250 µl/well of blocking buffer (5% milk/phosphate-buffered saline/0.05% Tween 20). Macaque sera, diluted in the ratio 1:20 in blocking buffer, were plated in six replica wells and incubated for 2 hours at room temperature. Anti-human CD74 mAb clone LN2 (Biolegend, San Diego, CA), diluted in the ratio 1:200, was added as positive control. Plates were then washed and incubated for 1 hour at room temperature with anti-monkey or anti-mouse immunoglobulin G Fc-specific conjugated alkaline phosphatase (both from Sigma-Aldrich, Milan, Italy), diluted 1:5,000 in blocking buffer. Alkaline phosphatase was revealed by incubation with p-nitrophenyl phosphate substrate solution (SigmaFast; Sigma-Aldrich, Milan, Italy). At various time intervals after development, the absorbance was measured by an automated enzyme-linked immunosorbent assay reader (Envision 2102 Multilabel reader, Perkin Elmer, Waltham, MA) at 405 and 620 nm. The mean + SD of the six replicates was calculated for each tested serum and expressed as optical densities.
Statistical analysis. Statistical analysis was conducted and graphs were prepared using Prism software (Graphpad, La Jolla, CA). Because populations were not always normally distributed, a two-tailed Mann–Whitney (nonparametric) test was used to compare two groups. Spearman correlation was used for comparison, as shown in Figure 2b.
Acknowledgments
The authors thank Peter Holst (University of Copenhagen) for sharing reagents and for continuous and useful discussion. The authors also thank BEI Resources for providing HCV peptides. The research leading to these results has received funding from the European Union's Seventh Framework Programme [FP7/2007–2013] under Grant Agreement No: 280873 ADITEC.
S.C., A.F., R.C., and A.N. are named inventors on patent applications covering HCV-vectored vaccines and chimpanzee adenovirus vectors [WO 2006133911 (A3) hepatitis C virus nucleic acid vaccine, WO 2005071093 (A3) chimpanzee adenovirus vaccine carriers, WO 03031588 (A2) hepatitis C virus vaccine].
Authors from Okairos are employees of and/or shareholders in Okairos. The other authors declare that they have no competing interests.
References
- Bassett JD, Swift SL, Bramson JL. Optimizing vaccine-induced CD8(+) T-cell immunity: focus on recombinant adenovirus vectors. Expert Rev Vaccines. 2011;10:1307–1319. doi: 10.1586/erv.11.88. [DOI] [PubMed] [Google Scholar]
- Appledorn DM, Patial S, Godbehere S, Parameswaran N, Amalfitano A. TRIF, and TRIF-interacting TLRs differentially modulate several adenovirus vector-induced immune responses. J Innate Immun. 2009;1:376–388. doi: 10.1159/000207194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman ZC, Kiang A, Everett RS, Serra D, Yang XY, Clay TM, et al. Adenovirus infection triggers a rapid, MyD88-regulated transcriptome response critical to acute-phase and adaptive immune responses in vivo. J Virol. 2007;81:1796–1812. doi: 10.1128/JVI.01936-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee EG, Blattman JN, Kasturi SP, Kelley RP, Kaufman DR, Lynch DM, et al. Multiple innate immune pathways contribute to the immunogenicity of recombinant adenovirus vaccine vectors. J Virol. 2011;85:315–323. doi: 10.1128/JVI.01597-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee EG, Kelley RP, Agarwal I, Lynch DM, La Porte A, Simmons NL, et al. TLR4 ligands augment antigen-specific CD8+ T lymphocyte responses elicited by a viral vaccine vector. J Virol. 2010;84:10413–10419. doi: 10.1128/JVI.00928-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karan D, Krieg AM, Lubaroff DM. Paradoxical enhancement of CD8 T cell-dependent anti-tumor protection despite reduced CD8 T cell responses with addition of a TLR9 agonist to a tumor vaccine. Int J Cancer. 2007;121:1520–1528. doi: 10.1002/ijc.22873. [DOI] [PubMed] [Google Scholar]
- Germain RN. Uncovering the role of invariant chain in controlling MHC class II antigen capture. J Immunol. 2011;187:1073–1075. doi: 10.4049/jimmunol.1101663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe HM, Lopes L, Ikeda Y, Bailey R, Barde I, Zenke M, et al. Immunization with a lentiviral vector stimulates both CD4 and CD8 T cell responses to an ovalbumin transgene. Mol Ther. 2006;13:310–319. doi: 10.1016/j.ymthe.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Grujic M, Holst PJ, Christensen JP, Thomsen AR. Fusion of a viral antigen to invariant chain leads to augmented T-cell immunity and improved protection in gene-gun DNA-vaccinated mice. J Gen Virol. 2009;90 Pt 2:414–422. doi: 10.1099/vir.0.002105-0. [DOI] [PubMed] [Google Scholar]
- Holst PJ, Sorensen MR, Mandrup Jensen CM, Orskov C, Thomsen AR, Christensen JP. MHC class II-associated invariant chain linkage of antigen dramatically improves cell-mediated immunity induced by adenovirus vaccines. J Immunol. 2008;180:3339–3346. doi: 10.4049/jimmunol.180.5.3339. [DOI] [PubMed] [Google Scholar]
- Sorensen MR, Holst PJ, Pircher H, Christensen JP, Thomsen AR. Vaccination with an adenoviral vector encoding the tumor antigen directly linked to invariant chain induces potent CD4(+) T-cell-independent CD8(+) T-cell-mediated tumor control. Eur J Immunol. 2009;39:2725–2736. doi: 10.1002/eji.200939543. [DOI] [PubMed] [Google Scholar]
- Jensen S, Steffensen MA, Jensen BA, Schlüter D, Christensen JP, Thomsen AR. Adenovirus-based vaccine against Listeria monocytogenes: extending the concept of invariant chain linkage. J Immunol. 2013;191:4152–4164. doi: 10.4049/jimmunol.1301290. [DOI] [PubMed] [Google Scholar]
- Holst PJ, Christensen JP, Thomsen AR. Vaccination against lymphocytic choriomeningitis virus infection in MHC class II-deficient mice. J Immunol. 2011;186:3997–4007. doi: 10.4049/jimmunol.1001251. [DOI] [PubMed] [Google Scholar]
- Mikkelsen M, Holst PJ, Bukh J, Thomsen AR, Christensen JP. Enhanced and sustained CD8+ T cell responses with an adenoviral vector-based hepatitis C virus vaccine encoding NS3 linked to the MHC class II chaperone protein invariant chain. J Immunol. 2011;186:2355–2364. doi: 10.4049/jimmunol.1001877. [DOI] [PubMed] [Google Scholar]
- Neumann-Haefelin C, Thimme R. Adaptive immune responses in hepatitis C virus infection. Curr Top Microbiol Immunol. 2013;369:243–262. doi: 10.1007/978-3-642-27340-7_10. [DOI] [PubMed] [Google Scholar]
- Swadling L, Klenerman P, Barnes E. Ever closer to a prophylactic vaccine for HCV. Expert Opin Biol Ther. 2013;13:1109–1124. doi: 10.1517/14712598.2013.791277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes E, Folgori A, Capone S, Swadling L, Aston S, Kurioka A, et al. Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci Transl Med. 2012;4:115ra1. doi: 10.1126/scitranslmed.3003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara GA, Duncan CJ, Ewer KJ, Collins KA, Elias SC, Halstead FD, et al. Clinical assessment of a recombinant simian adenovirus ChAd63: a potent new vaccine vector. J Infect Dis. 2012;205:772–781. doi: 10.1093/infdis/jir850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy SH, Duncan CJ, Elias SC, Biswas S, Collins KA, O'Hara GA, et al. Phase Ia clinical evaluation of the safety and immunogenicity of the Plasmodium falciparum blood-stage antigen AMA1 in ChAd63 and MVA vaccine vectors. PLoS One. 2012;7:e31208. doi: 10.1371/journal.pone.0031208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy SH, Duncan CJ, Elias SC, Collins KA, Ewer KJ, Spencer AJ, et al. Phase Ia clinical evaluation of the Plasmodium falciparum blood-stage antigen MSP1 in ChAd63 and MVA vaccine vectors. Mol Ther. 2011;19:2269–2276. doi: 10.1038/mt.2011.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capone S, D'Alise AM, Ammendola V, Colloca S, Cortese R, Nicosia A, et al. Development of chimpanzee adenoviruses as vaccine vectors: challenges and successes emerging from clinical trials. Expert Rev Vaccines. 2013;12:379–393. doi: 10.1586/erv.13.15. [DOI] [PubMed] [Google Scholar]
- Catanzaro AT, Koup RA, Roederer M, Bailer RT, Enama ME, Moodie Z, et al. Vaccine Research Center 006 Study Team Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 candidate vaccine delivered by a replication-defective recombinant adenovirus vector. J Infect Dis. 2006;194:1638–1649. doi: 10.1086/509258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priddy FH, Brown D, Kublin J, Monahan K, Wright DP, Lalezari J, et al. Merck V520-016 Study Group Safety and immunogenicity of a replication-incompetent adenovirus type 5 HIV-1 clade B gag/pol/nef vaccine in healthy adults. Clin Infect Dis. 2008;46:1769–1781. doi: 10.1086/587993. [DOI] [PubMed] [Google Scholar]
- Tatsis N, Ertl HC. Adenoviruses as vaccine vectors. Mol Ther. 2004;10:616–629. doi: 10.1016/j.ymthe.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchbinder SP, Mehrotra DV, Duerr A, Fitzgerald DW, Mogg R, Li D, et al. Step Study Protocol Team Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372:1881–1893. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElrath MJ, De Rosa SC, Moodie Z, Dubey S, Kierstead L, Janes H, et al. Step Study Protocol Team HIV-1 vaccine-induced immunity in the test-of-concept Step Study: a case-cohort analysis. Lancet. 2008;372:1894–1905. doi: 10.1016/S0140-6736(08)61592-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plauzolles A, Lucas M, Gaudieri S. Hepatitis C virus adaptation to T-cell immune pressure. ScientificWorldJournal. 2013;2013:673240. doi: 10.1155/2013/673240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, James I, Pfafferott K, Nolan D, Klenerman P, Cheng W, et al. Divergent adaptation of hepatitis C virus genotypes 1 and 3 to human leukocyte antigen-restricted immune pressure. Hepatology. 2009;50:1017–1029. doi: 10.1002/hep.23101. [DOI] [PubMed] [Google Scholar]
- Steffensen MA, Jensen BA, Holst PJ, Bassi MR, Christensen JP, Thomsen AR. Pre-existing vector immunity does not prevent replication deficient adenovirus from inducing efficient CD8 T-cell memory and recall responses. PLoS One. 2012;7:e34884. doi: 10.1371/journal.pone.0034884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AL, Mosbruger T, Lauer GM, Pardoll D, Thomas DL, Ray SC. Comprehensive analyses of CD8+ T cell responses during longitudinal study of acute human hepatitis C. Hepatology. 2005;42:104–112. doi: 10.1002/hep.20749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folgori A, Spada E, Pezzanera M, Ruggeri L, Mele A, Garbuglia AR, et al. Acute Hepatitis C Italian Study Group Early impairment of hepatitis C virus specific T cell proliferation during acute infection leads to failure of viral clearance. Gut. 2006;55:1012–1019. doi: 10.1136/gut.2005.080077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest. 2009;119:1745–1754. doi: 10.1172/JCI39133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194:1395–1406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedemeyer H, He XS, Nascimbeni M, Davis AR, Greenberg HB, Hoofnagle JH, et al. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J Immunol. 2002;169:3447–3458. doi: 10.4049/jimmunol.169.6.3447. [DOI] [PubMed] [Google Scholar]
- Baerlecken NT, Nothdorft S, Stummvoll GH, Sieper J, Rudwaleit M, Reuter S, et al. 2013Autoantibodies against CD74 in spondyloarthritis. Ann Rheum Disepub ahead of print). [DOI] [PubMed]
- Baraliakos X, Baerlecken N, Witte T, Heldmann F, Braun J.2013High prevalence of anti-CD74 antibodies specific for the HLA class II-associated invariant chain peptide (CLIP) in patients with axial spondyloarthritis. Ann Rheum Disepub ahead of print). [DOI] [PubMed]
- Capone S, Zampaglione I, Vitelli A, Pezzanera M, Kierstead L, Burns J, et al. Modulation of the immune response induced by gene electrotransfer of a hepatitis C virus DNA vaccine in nonhuman primates. J Immunol. 2006;177:7462–7471. doi: 10.4049/jimmunol.177.10.7462. [DOI] [PubMed] [Google Scholar]
- Colloca S, Barnes E, Folgori A, Ammendola V, Capone S, Cirillo A, et al. Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Sci Transl Med. 2012;4:115ra2. doi: 10.1126/scitranslmed.3002925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lullo G, Soprana E, Panigada M, Palini A, Agresti A, Comunian C, et al. The combination of marker gene swapping and fluorescence-activated cell sorting improves the efficiency of recombinant modified vaccinia virus Ankara vaccine production for human use. J Virol Methods. 2010;163:195–204. doi: 10.1016/j.jviromet.2009.09.016. [DOI] [PubMed] [Google Scholar]
- Di Lullo G, Soprana E, Panigada M, Palini A, Erfle V, Staib C, et al. Marker gene swapping facilitates recombinant Modified Vaccinia Virus Ankara production by host-range selection. J Virol Methods. 2009;156:37–43. doi: 10.1016/j.jviromet.2008.10.026. [DOI] [PubMed] [Google Scholar]