

Abstract
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is an autosomal recessive disorder caused by mutations in TYMP, enconding thymidine phosphorylase (TP). TP deficiency results in systemic accumulation of thymidine and deoxyuridine, which interferes with mitochondrial DNA (mtDNA) replication and leads to mitochondrial dysfunction. To date, the only treatment available for MNGIE patients is allogeneic hematopoietic stem cell transplantation, which is associated with high morbidity and mortality. Here, we report that AAV2/8-mediated transfer of the human TYMP coding sequence (hcTYMP) under the control of a liver-specific promoter prevents the biochemical imbalances in a murine model of MNGIE. hcTYMP expression was restricted to liver, and a dose as low as 2 × 1011 genome copies/kg led to a permanent reduction in systemic nucleoside levels to normal values in about 50% of treated mice. Higher doses resulted in reductions to normal or slightly below normal levels in virtually all mice treated. The nucleoside reduction achieved by this treatment prevented deoxycytidine triphosphate (dCTP) depletion, which is the limiting factor affecting mtDNA replication in this disease. These results demonstrate that the use of AAV to direct TYMP expression in liver is feasible as a potentially safe gene therapy strategy for MNGIE.
Introduction
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a genetic disorder whose main clinical features are severe gastrointestinal dysmotility, progressive external ophthalmoplegia, and peripheral neuropathy. Patients with this condition usually die of complications of their gastrointestinal problems and their critical nutritional status, with an average age at death of 37 years.1 MNGIE is inherited as an autosomal recessive trait, and is caused by mutations in the nuclear gene TYMP, encoding thymidine phosphorylase (TP).2 This enzyme catalyzes the first step of thymidine (dThd) and deoxyuridine (dUrd) catabolism. As a consequence of TP dysfunction, MNGIE patients accumulate dThd and dUrd systemically,3 which ultimately results in imbalances in the mitochondrial pool of deoxyribonucleoside triphosphates (dNTP). More precisely, increased deoxythymidine triphosphate (dTTP) and decreased dCTP levels in mitochondria due to dThd excess have been observed in vitro and in vivo. This dNTP imbalance interferes with mitochondrial DNA (mtDNA) replication and results in depletion, multiple deletions, and point mutations, which in turn cause mitochondrial dysfunction.4,5,6
Several therapeutic strategies attempted for MNGIE have focused on removing the systemic dThd and dUrd excess,7,8,9,10 but the only treatment that has achieved a sustained reduction in these nucleosides to undetectable or nearly undetectable levels and an improvement in the long-term clinical outcome is allogeneic hematopoietic stem cell transplantation.11 The TP enzyme present in donor-derived platelets and white blood cells acts as a powerful clearing agent that eliminates toxic nucleosides from the patients' blood and ultimately results in full clearance of these water-soluble compounds from tissues. However, allogeneic hematopoietic stem cell transplantation has serious limitations, including the difficulty of obtaining suitable donors, the toxicity of the conditioning regimen, and the risk of graft failure and graft-versus-host disease. In addition, MNGIE patients are generally in poor medical condition at the time of the diagnosis. As a consequence of all these factors, this treatment is associated with high morbidity and mortality rates in these patients.12
A murine model of MNGIE, the Tymp/Upp1 double knockout (KO), has been generated and characterized.5 This model recapitulates the biochemical imbalances and, in older animals, some molecular features of the disease. Nonetheless, certain differences in deoxyribonucleoside metabolism between humans and mice should be noted. Three enzymes (TP, uridine phosphorylase 1 and uridine phosphorylase 2) initiate dThd and dUrd catabolism in mice, but uridine phosphorylase 2 is not knocked out in this model. In humans, this role is played entirely by TP. In addition, the normal dThd and dUrd plasma levels are higher in mice (1–4 µmol/l) than in humans (below 0.05 µmol/l). These differences, together with the short life span of mice compared to humans, are probably accounting for the lack of MNGIE-like clinical features in this murine model.
We previously demonstrated, in this animal model, that gene therapy using a lentiviral vector and targeting hematopoietic stem cells is a feasible approach13; however, lentiviral vectors are integrative and thus a potential cause of insertional oncogenesis. Here, we used an adeno-associated virus (AAV) vector as an alternative. As compared to lentiviral vectors, AAV vectors have a safer profile as they are mainly found in episomal form inside the host cell, which minimizes the risk of insertional oncogenesis.14 Our results demonstrate that AAV-mediated liver-targeted TYMP expression normalizes nucleoside and mitochondrial nucleotide metabolism in the MNGIE animal model and constitutes a promising strategy to be tested in clinical studies.
Results
Effect of the treatment on nucleoside levels
We generated an AAV2/8 vector (Supplementary Figure S1) containing the human TYMP coding sequence (hcTYMP) under the control of the liver-specific thyroxine-binding globulin promoter (TBG). Eight- to 12-week-old male Tymp/Upp1 double KO mice (animal model of MNGIE) were treated with a single intravenous injection of AAV2/8-TBG-hcTYMP. Four groups of animals were established, receiving four different doses: 2 × 1011, 1012, 2 × 1012, and 1013 genome copies (gc)/kg. In order to facilitate matching the results of each mouse among different figures, the same symbol identifies a particular mouse, within each category (wt, KO, 2 × 1011, 1012, 2 × 1012, and 1013 gc/kg) in Figures 1,2,3,4 and Supplementary Figure S2.
Figure 1.

Plasma dThd and dUrd concentrations. Plasma dThd (panel a) and dUrd (panel b) in mice treated with different AAV-TBG-hcTYMP doses (indicated within each plot) during the monitoring period. Grey areas indicate the concentration range in wild-type (wt) mice. In order to facilitate matching the results of each mouse among different figures, the same symbol identifies a particular mouse, within each category (wt, KO, 2 × 1011, 1012, 2 × 1012, and 1013 gc/kg) in Figures 1–4 and Supplementary Figure S2.
Figure 2.
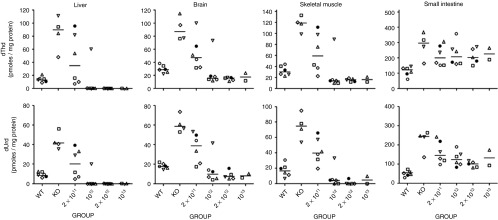
Nucleoside reduction in different tissues. dThd and dUrd concentration in liver, brain, skeletal muscle, and small intestine extracts at the end of the study (34 weeks after the treatment). Horizontal lines represent the median.
Figure 3.

Liver-specific thymidine phosphorylase (TP) restoration. (a) Representative western blot showing human TP and β-actin in liver homogenates (left panel) and densitometric analysis (right panel). Total amount of protein was quantified by densitometry and referred to a calibrator (see Materials and Methods). Arrows represent mobility of the molecular weight ladder. Bars represent mean (+SD) of relative human TP protein. Groups and doses (gc/kg) are indicted on the x-axis. (b) Adeno-associated virus genome copies per cell in liver DNA extracts measured by qPCR using a human TYMP cDNA-specific probe and referred to the single copy mouse gene, angiogenin-1. Horizontal lines represent the median. (c) TP activity measured in liver extracts. Horizontal lines represent the median.
Figure 4.

Thymidine phosphorylase (TP) activity in tissues other than liver. TP activity measured in blood cells, skeletal muscle, small intestine, lung, brain, spleen, kidney, and heart extracts of wild-type mice, nontreated KO mice, and treated KO mice 34 weeks after vector administration.
Blood samples were collected periodically over 28 weeks to measure nucleoside levels (Figure 1). At 3.5 weeks after AAV administration, plasma dThd concentration had decreased to wild-type (wt) values in six of eight (75%) animals treated with the lowest dose (2 × 1011 gc/kg), and three of them (37.5%) maintained these low levels over the 28 weeks of monitoring. Higher doses led to a reduction in plasma dThd below wt levels by 0.5 weeks after treatment in all but one animal (17 of 18), and the reduction was maintained over the entire time monitored. Only one mouse treated with 1012 gc/kg did not respond to the treatment (Figure 1a). Similar results were obtained for plasma dUrd levels (Figure 1b).
Eight months after the treatment (end of the study), a subgroup of mice was killed and tissues were collected for analysis. Nucleosides were measured in liver and three additional tissues affected in MNGIE patients (brain, skeletal muscle, and small intestine). In mice with reduced circulating dThd and dUrd concentrations after treatment (Figure 1 and Supplementary Figure S2), concomitant reductions were found in liver, brain, and skeletal muscle (Figure 2), demonstrating that TP activity in the liver efficiently clears dThd and dUrd from their entire distribution volume. Especially pronounced was the reduction in liver of animals receiving the higher AAV doses (undetectable dThd and dUrd in most cases), likely due to high local TP activity in the target tissue. In contrast, dThd and dUrd levels determined in small intestine were virtually unchanged in treated animals.
Transgenic TYMP expression was targeted to the liver
Human TP protein was undetectable by western blot analysis in liver in all untreated wt and KO mice analyzed (Figure 3a). Low but detectable levels were found in four of the six mice treated with the lowest AAV dose, and high levels were seen in all mice but one belonging to the groups receiving higher doses (Figure 3a). The only mouse lacking TP protein was the same animal that did not show a nucleoside reduction in plasma and tissues (see above). Real-time quantitative polymerase chain reaction (qPCR) analysis of hcTYMP copy number confirmed effective transduction in a dose-dependent manner. In the group of mice treated with the lowest vector dose, hcTYMP copy number was undetectable in animals that did not show a nucleoside reduction in blood collected at completion of the study (Figure 3b and Supplementary Figure S2). Similarly, in AAV-treated animals, liver TP activity increased in a dose-dependent manner (Figure 3c). For the lowest dose (2 × 1011 gc/kg), two animals reached normal TP activity, and the same animals also showed reduced nucleosides in blood and tissues (Figures 1 and 2; Supplementary Figure S2). Negligible or absent TP activities were found in tissues other than liver in treated mice (Figure 4), indicating highly specific hepatic transgene expression. In treated animals, AAV dose strongly correlated with hcTYMP copy number (P < 0.001), TP activity (P < 0.001), and TP protein (P = 0.001) levels in liver.
Immunostaining of hepatic tissue further confirmed that TP protein was present in liver (Figure 5). The protein showed a patchy distribution and the comparison with the nuclei staining indicated cytoplasmic localization, as expected for TP protein. The number of stained hepatocytes was consistent with the copy number results and the AAV dose.
Figure 5.

Liver immunostaining. Immunofluorescence of liver from wild-type (wt) mice, nontreated KO mice, and KO mice treated with 2 × 1011, 1012, 2 × 1012, and 1013 gc/kg. Liver cryosections were stained with an antibody against human thymidine phosphorylase (TP) (green). Each picture shows nine fields arranged in one image of a representative mouse (original magnification per field, ×20). For the 2 × 1011 and 1012 gc/kg doses, one mouse with low TP activity (left) and one mouse with high TP activity (right) are shown. Scale bar = 200 µm. Nuclei were stained with Hoechst 33342 (blue).
Hepatotoxicity was not detected in any of the animals, as assessed by monitoring plasma alanine aminotransferase activity, and no differences in weight were observed between treated mice and untreated KO or wt animals (Supplementary Figure S3).
AAV treatment prevents dCTP depletion
In MNGIE, dThd excess results in changes in the mitochondrial dNTP pools. We investigated whether the nucleoside decrease occurring in mice treated with the therapeutic vector (AAV2/8-TBG-hcTYMP) prevented mitochondrial dNTP imbalances in liver (Figure 6). Although the average dTTP value was slightly higher in KO mice than in wt mice, the difference did not reach statistical significance, in contrast to the results from a previous report.5 This is likely because our values had a rather wide variability. dCTP was significantly reduced in KO mice. At the end of the study, dCTP concentration was increased in treated mice and positively correlated with the dose (P < 0.05, Spearman's correlation test). A similar increase was observed in the deoxyguanosine triphosphate pool. In contrast, the treatment did not have an impact on dTTP levels.
Figure 6.
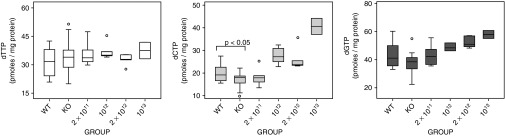
dNTPs in liver. Mitochondrial dNTP content in liver of wild-type (wt) mice (n = 16), nontreated KO mice (n = 14), and KO mice treated with 2 × 1011 (n = 6), 1012 (n = 6), 2 × 1012 (n = 5), and 1013 gc/kg (n = 2). Box plots represent the median (horizontal line), the interquartile range (box), the maximum and minimum (whiskers), and outliers (open circles). Statistical comparisons between wt and KO mice were performed with the Student t-test.
Discussion
AAV vectors have been extensively used in gene therapy clinical trials. The recent success of AAV-mediated gene transfer in hemophilia B has shown that the use of a liver-specific AAV vector provides sustained therapeutic levels of coagulation factor IX in humans.15 The effective AAV-mediated expression of the gene of interest in liver indicates a potential strategy for treatment of genetic metabolic disorders caused by systemic accumulation of toxic metabolites. The feasibility of this approach in ethylmalonic encephalopathy was shown in a preclinical study.16 This strategy is particularly appropriate for MNGIE, which is caused by systemic accumulation of dThd and dUrd. The molecular defect (TP deficiency) does not need to be corrected in a specific target tissue; instead, the enzyme can exert the catabolic role inside the cell because dThd and dUrd are small water-soluble molecules that readily diffuse across plasma membranes through equilibrative transporters.17 Our previous results have shown that TYMP expression does not require strict regulation, and correction of a limited number of cells suffices to clear the systemic overload of nucleosides.13
The results reported here with an AAV vector in vivo improve our previous results obtained with an ex vivo approach using lentiviral vectors targeted to the hematopoietic tissue. The use of an integrative vector was necessary to achieve permanent correction because of the high turnover of the hematopoietic cells. Although integrative vectors have been successfully used for clinical purposes, the risk of cancer by insertional mutagenesis, evidenced in some clinical trials, still constitutes a serious concern.18,19 Among the advantages of AAV vectors are their ability to transduce several tissues and cell types and the fact that they predominantly remain as episomes.20,21,22 Although a small percentage can integrate in the host genome, there is no evidence of AAV-induced malignancy.23,24 The vector used in the present study was designed to correct the enzyme deficiency in liver, since AAV2/8 tropism enables liver transduction among other target tissues25 and the TBG promoter used ensures liver-specific expression.26 In our study, a relatively low dose of vectors (2 × 1011 gc/kg) led to stable transduction in about half of treated mice, with a concomitant reduction in nucleoside levels to normal values in blood and tissues that was sustained over time. Higher doses resulted in over-reduction: nucleoside levels were lowered to values slightly below wt levels. hcTYMP copy number per cell and TP activity in liver correlated with the AAV dose. Therefore, very low AAV doses are effective in reducing nucleoside levels, and these doses could be reduced even further if the final therapeutic vector included additional improvements such as codon optimization or a self-complementary configuration.27 This is of particular importance for patients because low doses minimize any potential risk that may be associated with the treatment.
In contrast to the clear nucleoside reduction observed in liver, skeletal muscle, and brain, dThd and dUrd in small intestine did not reach wt levels in treated animals, and only a slight nonsignificant tendency to reduced nucleosides was observed. Absorption of nucleosides from the diet may be contributing to high concentrations of these compounds in transit through the epithelial cells in the intestinal lumen, which may be more difficult to clear than other tissues. This important point needs further investigation, as the gastrointestinal dysfunction is one of the hallmarks of the disease. Therefore, ensuring the effective nucleoside clearance in this organ may be needed. This animal model is not appropriate to answer this question because it does not recapitulate the gastrointestinal symptoms observed in MNGIE patients.5 However, the use of alternate promoters or AAV serotypes targeting TYMP expression also in the intestine in the animal model may be helpful to find out whether these approaches are more effective reducing nucleosides in this target tissue.
Interestingly, the reduction in nucleoside levels reached a sustained nadir at doses of 1012 gc/kg and higher (range of plasma dThd concentrations 0.2–2.3 µmol/l, regardless of AAV dose) suggesting that there may be some type of biochemical mechanism preventing a further decrease in dThd concentration below a certain level in vivo. Similar sustained nadirs were observed for dThd and dUrd content in brain and skeletal muscle. However, nucleoside levels were undetectable in liver of mice treated with higher doses (1012 and above), suggesting that the mechanism would be outweighed by the high local TP activities reached in this organ. It is important to note, however, that the animals showed no sign of hepatic toxicity during or at the end of the study.
In MNGIE, dThd excess leads to mitochondrial dNTP imbalance, namely dTTP excess and dCTP depletion.5,6 The nucleoside decrease achieved by AAV treatment prevented dCTP depletion. Because dThd concentrations were over-reduced in mice treated with the highest doses, a potential adverse effect would be induction of dTTP depletion, but this was not observed in our experiments, indicating that additional mechanisms may be activated to prevent dTTP over-reduction. Regulatory steps in dTTP synthesis, such as the TK2-deoxynucleotidase substrate cycle or thymidylate synthase upregulation, could be involved. However, the treatment induced an unexpected increase in mitochondrial deoxyguanosine triphosphate, paralleling dCTP increases. The reasons for this increase are unknown. Long-term study of these animals will clarify whether this deoxyguanosine triphosphate excess has any negative consequences. Overall, our results indicate that the treatment developed in this study makes it possible to influence the mitochondrial dNTP pool in vivo. Dose adjustment would likely allow us to modulate the effect to achieve a final desired balance of dTTP and dCTP.
Although the MNGIE mouse model only recapitulates the biochemical imbalances of the disease and some molecular features in very old animals, it is the best tool available to investigate therapy approaches for MNGIE, by enabling detailed study of the biochemical effects of these approaches. Our results clearly demonstrate that it is feasible to prevent the biochemical disturbances observed in MNGIE with a strategy based on AAV-mediated transfer of liver-targeted hcTYMP expression. We propose that this approach should be translated to clinical trials for patients who are not eligible for allogeneic hematopoietic stem cell transplantation.
Materials and methods
Vector construction, production, and titration. hcTYMP was PCR amplified, cloned into the Pcr2.1 TOPO vector (Invitrogen, Carlsbad, CA), sequence verified, and finally cloned into the single EagI restriction site of the AAV2/8-TBG vector. The virus was produced by the AAV Vector Core of the Telethon Institute of Genetics and Medicine by triple transfection of 293 cells, and was purified by CsCl gradients.28 Physical titers of the viral preparations (genome copies/ml) were determined by real-time qPCR29 (Applied Biosystems, Foster City, CA) and dot-blot analysis. The titers obtained with both methods were reasonably consistent (qPCR: 1.6 × 1013 genome copies (gc)/ml; dot-blot analysis: 2.0 × 1013 gc/ml). The mean of the two values was used for dosing the animals.
Animal procedures. All animal procedures were performed in accordance with protocols approved by our institutional review board and committee on animal care and use. Eight- to 12-week-old male Tymp/Upp1 KO mice5 were treated with a single intravenous injection (tail vein) of AAV2/8-TBG-hcTYMP. Four groups of animals were treated with four different doses: 2 × 1011, 1012, 2 × 1012, and 1013 genome copies (gc)/kg. Blood samples (ethylene diamine tetraacetic acid) were collected from the saphenous vein 1 week before treatment and every 2–4 weeks after treatment, from 0.5 weeks until 28 weeks after treatment.
TP activity and nucleoside determination. Plasma dThd and dUrd concentrations were analyzed by HPLC-UV, as previously described.13 TP activity and tissue nucleoside concentrations were measured in mice 34 weeks after treatment or in nontreated age-matched mice. After killing the mice by cervical dislocation, tissues were collected and immediately frozen in liquid nitrogen and stored at −80 °C until analysis. Frozen samples were homogenized in lysis buffer (50 mmol/l Tris–HCl, pH 7.2; 10 ml/l Triton X-100; 2 mmol/l phenylmethylsulfonyl fluoride; 0.2 ml/l 2-mercaptoethanol) in a Potter homogenizer. The homogenates were centrifuged at 20,000 x g for 30 minutes at 4 °C, and supernatants were separated into two aliquots. One aliquot was used as described elsewhere for protein determination30 and TP activity determination.31 The other aliquot was frozen until used to measure nucleosides by liquid chromatography coupled to tandem mass spectrometry. To prevent in vitro degradation of dThd and dUrd during the homogenization procedure, for samples with TP activities above 100 nmol/hour/mg prot (i.e., all liver samples of mice treated with doses equal or above 1012 gc/kg but one) and for all small intestine samples, a second piece of tissue was homogenized in the presence of the 100 µmol/l 5-bromouracil (TP inhibitor) for the determination of these nucleosides. Unfrozen supernatants were centrifuged at 20,000 x g for 10 minutes at 4 °C to eliminate any remaining particles, and clean supernatants were deproteinized by ultrafiltration (10 kDa Amicon Ultra filters; Merck Millipore, Billerica, MA) at 14,000 x g and 4 °C for 30 minutes. Five microliters of deproteinized homogenate were injected into an Acquity UPLC-MS/MS apparatus (Acquity UPLC-Xevo TQ Mass Spectrometer; Waters, Milford, MA) using an Acquity UPLC BEH C18 column (100 × 2.1 mm, 130 Å pore, 1.7 µm particle, Waters). The components of the sample were resolved at 0.5 ml/minute through a binary gradient-elution using a saline buffer (20 mmol/l ammonium acetate, pH 5.6) and acetonitrile as follows: 0 to 1.1 minutes, isocratic 100% saline buffer; 1.1 to 5 minutes, gradient from 0 to 13.6% acetonitrile; 5 to 5.1 minutes, gradient from 13.6 to 100% acetonitrile; 5.1 to 6.1 minutes, isocratic 100% acetonitrile; 6.1 to 7.2 minutes, isocratic 100% saline buffer. Eluate components were detected by multiple reaction monitoring, with positive electrospray for dThd (transition mass 242.8 to 127.1, cone voltage 10 V, collision energy 12 eV) and dUrd (transition mass 228.8-113.08, cone voltage 8 V, collision energy 12 eV). Calibration curves made with aqueous standards were processed in parallel, and concentrations were obtained from interpolation of the peak areas.
Western blot. Liver protein was extracted in TP activity lysis buffer supplemented with a protease inhibitor cocktail (100 µmol/l 4-(2-aminoethyl)benzenesulfonyl fluoride, 15 nmol/l aprotinin, 6.5 µmol/l bestatin, 50 µmol/l ethylene diamine tetraacetic acid, 700 nmol/l E-64, 50 nmol/l leupeptin; Sigma-Aldrich, St Louis, MO). In all, 10 µg of protein extracts were electrophoresed on a 10% sodium dodecyl sulfate–polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, and probed with an anti-TP rabbit polyclonal antibody (Abcam, Cambridge, UK) and an anti-β-actin mouse monoclonal antibody (Sigma-Aldrich), and then with peroxidase conjugated goat anti-rabbit immunoglobulins (Dako, Glostrup, Denmark). Bands were visualized by treating the membranes with the Immobilon Western chemiluminescent kit (Merck Millipore), and quantified using Image J software (NIH, Bethesda, MD). All the TP-to-β-actin ratios were referred to the corresponding ratio obtained from a control extract run in all gels and used as a calibrator.
Immunofluorescence. Immunofluorescent histological analysis of human TP was performed in 10 µm liver cryosections fixed with acetone:methanol (1:1 v/v). Sections were then blocked with BSA 2% (w/v) in TBS buffer (50 mmol/l Tris–HCl, pH 7.6; 125 mmol/l NaCl) for 30 minutes and incubated with 20 µg/ml anti-TP rabbit polyclonal antibody (Abcam) overnight at 4 °C. After washing, sections were stained with AlexaFluor 488 conjugated goat anti-rabbit IgG (Invitrogen). Nuclear staining was performed with 1 µg/ml Hoechst 33342.
Vector copy number. DNA was extracted from liver with the QIAamp DNA mini kit (Qiagen, Hilden, Germany). Detection and quantification of vector genome copies per cell were performed by qPCR in the ABI PRISM 7900 sequence detection system (Applied Biosystems). hcTYMP DNA was quantified using the predesigned TaqMan MGB gene expression assay Hs00157317_m1 (Applied Biosystems), and was referred to the single copy nuclear gene Ang1 using the predesigned TaqMan MGB gene expression assay Mm00833184_s1 (Applied Biosystems). The quantifications were based on a standard curve prepared with different dilutions of vectors containing hcTYMP DNA or a specific region of the Ang1 gene.
Liver mitochondrial dNTP quantification. Liver mitochondria were isolated as previously described.6 A volume (~0.1–0.3 ml) of suspension with isolated mitochondria containing 0.5 mg protein were treated with trichloroacetic acid (final concentration 0.5 mol/l) and centrifuged at 20,000 x g for 5 minutes at 4 °C. Supernatants were neutralized with 1.5 volumes of 0.5 mol/l tri-N-octylamine in Freon (1,1,2-trichlorotrifluoroethane) and centrifuged for 10 minutes at 10,000 x g at 4 °C. Half the aqueous upper phase was recovered and dried under speed vacuum. Dry dNTP extracts were dissolved in 125 µl of 40 mmol/l Tris-HCl (pH 7.4) and stored at −80 °C until measurement. For mitochondrial dNTP quantification, we used the polymerase-based assay previously described.6 In order to characterize the purity of the mitochondrial fractions obtained with our protocol, three additional independent mitochondrial isolations were performed to determine citrate synthase activity as mitochondrial marker,32 lactate dehydrogenase as cytosolic marker (LDH-L reagent; Thermo Scientific, Louisville, CO) and β-galactosidase and catalase as lysosomal and peroxisomal markers, respectively.33 Based on the determination of these markers in the raw homogenates and in the mitochondria-enriched fractions, we obtained a 4.3 ± 0.3-fold mitochondrial enrichment and negligible cytosolic contamination (0.78 ± 0.66%). Contamination by lysosomes (65 ± 34%) and peroxisomes (55 ± 10%) was rather high. Overall, the quality of our mitochondrial fraction was good enough for our purposes, because lysosomes and peroxisomes do not contain dNTPs, and cytosol, which contains dNTPs, was virtually eliminated from our preparations.
Statistical analysis. Statistical analysis was performed with the SPSS 15.0 software (IBM, Armonk, NY). The tests used are indicated in the figure legends. For statistical purposes, undetectable values were considered as zero.
SUPPLEMENTARY MATERIAL Figure S1. Schematic representation of AAV2/8-TBG-hcTYMP. Figure S2. Plasma nucleoside levels at the completion of the study. Figure S3. Absence of liver toxicity in AAV treated mice.
Acknowledgments
This work was supported by the Instituto de Salud Carlos III (grant PI12/00322 to R.M.), the United Mitochondrial Disease Foundation (postdoctoral grant 12–029 to J.T.), the French Muscular Dystrophy Association-Téléthon (AFMTéléthon, to Y.C.) the Pierfranco and Luisa Mariani Foundation Italy, Telethon-Italy (GPP10005 and GGP11011 to M.Z.), Cariplo (2011-0526 to M.Z.), European Research Council Grant (ERC-322424 to M.Z.), and the Italian Ministry of Health Research Grant (GR-2010-2306-756 to C.V). There is no conflict of interest to disclose.
Supplementary Material
References
- Garone C, Tadesse S, Hirano M. Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain. 2011;134 Pt 11:3326–3332. doi: 10.1093/brain/awr245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- Martí R, Nishigaki Y, Hirano M. Elevated plasma deoxyuridine in patients with thymidine phosphorylase deficiency. Biochem Biophys Res Commun. 2003;303:14–18. doi: 10.1016/s0006-291x(03)00294-8. [DOI] [PubMed] [Google Scholar]
- Pontarin G, Ferraro P, Valentino ML, Hirano M, Reichard P, Bianchi V. Mitochondrial DNA depletion and thymidine phosphate pool dynamics in a cellular model of mitochondrial neurogastrointestinal encephalomyopathy. J Biol Chem. 2006;281:22720–22728. doi: 10.1074/jbc.M604498200. [DOI] [PubMed] [Google Scholar]
- López LC, Akman HO, García-Cazorla A, Dorado B, Martí R, Nishino I, et al. Unbalanced deoxynucleotide pools cause mitochondrial DNA instability in thymidine phosphorylase-deficient mice. Hum Mol Genet. 2009;18:714–722. doi: 10.1093/hmg/ddn401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Vioque E, Torres-Torronteras J, Andreu AL, Martí R. Limited dCTP availability accounts for mitochondrial DNA depletion in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) PLoS Genet. 2011;7:e1002035. doi: 10.1371/journal.pgen.1002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavuz H, Ozel A, Christensen M, Christensen E, Schwartz M, Elmaci M, et al. Treatment of mitochondrial neurogastrointestinal encephalomyopathy with dialysis. Arch Neurol. 2007;64:435–438. doi: 10.1001/archneur.64.3.435. [DOI] [PubMed] [Google Scholar]
- Spinazzola A, Marti R, Nishino I, Andreu AL, Naini A, Tadesse S, et al. Altered thymidine metabolism due to defects of thymidine phosphorylase. J Biol Chem. 2002;277:4128–4133. doi: 10.1074/jbc.M111028200. [DOI] [PubMed] [Google Scholar]
- Moran NF, Bain MD, Muqit MM, Bax BE. Carrier erythrocyte entrapped thymidine phosphorylase therapy for MNGIE. Neurology. 2008;71:686–688. doi: 10.1212/01.wnl.0000324602.97205.ab. [DOI] [PubMed] [Google Scholar]
- Lara MC, Weiss B, Illa I, Madoz P, Massuet L, Andreu AL, et al. Infusion of platelets transiently reduces nucleoside overload in MNGIE. Neurology. 2006;67:1461–1463. doi: 10.1212/01.wnl.0000239824.95411.52. [DOI] [PubMed] [Google Scholar]
- Hirano M, Martí R, Casali C, Tadesse S, Uldrick T, Fine B, et al. Allogeneic stem cell transplantation corrects biochemical derangements in MNGIE. Neurology. 2006;67:1458–1460. doi: 10.1212/01.wnl.0000240853.97716.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halter J, Schüpbach WM, Casali C, Elhasid R, Fay K, Hammans S, et al. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach. Bone Marrow Transplant. 2011;46:330–337. doi: 10.1038/bmt.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Torronteras J, Gómez A, Eixarch H, Palenzuela L, Pizzorno G, Hirano M, et al. Hematopoietic gene therapy restores thymidine phosphorylase activity in a cell culture and a murine model of MNGIE. Gene Ther. 2011;18:795–806. doi: 10.1038/gt.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RH. Adeno-associated virus integration: virus versus vector. Gene Ther. 2008;15:817–822. doi: 10.1038/gt.2008.55. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meo I, Auricchio A, Lamperti C, Burlina A, Viscomi C, Zeviani M. Effective AAV-mediated gene therapy in a mouse model of ethylmalonic encephalopathy. EMBO Mol Med. 2012;4:1008–1014. doi: 10.1002/emmm.201201433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W, Engel K, Wang J. Mammalian nucleoside transporters. Curr Drug Metab. 2004;5:63–84. doi: 10.2174/1389200043489162. [DOI] [PubMed] [Google Scholar]
- Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Nakai H, Iwaki Y, Kay MA, Couto LB. Isolation of recombinant adeno-associated virus vector-cellular DNA junctions from mouse liver. J Virol. 1999;73:5438–5447. doi: 10.1128/jvi.73.7.5438-5447.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns WG, Afione SA, Fulmer SB, Pang MC, Erikson D, Egan M, et al. Recombinant adeno-associated virus (AAV-CFTR) vectors do not integrate in a site-specific fashion in an immortalized epithelial cell line. Gene Ther. 1996;3:748–755. [PubMed] [Google Scholar]
- Flotte TR, Afione SA, Zeitlin PL. Adeno-associated virus vector gene expression occurs in nondividing cells in the absence of vector DNA integration. Am J Respir Cell Mol Biol. 1994;11:517–521. doi: 10.1165/ajrcmb.11.5.7946381. [DOI] [PubMed] [Google Scholar]
- Donsante A, Vogler C, Muzyczka N, Crawford JM, Barker J, Flotte T, et al. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors. Gene Ther. 2001;8:1343–1346. doi: 10.1038/sj.gt.3301541. [DOI] [PubMed] [Google Scholar]
- Gauttier V, Pichard V, Aubert D, Kaeppel C, Schmidt M, Ferry N, et al. No tumour-initiating risk associated with scAAV transduction in newborn rat liver. Gene Ther. 2013;20:779–784. doi: 10.1038/gt.2013.7. [DOI] [PubMed] [Google Scholar]
- Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- Tessitore A, Faella A, O'Malley T, Cotugno G, Doria M, Kunieda T, et al. Biochemical, pathological, and skeletal improvement of mucopolysaccharidosis VI after gene transfer to liver but not to muscle. Mol Ther. 2008;16:30–37. doi: 10.1038/sj.mt.6300325. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Gray JT, Ng CY, Zhou J, Spence Y, Waddington SN, et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood. 2006;107:2653–2661. doi: 10.1182/blood-2005-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Chirmule N, Berta SC, McCullough B, Gao G, Wilson JM. Gene therapy vectors based on adeno-associated virus type 1. J Virol. 1999;73:3994–4003. doi: 10.1128/jvi.73.5.3994-4003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Qu G, Burnham MS, Huang J, Chirmule N, Joshi B, et al. Purification of recombinant adeno-associated virus vectors by column chromatography and its performance in vivo. Hum Gene Ther. 2000;11:2079–2091. doi: 10.1089/104303400750001390. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Valentino ML, Martí R, Tadesse S, López LC, Manes JL, Lyzak J, et al. Thymidine and deoxyuridine accumulate in tissues of patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) FEBS Lett. 2007;581:3410–3414. doi: 10.1016/j.febslet.2007.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medja F, Allouche S, Frachon P, Jardel C, Malgat M, Mousson de Camaret B, et al. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion. 2009;9:331–339. doi: 10.1016/j.mito.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Graham JM.2001Purification of a crude mitochondrial fraction by density-gradient centrifugation Curr Protoc Cell BiolChapter 3: Unit 3 4. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.