

The recessively inherited, adult onset, quadriceps sparing myopathy with a predilection for distal muscles has received multiple historic names. The disorder was described in 1981 in Japanese patients and termed Nonaka Distal Myopathy [1], later commonly referred to as Distal Myopathy with Rimmed Vacuoles (DMRV) (OMIM#605820). In 1984, the disorder was described as vacuolar myopathy sparing the quadriceps in Iranian-Jewish patients [2], later commonly referred to as Inclusion Body Myopathy 2 (IBM2) or Hereditary Inclusion Body Myopathy (HIBM) (OMIM#600737). Mapping of the causative gene to the same locus on chromosome 9 in different cohorts of patient [3, 4], and ultimately identification of mutations in the causative gene GNE in all cohorts [5, 6], confirmed that these myopathies are in fact the same condition.
However, since identification of GNE as the common causative gene, the multiple historic names for the disorder continue to be used by research groups worldwide. This disease nomenclature becomes increasingly confusing for clinicians, patients and researchers. Therefore, an international consortium (of which the authors are also members) has recently proposed to rename the disorder “GNE myopathy”, substituting all previous disease definitions. We all are now using this new name and hope that it will become the only term worldwide.
After initial discovery of GNE gene defects to be causative for GNE myopathy, eight different GNE mRNA splice variants were identified, encoding (at least theoretically) eight protein isoforms [7]. The human GNE gene (GenBank Gene ID: 10020, NC_000009; ENSEMBL ENSG00000159921) consists of 13 exons, but each of the individual GNE mRNA splice variants consists of fewer exons. However, for mutation annotation purposes, only two major transcripts are relevant, which together span all 13 exons (Figure 1) [7]. We provide NCBI GenBank accession numbers for the two major isoforms hGNE1 and hGNE2 in the text below, and provide their ENSEMBL IDs in Table 1.
Figure 1.
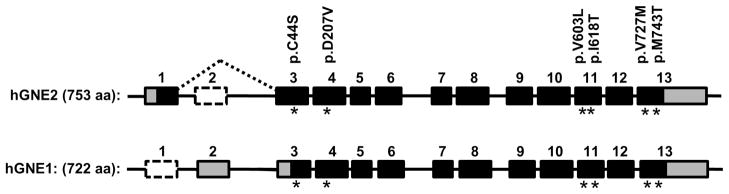
Human GNE mRNA transcripts and isoforms.
Structures of the two main human GNE mRNA transcripts (not to scale) and the human GNE isoforms (hGNE1 and hGNE2) are illustrated. Note that mRNA variant 1 (the longest splice form) encodes the hGNE2 protein, while mRNA variant 2 encodes the hGNE1 protein (traditionally known and studied as the sole translated GNE protein). Black boxes: open reading frame; Gray boxes: untranslated mRNA regions; White dotted lined boxes: skipped exons. Locations of selected GNE myopathy-associated mutations (see Table 1) are indicated by stars. GenBank Accession numbers and translated amino acids (aa) are provided. Modified and updated from [7].
Table 1.
Most frequent GNE myopathy-associated GNE variants
| New Nomenclature1 | Previous Nomenclature | |||||
|---|---|---|---|---|---|---|
| hGNE2 isoform | mRNA transcript | Exon | hGNE1 isoform | mRNA transcript | Exon | Ethnicity2 |
| GenBank hGNE2 NP_001121699 |
GenBank Variant 1 NM_001128227 |
GenBank GNE gDNA NC_000009 |
GenBank hGNE1 NP_005467 |
GenBank Variant 2 NM_005476 |
||
| ENSEMBL GNE-003 ENSP 00000379839 |
ENSEMBL GNE-003 ENST 00000396594 |
ENSEMBL GNE gDNA ENSG 00000159921 |
ENSEMBL GNE-001 ENSP 00000367134 |
ENSEMBL GNE-001 ENST 00000377902 |
||
| p.C44S | c.131G>C | 3 | p.C13S | c.38G>C | 2 | Japanese |
| p.D207V | c.620A>T | 4 | p.D176V | c.527A>T | 3 | Japanese |
| p.V603L | c.1807G>C | 11 | p.V572L | c.1714G>C | 10 | Japanese |
| p.I618T | c.1853T>C | 11 | p.I587T | c.1760T>C | 10 | Cajun, Roma Gypsies |
| p.V727M | c.2179G>A | 13 | p.V696M | c.2086G>A | 12 | Indian |
| p.M743T | c.2228T>C | 13 | p.M712T | c.2135T>C | 12 | Middle Eastern |
Nomenclature according to universally adapted gene/protein nomenclature rules.
Ethnicity in which the variant is mostly reported.
hGNE1 (GenBank NP_005467) is the originally described GNE protein which covers 722 amino acids [5] and is, confusingly, encoded in GenBank by mRNA transcript variant 2 (NM_005476). The hGNE2 isoform (NP_001121699) covers 753 amino acids and is encoded by the longest GNE mRNA transcript, variant 1 (NM_001128227).
The discovery of the additional N-terminal sequence (and novel exon 1) [8] encoding hGNE2, is potentially confusing since most previous molecular and biochemical studies (including all mutation reports) refer to the hGNE1 isoform, while according to universally adapted gene/protein nomenclature rules the longest mRNA splice form ought to be used for annotating nucleotide/amino acid locations (http://www.hgvs.org/mutnomen/refseq.html). Hence, amino acid numbering of previously reported GNE studies (based on hGNE1 nomenclature), including patient mutation reports, should be supplemented with 31 amino acids to adhere to the current (hGNE2) nomenclature guidelines, and nucleotide numbering should be supplemented with 93 bases. For exon numbering, the numbering according to the entire 13 exon GNE gDNA gene ought to be used, which means that exon numbering of previously reported GNE studies (based on hGNE1 nomenclature) have to be supplemented with one exon.
Adaptation to the hGNE2 nomenclature can initially be confusing; however, we strongly support adaptation of this ‘new’ nomenclature. Laboratories/researchers not familiar with the GNE myopathy field and disease/gene history will report patient mutations and research tools (antibodies, enzyme activities, siRNA, nextgen sequence databases, etc.) according to current universally adapted nomenclature rules. Moreover, although there are no variants reported yet in the additional 31 amino acids of hGNE2 (perhaps because this region has not been considered for mutation analysis in many patients), future variants in this region could not be accurately named using hGNE1 as a reference. To illustrate the new terminology, we list both hGNE2 (‘new’ nomenclature) and hGNE1 (previous nomenclature) classifications and up to date exon numbers of the most frequent GNE mutations associated with GNE myopathy in Table 1. However, since history will leave its tracks, we strongly suggest accompanying all future references to GNE with the appropriate GenBank accession numbers.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nonaka I, Sunohara N, Ishiura S, Satoyoshi E. Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J Neurol Sci. 1981;51:141–55. doi: 10.1016/0022-510x(81)90067-8. [DOI] [PubMed] [Google Scholar]
- 2.Argov Z, Yarom R. “Rimmed vacuole myopathy” sparing the quadriceps. A unique disorder in Iranian Jews. J Neurol Sci. 1984;64:33–43. doi: 10.1016/0022-510x(84)90053-4. [DOI] [PubMed] [Google Scholar]
- 3.Mitrani-Rosenbaum S, Argov Z, Blumenfeld A, Seidman CE, Seidman JG. Hereditary inclusion body myopathy maps to chromosome 9p1-q1. Hum Mol Genet. 1996;5:159–63. doi: 10.1093/hmg/5.1.159. [DOI] [PubMed] [Google Scholar]
- 4.Ikeuchi T, Asaka T, Saito M, et al. Gene locus for autosomal recessive distal myopathy with rimmed vacuoles maps to chromosome 9. Ann Neurol. 1997;41:432–7. doi: 10.1002/ana.410410405. [DOI] [PubMed] [Google Scholar]
- 5.Eisenberg I, Avidan N, Potikha T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29:83–7. doi: 10.1038/ng718. [DOI] [PubMed] [Google Scholar]
- 6.Nishino I, Noguchi S, Murayama K, et al. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002;59:1689–93. doi: 10.1212/01.wnl.0000041631.28557.c6. [DOI] [PubMed] [Google Scholar]
- 7.Yardeni T, Choekyi T, Jacobs K, et al. Identification, tissue distribution, and molecular modeling of novel human isoforms of the key enzyme in sialic acid synthesis, UDP-GlcNAc 2-epimerase/ManNAc kinase. Biochemistry. 2011;50:8914–25. doi: 10.1021/bi201050u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watts GDJ, Thorne M, Kovach MJ, Pestronk A, Kimonis VE. Clinical and genetic heterogeneity in chromosome 9p associated hereditary inclusion body myopathy: exclusion of GNE and three other candidate genes. Neuromuscul Disord. 2003;13:559–67. doi: 10.1016/s0960-8966(03)00070-1. [DOI] [PubMed] [Google Scholar]