

Abstract
Intercellular adhesion molecule-1 (ICAM-1) plays an important role in leukocyte trafficking, immunological synapse formation and, numerous cellular immune responses. Although considered a single glycoprotein, there are multiple membrane bound and soluble ICAM-1 isoforms which arise from alternative splicing and proteolytic cleavage during inflammatory responses. The function and expression of these isoforms on various cell types is poorly understood. In the generation of ICAM-1-deficient mice, two isoform-deficient ICAM-1 mutants were inadvertently produced due to alternative splicing. These mice along with true ICAM-1-deficient mice and newly generated ICAM-1 transgenic mice have provided the opportunity to begin examining the role of ICAM-1 isoforms (singly or in combination) in various disease settings. In this review we highlight the sharply contrasting disease phenotypes using ICAM-1 isoform mutant mice. These studies demonstrate that ICAM-1 immunobiology is highly complex but that individual isoforms, aside from the full-length molecule, make significant contributions to disease development and pathogenesis.
Keywords: adhesion molecules, alternative splicing, cerebral malaria, demyelinating disease, isoforms
Introduction
ICAM-1 (CD54) is a membrane-bound glycoprotein that plays a central role in leukocyte trafficking, activation of lymphocytes, and numerous additional immune functions (reviewed in 1–5). Originally cloned and sequenced from the neutrophilic promyelocyte cell line, HL-60, and umbilical vein endothelial cells (6), ICAM-1 is now known to be expressed on essentially all leukocyte subsets, endothelial cells, platelets, fibroblasts, epithelial cells, glial cells, and others (7–13). On most cell types and under non-inflammatory conditions, ICAM-1 expression is constitutively low, generally detectable only on endothelial cells (6, 14, 15), however TNF-α, IL-1β, IFN-γ and other cytokines elicit increased expression in a cell- and cytokine-specific fashion (7, 14, 16–18). ICAM-1 up-regulation is a signature event during inflammation, particularly on endothelium where expression may remain elevated for extended periods of time (11, 15)
ICAM-1 is a member of the immunoglobulin super-gene family and the full-length isoform is composed of five Ig domains, a transmembrane domain, and a short cytoplasmic tail with multiple threonine residues (Fig. 1A) (6). The cytoplasmic tail interacts with the actin cytoskeleton and the lipid raft associated ERM complex (ezrin/radixin/moesin) (19, 20). ICAM-1 is heavily glycosylated containing multiple N-glycosylation sites (8 in human, 10 in mice) and N-glycans are required for surface expression of the protein (reviewed in (21). Recently a functional high-mannose ICAM-1 glycoform, expressed on endothelial cells, has been characterized (20). The full-length isoform can form a homodimer in which each subunit bends at the junction between Ig domains three and four (Fig. 1B). The binding between subunits in the homodimer occurs through residues exposed on structural rearrangement of Ig domain four (22, 23). Dimerization of ICAM-1 subunits significantly increases the affinity for LFA-1, which may impact the ligand's ability to promote intracellular signaling (24). In addition to LFA-1, ICAM-1 binds to multiple ligands including other β2-integrins (Mac-1 and p150,95), fibrinogen, hyaluronan, PfEMP1 (a Plasmodium falciparum RBC membrane protein), and major group rhinoviruses (2, 25–30). The majority of ICAM-1 ligands bind to the first Ig domain of ICAM-1 (Fig. 1A) although Mac-1 and p150,95 have been shown to bind to other regions (Ig domains 3 and 3/4, respectively) (28–33). Studies have demonstrated that crosslinking ICAM-1 results in association with the actin cytoskeleton and activation of several intracellular signaling pathways that contribute to cytokine production and cellular trafficking events (reviewed in 19, 34).
Figure 1.
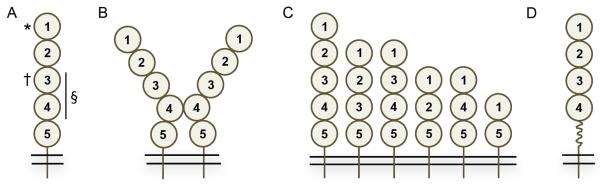
Schematic representation of ICAM-1 isoforms. (A) Full length ICAM-1 isoform with five Ig domains, transmembrane and intracellular domains, * denotes the binding site for LFA-1 and other ligands (see text for details) in the first Ig domain, † denotes the Mac-1 binding site in the third Ig domain, § denotes the p150,95 binding site in the third and fourth domains. (B) ICAM-1 full-length isoforms dimerize by interaction through the fourth Ig domains. (C) Shown are the six membrane ICAM-1 isoforms expressed on endothelial and other cell types. (D) Alternative spicing in exon 6 leads to truncation of the fifth Ig domain 5 in the full length isoform under inflammatory conditions. In each panel the ovals represents an Ig-like domain. The numbering of each Ig domain is based on the full-length isoform containing five Ig-like domains.
Alternative splicing gives rise to multiple ICAM-1 Isoforms
Alternative splicing is a common post-transcriptional mechanism, occurring in over 90% of multi-exon genes that broadly serves to regulate gene expression (reviewed in 35–37). ICAM-1 undergoes alternative splicing, similar to many genes in the immune system, including cell surface receptors, transcriptional regulators, and intracellular signaling molecules (38). Alternative splicing generates at least six membrane-bound forms and one soluble form of ICAM-1 (Fig. 1C) (39–44). The membrane bound isoforms identified to date contain two, three, four or five Ig domains (see Fig. 1A for Ig domain numbering) and all isoforms contain at least Ig domains one and five. The ability of these isoforms to bind LFA-1 in vitro was previously shown to be highly variable based on which Ig domains are present (40). The known soluble form of ICAM-1 is a full-length isoform and is derived from an mRNA transcript lacking a transmembrane domain. Expression of soluble ICAM-1 is, in part, cell-specific (endothelial and peripheral blood mononuclear cells) and modulated by cytokines such as IFN-1α (39). Soluble ICAM-1 is also generated by proteolytic cleavage of the membrane bound form by neutrophil elastase, cathepsin G, and bacteria-derived enzymes (42, 45, 46). Interestingly, the smaller ICAM-1 isoforms are more susceptible to proteolytic cleavage than the full-length isoform (42). In addition to the six membrane-bound isoforms, studies have demonstrated a splice variant in which the fifth Ig domain is truncated by 24 amino acids with a corresponding loss of Ig domain structure (Fig. 1D) (47). This variant was detected at significantly lower levels than the full-length isoform, in multiple tissues including lung, spleen, and kidney after pulmonary LPS challenge. Truncation of the fifth Ig domain in the remaining membrane bound ICAM-1 isoforms appears to be minimal, suggesting that this modification is largely restricted to the full-length isoform, at least in the model system employed in the study. Whether the utilization of this alternative splice site occurs in any of the other isoforms, as a function of infection or disease-induced inflammation, remains unexplored. The functional effects of this in-frame exon truncation event have not been examined, but could affect ICAM-1 dimerization, inter- and intracellular signaling and enzymatic cleavage of membrane forms of the protein (42). The result of these post-transcriptional and enzymatic modifications is at least seven membrane-bound versions of ICAM-1 arising from alternative splicing through cassette exon and alternative splice-site usage and numerous potential soluble forms of ICAM-1. Understanding how these isoforms contribute to ICAM-1-mediated immune responses has been problematic due to multiple factors, such as limited awareness of multiple ICAM-1 isoforms in mammals, limited reagents to identify the specific isoforms expressed on different cell types, changes in isoform expression during the immune response, and how these isoforms function during different disease states.
Alternative splicing and the generation of isoform-deficient ICAM-1 mice
Shortly after the characterization of ICAM-1, two groups independently undertook efforts to generate ICAM-1-deficient mice. Unaware that ICAM-1 was alternatively spliced, both groups utilized replacement constructs designed to insert a neomycin selection gene into a single exon to generate a truncated and non-functional molecule. Deletion of exon 4, which encodes Ig domain 3, led to the production of Icam1tm1Jcgr mice, while deletion of exon 5, which encodes Ig domain 4, led to the production of Icam1tm1Bay mice (48, 49). These ICAM-1 isoform-deficient mice both express two of the small isoforms, one composed of Ig domains 1, 2, and 5 and the other composed of Ig domains 1 and 5. The Icam1tm1Jcgr mice also express the isoform composed of Ig domains 1, 4, and 5 (Fig. 2A), while the Icam1tm1Bay mice lack this isoform and instead express an isoform containing Ig domains 1, 2, 3 and 5 (Fig. 2B). Neither, the Icam1tm1Jcgr or the Icam1tm1Bay mutant mouse, expresses the full-length isoform. At the time of their construction, these mice were considered ICAM-1 null mutants based on screening strategies that examined for alterations in genomic DNA surrounding the neomycin gene insertion site and the low expression of their isoforms under basal or inflammatory conditions as assessed by immunohistochemistry or flow cytometry (48, 49). Although subsequent studies demonstrated that these mice did express the unique subsets of alternatively spliced isoforms described above (40, 43), numerous investigators performed studies in various disease or injury model systems assuming they were deficient in all ICAM-1 isoforms. Thus, the majority of the literature describing results using ICAM-1-deficient mice reports on findings using specific isoform-deficient mice, rather than a true ICAM-1 knockout. Mice deficient in all ICAM-1 isoforms (ICAM-1null or Icam1tm1Alb) were generated a few years later, using a cloning strategy that deleted the entire coding region (50, 51). Icam1tm1Alb mice are phenotypically normal, fertile, and have no apparent abnormalities. Recently, two transgenic ICAM-1 mice were generated to address specific functions of ICAM-1 isoforms (52–54). These transgenic mice express either the full-length isoform (CD2-Icam1fl/Icam1null, Fig. 2C) or the isoform containing Ig domains 1, 2, 3 and 5 (CD2-Icam1D4del/Icam1null, Fig. 2D), under the control of the CD2 promoter on an Icam1tm1Alb background.
Figure 2.
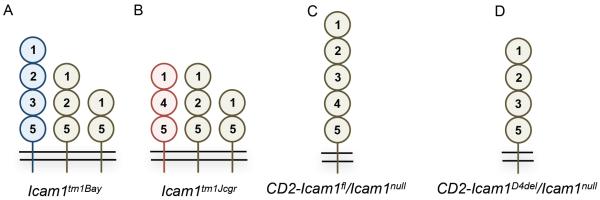
Schematic representation of ICAM-1 isoform structure and expression in ICAM-1 mutant mice. (A) Icam1tm1Bay, (B) Icam1tm1Jcgr, (C) CD2-Icam1fl/Icam1null, and (D) CD2-Icam1D4del/Icam1null mice. Numbering is the same as described in the legend to figure 1.
Does Differential ICAM-1 Isoform Expression Alter Disease Phenotype?
Determining how ICAM-1 isoforms contribute to immune-mediated pathophysiology is a complex question. The ICAM-1 mutant mice described above provide a rich experimental resource to begin deciphering their functions. In the following sections, we have highlighted a small number of models comparing these multiple isoform mutant mice used under the same experimental conditions or examined in the same study, allowing better insight into ICAM isoform function.
Endotoxin shock model
The first study to examine the role of ICAM-1 in LPS-induced shock used Icam1tm1Jcgr mice (49). Mice were treated with a lethal dose of LPS and only 20% of wild type mice survived to 72 hrs, whereas 100% of Icam1tm1Jcgr mice survived (Table 1). Interestingly, Icam1tm1Jcgr mice had little to no neutrophil infiltration in the liver compared to wild type mice. These findings were confirmed and extended in a second study in which Icam1tm1Jcgr and Icam1tm1Bay mice were simultaneously compared to wild type mice for LPS sensitivity (42). Using a similar dose of LPS, Icam1tm1Jcgr mice were again found to be resistant to endotoxic shock compared to wild type mice (0% vs. 50% mortality, respectively). In contrast, Icam1tm1Bay mice were highly susceptible to the effects of LPS treatment with high mortality (~80%) and significant neutrophil infiltration into the liver. Although these results demonstrate remarkably different outcomes, they provide little insight into isoform-specific mechanisms behind the phenotypes. This is due, in part, to the limited analysis of the mice with respect to inflammatory mediators such as changes in acute phase protein and cytokine levels and infiltration and activation of leukocytes. Studies using additional ICAM-1 mutant mice are required to determine how isoforms contribute to this acute inflammatory condition.
Table 1.
Comparison of EAE and ECM clinical signs between wild type and ICAM-1 mutant mice.
| ICAM-1 genotype | EAE Phenotype | ECM Phenotype |
|---|---|---|
| Wild Type | Acute onset, chronic disease | Acute onset, uniformly fatal |
| Icam1tm1Alb | Little to no disease | Little to no disease |
| Icam1tm1Bay | More severe than wild type | Delayed onset, significant survival |
| Icam1tm1Jcgr | Delayed onset, attenuated disease | Delayed onset, significant survival |
| CD2-Icam1fl/Icam1null | Delayed onset, attenuated disease | Acute onset, uniformly fatal |
| CD2-Icam1D4del/Icam1null | Acute onset, chronic disease | Delayed onset, significant survival |
Experimental autoimmune encephalomyelitis
Experimental autoimmune encephalomyelitis (EAE) is the animal model for the human disease multiple sclerosis (55, 56). ICAM-1 is an iconic marker of inflammation in EAE and multiple sclerosis with elevated expression at lesion sites on endothelial cells and on infiltrating and resident CNS cells (7, 57, 58). Although EAE can be induced in many animal species using myelin, or a variety of myelin-derived proteins or peptides, currently the most commonly used and well-characterized model employs C57BL/6 mice and a myelin oligodendrocyte glycoprotein peptide (amino acids 35-55; MOG35-55) (59, 60). This is the experimental paradigm used in the EAE studies described below. Given the multiple ICAM-1 ligands expressed by infiltrating cells, its absence would predictably result in less severe EAE. In fact, Icam1tm1Alb mice (deficient in all isoforms) failed to develop EAE whether actively-induced with MOG35-55 peptide or by transfer of encephalitogenic T cells (51) (Table 1). Not surprisingly, leukocyte infiltration and demyelination were also significantly reduced in Icam1tm1Alb mice, compared to wild type mice. Interestingly, antigen-specific T cell proliferation assays, using various combinations of wild type and ICAM-1-deficient T cells and APCs, demonstrated that ICAM-1 expression on T cells but not APCs, is required for T cell proliferation (51). These studies indicated that ICAM-1 expression on multiple cell types is required for the development of EAE, but provided little insight into isoform-specific functions.
In contrast, Icam1tm1Bay and Icam1tm1Jcgr mice clearly demonstrate that different combinations of isoforms (see Fig. 2A and B) lead to different EAE phenotypes. First to be studied were Icam1tm1Bay mice, which unexpectedly developed EAE more severe than that observed in wild type mice (Table 1) with concomitant elevated leukocyte infiltration, demyelination, and mortality (61, 62). Transfer of Icam1tm1Bay encephalitogenic T cells to wild type or Icam1tm1Alb mice also resulted in significantly worse disease compared to wild type-to-wild type transfers (62). Icam1tm1Jcgr mice however, develop a significantly milder EAE disease course compared to wild type mice (Table 1). Transfer of encephalitogenic T cells from Icam1tm1Jcgr mice to wild type recipients also produces a mild disease phenotype. Collectively the results with Icam1tm1Bay and Icam1tm1Jcgr mice show that 1) alternatively spliced ICAM-1 isoforms other than the full-length isoform are functional, 2) the full length ICAM-1 isoform is not required for disease development, 3) ICAM-1 expression on endothelial cells is not required for disease development based on Icam1tm1Bay to Icam1tm1Alb transferred EAE and, 4) Icam1tm1Bay T cells are more encephalitogenic by virtue of the combination of ICAM-1 isoforms they express. It is unclear if Icam1tm1Bay isoforms expressed on leukocytes, APCs and other cell types generate a highly potent T cell priming environment or if, their expression by T cells accounts for the observed enhancement in disease pathogenesis.
The results of EAE studies with Icam1tm1Bay and Icam1tm1Jcgr mice raised questions regarding the role of ICAM-1 isoforms on leukocytes. This led to the generation of CD2-Icam1fl/Icam1null and CD2-Icam1D4del/Icam1null mice, which express a single ICAM-1 isoform under the control of the CD2 promoter on an ICAM-1 null background, thereby allowing the study of single isoform immunobiology (see Fig. 2C and D) (52, 54). Expression of either isoform is comparable to wild type levels on CD4+ and CD8+ T cells and at significantly lower levels on other leukocyte subsets (γδ T cells, B cells and NK cells) (52, 54). Both mice develop EAE, but with unique phenotypes (Table 1) (54, unpublished observations). CD2-Icam1fl/Icam1null mice present with significantly delayed and attenuated disease while CD2-Icam1D4del/Icam1null mice present with robust disease comparable to that seen in wild type mice. These findings are remarkable in that they demonstrate the ability of single ICAM-1 isoforms expressed predominately on T cells, but not endothelial cells, to drive disease development. Furthermore, these studies indicate that the isoform missing Ig domain 4 can promote severe disease pathogenesis in isolation. Future studies are necessary to determine how other ICAM-1 isoforms may contribute to disease development.
Experimental cerebral malaria
Experimental cerebral malaria is a commonly used model for the most severe and deadly form of the human disease, cerebral malaria (63, 64). Sequestration of infected RBCs, leukocytes and platelets on inflamed endothelium due to increased expression of adhesion molecules including ICAM-1 is thought to be a major contributor to disease development (65–68). ICAM-1 binds to Plasmodium falciparum erythrocyte membrane protein 1, expressed on the surface of infected RBCs (iRBC), at a site on the amino-terminal Ig domain (28–31). In rodents, innoculation of C567BL/6 mice with Plasmodium berghei ANKA iRBCs has recently become a widely used model for ECM (69–71). Additional evidence to support a role for ICAM-1 in ECM comes from studies demonstrating that anti-ICAM-1 antibody treatment abrogates iRBC adherence and rolling in both in vitro and in vivo model systems (66, 68, 72). However, direct evidence implicating an important role for ICAM-1 and its isoforms in ECM has been lacking until recently.
In studies simultaneously comparing the course of ECM in different lines of ICAM-1 mutant mice, it has now been shown that the ECM phenotype varies directly with the combination of isoforms expressed (53). Not surprisingly, Icam1tm1Alb mice were highly resistant to ECM with over 90% of mice surviving ten days post-infection, a time at which all wild type mice had succumbed to disease (Table 1). Both Icam1tm1Bay and Icam1tm1Jcgr mice were more susceptible to ECM with Icam1tm1Bay mice presenting with a more protective phenotype (60% vs. 40% survival, respectively; Table 1). These results are an interesting reversal of the disease phenotype seen for Icam1tm1Bay and Icam1tm1Jcgr mice in EAE where Icam1tm1Bay mice developed exacerbated disease compared to wild type mice (62). CD2-Icam1fl/Icam1null and CD2-Icam1D4del/Icam1null mice presented with remarkably different ECM phenotypes. CD2-Icam1fl/Icam1null were fully susceptible to disease similar to wild type mice, however the time to 50% survival was two days slower than wild type mice. In contrast, CD2-Icam1D4del/Icam1null mice were significantly more resistant to ECM with 60% of the mice surviving ten days post-infection (Table 1). This disease course is very similar to that observed for Icam1tm1Bay mice with ECM and suggests that expression of the Icam1D4del isoform on leukocytes is sufficient to drive this disease phenotype. These studies demonstrate that ICAM-1 expression on the CNS microvascular endothelium is not required for development of ECM and suggests an important contribution for ICAM-1-mediated sequestration within the microvascular space.
Conclusions
Despite reports in the early to mid 1990s, alternatively spliced forms of different adhesion molecules, such as VCAM-1, PECAM-1, MAdCAM-1, ICAM-1, have virtually been ignored, and the majority of the scientific field has considered these molecules to exist as single proteins. However, with expansion of genomic technologies, it is clear that the majority of genes, including those that encoding for the adhesion molecules, undergo alternative splicing and have the potential to produce multiple isoforms. The expression patterns, ligand interactions, and functions of these isoforms still remain largely undefined. Future studies are needed to understand how these isoforms contribute to immune and inflammatory responses, as well as potentially modulate disease phenotypes.
Studies of the alternatively spliced forms of ICAM-1 have significantly benefited from the generation of multiple lines of ICAM-1 deficient mice. In fact, their initial discovery was facilitated by the identification of ICAM-1 isoforms expressed in the Icam1tm1Bay mice. It is now evident from numerous investigations of these mice that unique combinations of ICAM-1 isoforms contribute to disease-specific outcomes. How do these ICAM-1 isoforms mediate such remarkably different pathology? In our view, there are overlapping possibilities that include 1) differential isoform expression, 2) structural changes of the molecule (due to presence or absence of different Ig domains), and 3) alterations in ICAM-1 facilitated intracellular signaling. The potential number of combinations of ICAM-1 isoforms expressed even on a single cell type are staggering. This complexity is further increased when considering the numerous cell types, which express ICAM-1, thus creating a daunting task for determining isoform function. The key first step is the application of genomic and proteomic approaches to provide insight into these expression patterns.
Alternative splicing could lead to significant structural changes in the ICAM-1 molecule that may then alter ligand interactions and functional activity. In addition to potential effects on dimerization, there may be preferential interactions of ICAM-1 isoforms that serve to regulate ligand accessibility, thereby modulating adhesion strength. Such interactions may also control access to the actin cytoskeleton and adapter protein complexes in lipid rafts, altering intracellular signaling and disease outcome. Altered glycosylation due to the loss of Ig domains in alternatively spliced in ICAM-1 isoforms may also contribute to differential function of these molecules. The extent of glycosylation on both membrane-bound and soluble ICAM-1 can regulate ligand binding (21). In addition, recent studies have demonstrated that the high-mannose form of ICAM-1 is functionally distinct from the complex N-glycan form of ICAM-1 with respect to monocyte rolling and adhesion and cytoskeleton interaction (20). Although these findings indicate that glycosylation plays a fundamental role in modulating the role of ICAM-1, the effects of these modifications on isoform activity remains unexplored.
Intracellular signaling through ICAM-1 in T cells, APC's and, endothelial cells is considered co-stimulatory in nature, similar to CD80/86, CD28 and other costimulatory molecules. ICAM-1 dimerization is believed to be required for signaling through an ITIM-like motif that interacts with protein phosphatases containing SH2 domains. Upon ligand binding, ICAM-1 migrates to lipid rafts leading to the activation of ERK, Akt, and JNK pathways and the subsequent production of cytokines, chemokines and changes in the expression of adhesion molecules (19, 20). These studies have assumed signaling occurs through the full-length ICAM-1 isoform, however, there is currently no evidence to indicate a requirement for signaling through this isoform. The distinct disease phenotypes and level of T cell activation (as determined by antigen recall assays) in ICAM-1 mutant mice (51, 54, 62) suggests that discrete combinations of isoforms contribute to intracellular signaling events and ultimately to disease severity. Specifically, the severe disease phenotype observed in Icam1tm1Bay mice compared to Icam1tm1Jcgr and CD2-Icam1fl/Icam1null mice suggest that the isoform containing Ig domains 1, 2, 3 and 5 is more critical for pro-inflammatory ICAM-1 signaling events than the full-length isoform. Understanding how these isoforms modulate intracellular signaling and their downstream functions requires additional study. The currently available repertoire of ICAM-1 mutant mice and the generation of new ICAM-1 mutant mice, expressing single or unique combinations of ICAM-1 isoforms, will be useful in addressing key questions regarding ICAM-1 interactions.
Acknowledgements
The authors acknowledge the support of Drs. Julian Rayner and Oliver Billker at the Wellcome Trust Sanger Institute and the guidance and technical expertise of Dr. Robert Kesterson and his staff in UAB Transgenic Mouse Facility. We acknowledge Albert and Hank Barnum for continuing inspiration.
This work was supported by NIH grants T32 AI07051 (to TNR), F31NS077811 (to TNR), P30 CA13148, P30 AR048311 and NMSS PP1058 (to SRB and DCB).
Abbreviations used in this article
- CM
cerebral malaria
- EAE
experimental autoimmune encephalomyelitis
- ECM
experimental cerebral malaria
- iRBCs
infected RBCs
- PbA
Plasmodium berghei ANKA
- PfEMP1
Plasmodium falciparum erythrocyte membrane protein 1
Footnotes
Disclosures The authors have no financial conflicts
References
- 1.Harlan JM, Winn RK, Vedder NB, Doerschuk CM, Rice CL. Adhesion: Its Role in Inflammatory Disease. W. H. Freeman and Company; New York: 1992. [Google Scholar]
- 2.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 3.Liu L, Kubes P. Molecular mechanisms of leukocyte recruitment: organ-specific mechanisms of action. Thrombosis and haemostasis. 2003;89:213–220. [PubMed] [Google Scholar]
- 4.Steeber DA, Venturi GM, Tedder TF. A new twist to the leukocyte adhesion cascade: intimate cooperation is key. Trends in immunology. 2005;26:9–12. doi: 10.1016/j.it.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 5.Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, Dvorak HF, Dvorak AM, Springer TA. Transcellular diapedesis is initiated by invasive podosomes. Immunity. 2007;26:784–797. doi: 10.1016/j.immuni.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staunton DE, Marlin SD, Stratowa C, Dustin ML, Springer TA. Primary structure of ICAM-1 demonstrates interaction between members of the immunoglobulin and integrin supergene families. Cell. 1988;52:925–933. doi: 10.1016/0092-8674(88)90434-5. [DOI] [PubMed] [Google Scholar]
- 7.Lee SJ, Benveniste EN. Adhesion molecule expression and regulation on cells of the central nervous system. Journal of neuroimmunology. 1999;98:77–88. doi: 10.1016/s0165-5728(99)00084-3. [DOI] [PubMed] [Google Scholar]
- 8.Roebuck KA, Finnegan A. Regulation of intercellular adhesion molecule-1 (CD54) gene expression. Journal of leukocyte biology. 1999;66:876–888. doi: 10.1002/jlb.66.6.876. [DOI] [PubMed] [Google Scholar]
- 9.Limb GA, Webster L, Soomro H, Janikoun S, Shilling J. Platelet expression of tumour necrosis factor-alpha (TNF-alpha), TNF receptors and intercellular adhesion molecule-1 (ICAM-1) in patients with proliferative diabetic retinopathy. Clinical and experimental immunology. 1999;118:213–218. doi: 10.1046/j.1365-2249.1999.01067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hubbard AK, Rothlein R. Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radic Biol Med. 2000;28:1379–1386. doi: 10.1016/s0891-5849(00)00223-9. [DOI] [PubMed] [Google Scholar]
- 11.Koning GA, Schiffelers RM, Storm G. Endothelial cells at inflammatory sites as target for therapeutic intervention. Endothelium : journal of endothelial cell research. 2002;9:161–171. doi: 10.1080/10623320213631. [DOI] [PubMed] [Google Scholar]
- 12.Muro S, Muzykantov VR. Targeting of antioxidant and antithrombotic drugs to endothelial cell adhesion molecules. Current pharmaceutical design. 2005;11:2383–2401. doi: 10.2174/1381612054367274. [DOI] [PubMed] [Google Scholar]
- 13.Tailor A, Cooper D, Granger DN. Platelet-vessel wall interactions in the microcirculation. Microcirculation. 2005;12:275–285. doi: 10.1080/10739680590925691. [DOI] [PubMed] [Google Scholar]
- 14.Almenar-Queralt A, Duperray A, Miles LA, Felez J, Altieri DC. Apical topography and modulation of ICAM-1 expression on activated endothelium. The American journal of pathology. 1995;147:1278–1288. [PMC free article] [PubMed] [Google Scholar]
- 15.Scholz D, Devaux B, Hirche A, Potzsch B, Kropp B, Schaper W, Schaper J. Expression of adhesion molecules is specific and time-dependent in cytokine-stimulated endothelial cells in culture. Cell and tissue research. 1996;284:415–423. doi: 10.1007/s004410050602. [DOI] [PubMed] [Google Scholar]
- 16.Pober JS, Gimbrone MA, Jr., Lapierre LA, Mendrick DL, Fiers W, Rothlein R, Springer TA. Overlapping patterns of activation of human endothelial cells by interleukin 1, tumor necrosis factor, and immune interferon. The Journal of Immunology. 1986;137:1893–1896. [PubMed] [Google Scholar]
- 17.Dustin ML, Rothlein R, Bhan AK, Dinarello CS, Springer TA. Induction by IL-1 and interferon, tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1) Journal of Immunolgy. 1986;137:245–254. [PubMed] [Google Scholar]
- 18.Hua S. Targeting sites of inflammation: intercellular adhesion molecule-1 as a target for novel inflammatory therapies. Frontiers in pharmacology. 2013;4:127. doi: 10.3389/fphar.2013.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lebedeva T, Dustin ML, Sykulev Y. ICAM-1 co-stimulates target cells to facilitate antigen presentation. Curr Opin Immunol. 2005;17:251–258. doi: 10.1016/j.coi.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 20.Scott DW, Dunn TS, Ballestas ME, Litovsky SH, Patel RP. Identification of a high-mannose ICAM-1 glycoform: effects of ICAM-1 hypoglycosylation on monocyte adhesion and outside in signaling. American journal of physiology. Cell physiology. 2013;305:C228–237. doi: 10.1152/ajpcell.00116.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott DW, Patel RP. Endothelial heterogeneity and adhesion molecules N-glycosylation: implications in leukocyte trafficking in inflammation. Glycobiology. 2013;23:622–633. doi: 10.1093/glycob/cwt014. [DOI] [PubMed] [Google Scholar]
- 22.Kirchhausen T, Staunton DE, Springer TA. Location of the domains of ICAM-1 by immunolabeling and single-molecule electron microscopy. Journal of leukocyte biology. 1993;53:342–346. doi: 10.1002/jlb.53.3.342. [DOI] [PubMed] [Google Scholar]
- 23.Chen X, Kim TD, Carman CV, Mi LZ, Song G, Springer TA. Structural plasticity in Ig superfamily domain 4 of ICAM-1 mediates cell surface dimerization. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15358–15363. doi: 10.1073/pnas.0707406104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller J, Knorr R, Ferrone M, Houdei R, Carron CP, Dustin ML. Intercellular adhesion molecule-1 dimerization and its consequences for adhesion mediated by lymphocyte function associated-1. The Journal of experimental medicine. 1995;182:1231–1241. doi: 10.1084/jem.182.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diamond MS, Staunton DE, Marlin SD, Springer TA. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglubulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991;65:961–971. doi: 10.1016/0092-8674(91)90548-d. [DOI] [PubMed] [Google Scholar]
- 26.McCourt PAG, Ek B, Forsberg N, Gustafson S. Intercellular adhesion molecule-1 is a cell surface receptor for hyaluronan. The Journal of biological chemistry. 1994;269:30081–30084. [PubMed] [Google Scholar]
- 27.Languino LR, Duperray A, Joganic KJ, Fornaro M, Thornton GB, Altieri DC. Regulation of leukocyte-endothelium interaction and leukocyte transendothelial migration by intercellular adhesion molecule 1-fibrinogen recognition. Proc Natl Acad Sci USA. 1995;92:1505–1509. doi: 10.1073/pnas.92.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ockenhouse CF, Betageri R, Springer TA, Staunton DE. Plasmodium falciparum-infected erythrocytes bind ICAM-1 at a site distinct from LFA-1, Mac-1, and human rhinovirus. Cell. 1992;68:63–69. doi: 10.1016/0092-8674(92)90206-r. [DOI] [PubMed] [Google Scholar]
- 29.Smith JD, Craig AG, Kriek N, Hudson-Taylor D, Kyes S, Fagan T, Pinches R, Baruch DI, Newbold CI, Miller LH. Identification of a Plasmodium falciparum intercellular adhesion molecule-1 binding domain: a parasite adhesion trait implicated in cerebral malaria. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1766–1771. doi: 10.1073/pnas.040545897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tse MT, Chakrabarti K, Gray C, Chitnis CE, Craig A. Divergent binding sites on intercellular adhesion molecule-1 (ICAM-1) for variant Plasmodium falciparum isolates. Mol Microbiol. 2004;51:1039–1049. doi: 10.1046/j.1365-2958.2003.03895.x. [DOI] [PubMed] [Google Scholar]
- 31.Brown A, Turner L, Christoffersen S, Andrews KA, Szestak T, Zhao Y, Larsen S, Craig AG, Higgins MK. Molecular architecture of a complex between an adhesion protein from the malaria parasite and intracellular adhesion molecule 1. The Journal of biological chemistry. 2013;288:5992–6003. doi: 10.1074/jbc.M112.416347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frick C, Odermatt A, Zen K, Mandell KJ, Edens H, Portmann R, Mazzucchelli L, Jaye DL, Parkos CA. Interaction of ICAM-1 with beta 2-integrin CD11c/CD18: characterization of a peptide ligand that mimics a putative binding site on domain D4 of ICAM-1. European journal of immunology. 2005;35:3610–3621. doi: 10.1002/eji.200425914. [DOI] [PubMed] [Google Scholar]
- 33.Choi J, Choi J, Nham SU. Characterization of the residues of alphaX I-domain and ICAM-1 mediating their interactions. Molecules and cells. 2010;30:227–234. doi: 10.1007/s10059-010-0111-2. [DOI] [PubMed] [Google Scholar]
- 34.Greenwood J, Heasman SJ, Alvarez JI, Prat A, Lyck R, Engelhardt B. Review: leucocyte-endothelial cell crosstalk at the blood-brain barrier: a prerequisite for successful immune cell entry to the brain. Neuropathology and applied neurobiology. 2011;37:24–39. doi: 10.1111/j.1365-2990.2010.01140.x. [DOI] [PubMed] [Google Scholar]
- 35.Lynch KW. Consequences of regulated pre-mRNA splicing in the immune system. Nature reviews. Immunology. 2004;4:931–940. doi: 10.1038/nri1497. [DOI] [PubMed] [Google Scholar]
- 36.Heyd F, Lynch KW. Degrade, move, regroup: signaling control of splicing proteins. Trends in biochemical sciences. 2011;36:397–404. doi: 10.1016/j.tibs.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinez NM, Lynch KW. Control of alternative splicing in immune responses: many regulators, many predictions, much still to learn. Immunological reviews. 2013;253:216–236. doi: 10.1111/imr.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ergun A, Doran G, Costello JC, Paik HH, Collins JJ, Mathis D, Benoist C, ImmGen C. Differential splicing across immune system lineages. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:14324–14329. doi: 10.1073/pnas.1311839110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giorelli M, De Blasi A, Defazio G, Avolio C, Iacovelli L, Livrea P, Trojano M. Differential regulation of membrane bound and soluble ICAM 1 in human endothelium and blood mononuclear cells: effects of interferon beta-1a. Cell Commun Adhes. 2002;9:259–272. doi: 10.1080/15419060216305. [DOI] [PubMed] [Google Scholar]
- 40.King PD, Sandberg ET, Selvakumar A, Fang P, Beaudet AL, Dupont B. Novel isoforms of murine intercellular adhesion molecule-1 generated by alternative RNA splicing. Journal of Immunology. 1995;154:6080–6093. [PubMed] [Google Scholar]
- 41.Ochietti B, Lemieux P, Kabanov AV, Vinogradov S, St-Pierre Y, Alakhov V. Inducing neutrophil recruitment in the liver of ICAM-1-deficient mice using polyethyleneimine grafted with Pluronic P123 as an organ-specific carrier for transgenic ICAM-1. Gene Ther. 2002;9:939–945. doi: 10.1038/sj.gt.3301716. [DOI] [PubMed] [Google Scholar]
- 42.Robledo O, Papaioannou A, Ochietti B, Beauchemin C, Legault D, Cantin A, King PD, Daniel C, Alakhov VY, Potworowski EF, St-Pierre Y. ICAM-1 isoforms: specific activity and sensitivity to cleavage by leukocyte elastase and cathepsin G. European journal of immunology. 2003;33:1351–1360. doi: 10.1002/eji.200323195. [DOI] [PubMed] [Google Scholar]
- 43.van Den Engel NK, Heidenthal E, Vinke A, Kolb H, Martin S. Circulating forms of intercellular adhesion molecule (ICAM)-1 in mice lacking membranous ICAM-1. Blood. 2000;95:1350–1355. [PubMed] [Google Scholar]
- 44.Wakatsuki T, Kimura K, Kimura F, Shinomiya N, Ohtsubo M, Ishizawa M, Yamamoto M. A distinct mRNA encoding a soluble form of ICAM-1 molecule expressed in human tissues. Cell Adhes Commun. 1995;3:283–292. doi: 10.3109/15419069509081014. [DOI] [PubMed] [Google Scholar]
- 45.Champagne B, Tremblay P, Cantin A, St Pierre Y. Proteolytic cleavage of ICAM-1 by human neutrophil elastase. J Immunol. 1998;161:6398–6405. [PubMed] [Google Scholar]
- 46.Grenier D, Bodet C. Streptococcus suis stimulates ICAM-1 shedding from microvascular endothelial cells. FEMS immunology and medical microbiology. 2008;54:271–276. doi: 10.1111/j.1574-695X.2008.00476.x. [DOI] [PubMed] [Google Scholar]
- 47.Mizgerd JP, Spieker MR, Lupa MM. Exon truncation by alternative splicing of murine ICAM-1. Physiol Genomics. 2002;12:47–51. doi: 10.1152/physiolgenomics.00073.2002. [DOI] [PubMed] [Google Scholar]
- 48.Sligh JE, Ballantyne CM, Rich SS, Hawkins HK, Smith CW, Bradley A, Beaudet AL. Inflammatory and immune responses are impaired in mice deficient in intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. 1993;90:8529–8533. doi: 10.1073/pnas.90.18.8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu H, Gonzalo JA, St Pierre Y, Williams IR, Kupper TS, Cotran RS, Springer TA, Gutierrez-Ramos JC. Leukocytosis and resistance to septic shock in intercellular adhesion molecule 1-deficient mice. The Journal of experimental medicine. 1994;180:95–109. doi: 10.1084/jem.180.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dunne JL, Collins RG, Beaudet AL, Ballantyne CM, Ley K. Mac-1, but not LFA-1, uses intercellular adhesion molecule-1 to mediate slow leukocyte rolling in TNF-alpha-induced inflammation. J Immunol. 2003;171:6105–6111. doi: 10.4049/jimmunol.171.11.6105. [DOI] [PubMed] [Google Scholar]
- 51.Bullard DC, Hu X, Schoeb TR, Collins RG, Beaudet AL, Barnum SR. Intercellular adhesion molecule-1 expression is required on multiple cell types for the development of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:851–857. doi: 10.4049/jimmunol.178.2.851. [DOI] [PubMed] [Google Scholar]
- 52.Cox MA, Barnum SR, Bullard DC, Zajac AJ. ICAM-1-dependent tuning of memory CD8 T-cell responses following acute infection. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1416–1421. doi: 10.1073/pnas.1213480110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramos TN, Bullard DC, Darley MM, McDonald K, Crawford DF, Barnum SR. Experimental cerebral malaria develops independently of endothelial expression of intercellular adhesion molecule-1 (icam-1) The Journal of biological chemistry. 2013;288:10962–10966. doi: 10.1074/jbc.C113.457028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bullard DC, Hu X, Crawford D, McDonald K, Ramos TN, Barnum SR. Expression of a single ICAM-1 isoform on T cells is sufficient for development of experimental autoimmune encephalomyelitis. European journal of immunology. 2014 doi: 10.1002/eji.201344023. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baxter AG. The origin and application of experimental autoimmune encephalomyelitis. Nature reviews. Immunology. 2007;7:904–912. doi: 10.1038/nri2190. [DOI] [PubMed] [Google Scholar]
- 56.Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS) British journal of pharmacology. 2011;164:1079–1106. doi: 10.1111/j.1476-5381.2011.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schulz M, Engelhardt B. The circumventricular organs participate in the immunopathogenesis of experimental autoimmune encephalomyelitis. Cerebrospinal fluid research. 2005;2:8. doi: 10.1186/1743-8454-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Linker RA, Reinhardt M, Bendszus M, Ladewig G, Briel A, Schirner M, Maurer M, Hauff P. In vivo molecular imaging of adhesion molecules in experimental autoimmune encephalomyelitis (EAE) Journal of autoimmunity. 2005;25:199–205. doi: 10.1016/j.jaut.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 59.Batoulis H, Recks MS, Addicks K, Kuerten S. Experimental autoimmune encephalomyelitis--achievements and prospective advances. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica. 2011;119:819–830. doi: 10.1111/j.1600-0463.2011.02794.x. [DOI] [PubMed] [Google Scholar]
- 60.Jones MV, Nguyen TT, Deboy CA, Griffin JW, Whartenby KA, Kerr DA, Calabresi PA. Behavioral and pathological outcomes in MOG 35-55 experimental autoimmune encephalomyelitis. Journal of neuroimmunology. 2008;199:83–93. doi: 10.1016/j.jneuroim.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 61.Samoilova EB, Horton JL, Chen Y. Experimental autoimmune encephalomyelitis in intercellular adhesion molecule-1-deficient mice. Cell Immunol. 1998;190:83–89. doi: 10.1006/cimm.1998.1395. [DOI] [PubMed] [Google Scholar]
- 62.Hu X, Barnum SR, Wohler JE, Schoeb TR, Bullard DC. Differential ICAM-1 isoform expression regulates the development and progression of experimental autoimmune encephalomyelitis. Molecular immunology. 2010;47:1692–1670. doi: 10.1016/j.molimm.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 2006;22:503–508. doi: 10.1016/j.pt.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 64.Haldar K, Murphy SC, Milner DA, Taylor TE. Malaria: mechanisms of erythrocytic infection and pathological correlates of severe disease. Annu Rev Pathol. 2007;2:217–249. doi: 10.1146/annurev.pathol.2.010506.091913. [DOI] [PubMed] [Google Scholar]
- 65.Berendt AR, Simmons DL, Tansey J, Newbold CI, Marsh K. Intercellular adhesion molecule-1 is an endothelial cell adhesion receptor for Plasmodium falciparum. Nature. 1989;341:57–59. doi: 10.1038/341057a0. [DOI] [PubMed] [Google Scholar]
- 66.Willimann K, Matile H, Weiss NA, Imhof BA. In vivo sequestration of Plasmodium falciparum-infected human erythrocytes: a severe combined immunodeficiency mouse model for cerebral malaria. The Journal of experimental medicine. 1995;182:643–653. doi: 10.1084/jem.182.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Newbold C, Warn P, Black G, Berendt A, Craig A, Snow B, Msobo M, Peshu N, Marsh K. Receptor-specific adhesion and clinical disease in Plasmodium falciparum. The American journal of tropical medicine and hygiene. 1997;57:389–398. doi: 10.4269/ajtmh.1997.57.389. [DOI] [PubMed] [Google Scholar]
- 68.Yipp BG, Hickey MJ, Andonegui G, Murray AG, Looareesuwan S, Kubes P, Ho M. Differential roles of CD36, ICAM-1, and P-selectin in Plasmodium falciparum cytoadherence in vivo. Microcirculation. 2007;14:593–602. doi: 10.1080/10739680701404705. [DOI] [PubMed] [Google Scholar]
- 69.Sinden RE, Butcher GA, Beetsma AL. Maintenance of the Plasmodium berghei life cycle. Methods Mol Med. 2002;72:25–40. doi: 10.1385/1-59259-271-6:25. [DOI] [PubMed] [Google Scholar]
- 70.Engwerda C, Belnoue E, Gruner AC, Renia L. Experimental models of cerebral malaria. Curr Top Microbiol Immunol. 2005;297:103–143. [PubMed] [Google Scholar]
- 71.Lamb TJ, Brown DE, Potocnik AJ, Langhorne J. Insights into the immunopathogenesis of malaria using mouse models. Expert Rev Mol Med. 2006;8:1–22. doi: 10.1017/S1462399406010581. [DOI] [PubMed] [Google Scholar]
- 72.Ho M, Hickey MJ, Murray AG, Andonegui G, Kubes P. Visualization of Plasmodium falciparum-endothelium interactions in human microvasculature: mimicry of leukocyte recruitment. The Journal of experimental medicine. 2000;192:1205–1211. doi: 10.1084/jem.192.8.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]