

Abstract
The manufacturing of human mesenchymal stem cells (hMSCs) as cell-based products for clinical use should be performed with appropriate controls that ensure its safety and quality. The use of hMSCs in cell therapy has increased considerably in the past few years. In line with this, the assessment and management of contamination risks by microbial agents that could affect the quality of cells and the safety of patients have to be considered. It is necessary to implant a quality control program (QCP) covering the entire procedure of the ex vivo expansion, from the source of cells, starting materials, and reagents, such as intermediate products, to the final cellular medicine. We defined a QCP to detect microbiological contamination during manufacturing of autologous hMSCs for clinical application. The methods used include sterility test, Gram stain, detection of mycoplasma, endotoxin assay, and microbiological monitoring in process according to the European Pharmacopoeia (Ph. Eur.) and each analytical technique was validated in accordance with three different cell cultures. Results showed no microbiological contamination in any phases of the cultures, meeting all the acceptance criteria for sterility test, detection of mycoplasma and endotoxin, and environmental and staff monitoring. Each analytical technique was validated demonstrating the sensitivity, limit of detection, and robustness of the method. The quality and safety of MSCs must be controlled to ensure their final use in patients. The evaluation of the proposed QCP revealed satisfactory results in order to standardize this procedure for clinical use of cells.
Introduction
Cell-based therapy has led to the development of new biological medicines to repair, replace, or recover the biological function of damaged tissues and organs [1]. Among cell types used for this propose, human mesenchymal stem cells (hMSCs) are considered as cell-based therapy medical product (CTMP) and should be handled with appropriate controls to ensure their safety, quality, and efficacy as a final medicine [2–6].
The manufacture of hMSCs involves an ex vivo expansion for a relatively long period of time, which leads to a risk of contamination by microbiological agents that could affect the quality and safety of the cells [7]. Contamination of a CTMP can cause adverse reactions in patients (eg, fever, chills, infections, and irreversible septic shock) and even death [7,8]. Therefore it will be necessary to standardize and validate all procedures and analytical techniques involved in the manufacture of CTMP [9], posing a quality control program (QCP).
A QCP should ensure that cells have been manufactured in aseptic conditions, under GMP conditions to minimize the contamination risk of the cell medicine and, thus, to ensure the safety of patients and the quality of the medicine. This program will comprise the whole process of ex vivo expansion, starting from type of cells, source of materials, reagents, and intermediate products (subcultures), to CTMP, the final cellular medicine [10,11]. Chiefly, because the cells must be viable for their administration and should not be sterilized by physicochemical methods, in this scenario a risk analysis must be performed to determine the possibilities of microbiological contamination before designing a QCP. For a QCP applied to a CTMP, each analytical technique should be justified, and the amount and type of evidence required for microbiological quality control were defined according to the different pharmacopoeias, as well as, the guidelines issued by regulatory agencies and International Conference on Harmonisation (ICH), in particular, quality guidelines [12,13]. Validation studies must be performed for each analytical technique to demonstrate and verify that the procedure adopted at each site laboratory does not alter the method and consequently the result [14].
The aim of this study was to develop a microbial QCP of a CTMP (Fig. 1), for the long-term expansion of human adipose-derived MSCs. In particular, the manufactured medicine was an injectable cell suspension, elaborated by suspending the active principle (hMSCs) and other additives (culture medium or packing medium), packaged in a suitable container to be administered parenterally (intramuscular, intravenous, and intra-arterial). Contamination by bacteria, fungi, and mycoplasma and bacterial endotoxin concentration were analyzed in line with QCP proposed in different phases, such as Master Cell Bank (MCB), Working Cell Bank (WCB), and in the final cellular medicine. Each analytical technique was validated on three different cultures.
FIG. 1.

Scheme of manufacturing process of autologous stem cell mesenchymal as a cell therapy medicinal product.
Materials and Methods
This study was performed in the quality control unit of the CABIMER's GMP facility authorized by the Spanish Agency of Medicines and Medical Devices and regularly inspected by the Spanish competent authorities.
Isolation and culture of hMSCs
Autologous hMSCs were isolated from abdominal adipose tissue biopsies of patients enrolled in a Phase I/II clinical trial. All donors provided informed consent that was formerly approved by local and regional medical research ethics committees. Each patient was appropriately screened and tested for human pathogens. In particular, the presence of human immunodeficiency virus (HIV), hepatitis B virus, and hepatitis C virus was analyzed. On the other hand, all starting materials and reagents required for the expansion were analyzed to certify that they were sterile and endotoxin free.
Cells were isolated from human adipose tissue by enzymatic digestion with collagenase (2 mg collagenase/1 g adipose tissue; Roche Farma, Reinach, BL, Switzerland), centrifuged at 400 g for 10 min, and filtered and washed with phosphate-buffered saline (PBS; Sigma-Aldrich, St. Louis, MO) to obtain the stromal cells. These cells were suspended and plated at medium density of 12–20×104 cells/cm2 in culture flasks (Nunc, Roskilde, Roskilde-DK4 000, Denmark) with expansion medium composed by Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 1% of 10,000 IU/mL penicillin and 10 mg/mL streptomycin, 2% of 200 mM L-alanine solution, and 1% of 200 mM L-glutamina (all from Sigma-Aldrich). After 24 h, nonadherent cells were removed by replacing the expansion medium. The medium was replaced every 2 or 3 days a week. Cells were harvested upon reaching 80% confluence, and subcultured using 0.25% trypsin (Gibco, Invitrogen, Grand Island, NY) in expansion medium, and plated at medium density, 3500–5500 cells/cm2. Cells were cultured under 95% relative humidity, at 37°C, and 5% CO2. Three different processes of ex vivo expansion of hMSCs were carried out (named 1, 2, and 3) from passages 3–4 to analyze their microbiological quality. For the final product, cells were packed at concentration of 1×106 cells/mL with 1 mL of packaging medium composed by 50% of glucose 5%, 45% of lactated Ringer's solution, and 55% of albumin 20% (Grifols, Barcelona, Spain). hMSCs were packed in 10-mL Luer-lock syringes (Becton Dickinson and Company, Franklin Lakes, NJ).
Sterility test and Gram stain
The test was carried out in different phases (MCB, WCB, and final cellular medicine) on three different cultures, by direct inoculation of 1 mL of intermediate product (supernatant) or final suspension in the microbiology medium, so that volume of the product is not more than 10% of the volume of the medium but not less than 1 mL [15]. Two media were used: Thioglycollate Penase Broth 125 mL (TPB; BDH Prolabo, Mexico D.F., Mexico), to detect anaerobic and aerobic bacteria, and Tryptic Soy Penase Broth 125 mL (TSPB; BDH Prolabo), which is a soybean casein digest medium to detect fungi and aerobic bacteria. For each media (TPB and TSPB), both sterility test and growth promotion test of aerobes, anaerobes, and fungi were verified before testing; the strains used in the growth promotion test are indicated in the Table 1. A negative control was included inoculating 1 mL of 0.9% sterile NaCl (bioMérieux, Marcy l'Etoile, France) for each medium. The inoculated media were incubated for 14 days at 35°C and 22°C for TPB and TSPB, respectively. If microbial growth appears after 14 days, then the medium will show turbidity.
Table 1.
Microbial Strains Needed to the Growth Promotion Test and the Validation of Sterility Test
| Culture medium | Microorganisms | Strain |
|---|---|---|
| Thioglycollate Penase Broth | Clostridium sporogenes | ATCC 11437 |
| Pseudomonas aeruginosa | ATCC 9027 | |
| Staphylococcus aureus | ATCC 6538 | |
| Aspegillus niger | ATCC 16404 | |
| Tryptic Soy Penase Broth | Bacillus subtilis | ATCC 6633 |
| Candida albicans | ATCC 10231 |
ATCC, American Type Culture Collection.
Sterility test was performed as soon as possible after sample collection, which was stored at 5°C±3°C for up to 24 h [16] to avoid phagocytosis of microorganisms by cells present in the sample. This assay was performed in aseptic conditions with an isolator HPI-4PI-S (Esco Technologies, Inc., Hatboro, PA).
Additionally, in each final cellular medicine for the three cultures, the presence/absence of colony forming unit (CFU) was examined by standard procedure of Gram staining [17]. Spanish Agency of Medicines and Medical Devices recommends that Gram staining should be performed before releasing the medicine to verify that there is no contamination. If the Gram stain is positive, then the cells will not be administered.
Pictures of Gram stains were taken using brightfield microscope (Zeiss Axioskop 40; Carl Zeiss Mediatec AG, Jena, Germany). The stains were photographed by a microscope Zeiss Axioskop 40 (Carl Zeiss Mediatec AG), data not shown.
Detection of mycoplasma
The test was carried out in different phases (MCB, WCB, and final cellular medicine) on three different cultures, cells were centrifuged and resuspended at concentration of 1×106 cells/mL with 1 mL of expansion medium, and the detection of mycoplasma in the final cellular medicine was as previously described [18].
To perform this assay, 5×105 cells in 500 μL of supernatant were used. Briefly, the sample was introduced in the dry-block thermostat (TDB-120; Biosan Ltd., Riga, Latvia) to inactivate DNAses at 95°C for 10 min to avoid mycoplasma DNA degradation. Then, genomic DNA extraction was performed by the method of MB DNA (Minerva Biolabs, Berlin, Germany), which is sensitive for mycoplasma genomes. DNA was amplified by the nucleic acid amplification technique (NAT) with a PCR mycoplasma detection kit (VenorGeM; Minerva Biolabs). The DNA polymerase used for the amplification was MB Taq DNA polymerase (Minerva Biolabs). The PCR device utilized was a Life Express thermal cycler (Bioer Technology Co., Hangzhou, P.R. China). The amplification products were analyzed by gel electrophoresis in the FlashGel® DNA System containing ethidium bromide (Lonza, Walkersville, MD). The bands of any PCR amplicons were visualized by UV transillumination. They were identified by comparing them with the bands visible in the positive control and the negative control reaction. The presence of mycoplasma was indicated by an amplification product at ∼267 bp. A 191-bp band in every lane was indicated a successfully performed PCR without polymerase inhibition. The samples were analyzed before and until 24 h after their collection, which after inactivation were stored for 6 days from 2°C to 8°C [19].
Endotoxin assay
The bacterial endotoxin test (BET) method was used to detect the endotoxin unit (EU) by the gel-clot technique limulus amebocyte lysate (LAL; Pyrogent Ultra; Lonza) [20,21] from the sample in different phases (MCB, WCB, and final cellular medicine on three different cultures). For the endotoxin assay in the final cellular medicine, cells are centrifuged and the resulting pellet is resuspended at concentration of 1×106 cells/mL with 1 mL of expansion medium. A 1/10 dilution of sample was necessary (100 μL of sample in 900 μL of water for BET); this dilution did not exceed the maximum valid dilution (MVD). Then, 100 μL of LAL and 100 μL of the diluted sample were mixed in a pyrogen-free tube (Lonza). Each sample also included a negative control (100 μL of LAL and 100 μL water for BET), and two different positive controls (100 μL of LAL and 100 μL of 2λ/diluted test solution or 2λ/water for BET). Each dilution was assayed in duplicate. All tubes were incubated at 37°C for 1 h in a water bath (Grant Instruments Ltd., Cambridge, United Kingdom). Each tube was examined to observe the presence/absence of gelation. The storage conditions of the samples in this assay are not defined, so each laboratory should define and validate those conditions.
Storage stability testing
To determine sample storage conditions, a study was performed using a purified standard control of endotoxin from Escherichia coli (ATCC 12014; 10 ng/vial) with a potency of 4 EU/ng (Lonza). The samples were prepared adding 200 μL of standard control and 1800 μL of sample obtaining a solution of 0.8 EU/mL. About 2 mL of solution was stored at 4°C and −20°C for 6 months in duplicate, and analyzed at different times (24 h, 48 h, 7 days, 14 days, 28 days, 2 months, 3 months, 4 months, 5 months, and 6 months). For each assay the storage sample was vigorously vortexed for 15 min. Then, 100 μL of stored sample was mixed with 900 μL of water for BET obtaining a dilution of 1:10. Equally, a negative control was evaluated in each assay.
Microbiological monitoring in process
An environmental microbiological monitoring plan was carried out to control the air, surfaces, and staff in accordance with the European Union standards: EudraLex—Volume 4 GMP guidelines.
The environmental and staff monitoring were conducted during the manufacturing process and the sterility assay in work areas (laminar-flow cabin and isolator, respectively) with settle plates. Two media were used: Trypcase Soja Agar (TSA; bioMérieux) for detection of bacteria and Sabouraud Dextrose Chloramphenicol (SDC; bioMérieux) for fungi. The plates were incubated for 2 days at 35°C for TSA and 5 days at 22°C for SDC [22].
For environmental monitoring, two settle plates (TSA and SDC) were exposed during each handling near the activity for a maximum of 4 h. For staff monitoring, when work shift finished, the operator gloves' print (five fingers) was monitored in each media (TSA and SDC).
Analytical method validation
Analytical methods of sterility, mycoplasma, and endotoxin are “limit tests” analytical procedures, for this, an investigation of specificity; limit of detection (LOD) should be conducted during their validation according to ICH Q2 [23]. In the development phase of an analytical procedure, robustness should also be considered GMP [24].
The validation of sterility test was carried out in the intermediate product (cells with expansion medium, phase of MCB) and in the final cellular medicine (cells and packaging medium), each in triplicate (cultures 1, 2, and 3).
Validation detection of mycoplasma and endotoxin assay was carried out with three samples of intermediate product (phase of MCB) of each culture (1, 2, and 3).
Validation of sterility test
Validation was carried out to verify whether the antimicrobial activity of the intermediate product and final cellular medicine had been satisfactorily eliminated under the conditions of the test [15,16]. Specificity for sterility test was analyzed by absence of false positive results [25]; to determine LOD, a visual evaluation of turbidity was performed [15] and the robustness of these methods was studied by the reproducibility of the procedures.
The test was validated with respect to LOD accepted by Pharmacopoeia, with an inoculum not more than 100 CFU (BioBall SingleShot; bioMérieux). BioBall is an accredited reference material that contains a precise number of between 28 and 30 CFU with a standard deviation of 3 CFU [15]. The validation was based on the inoculation, in a laminar-flow cabinet BIOII-A (Telstar S.A., Madrid, Spain), of six different viable microorganisms, in each medium (TSB and TSPB) with 1 mL of supernatant or final suspension. The microorganisms used for the validation are indicated in Table 1.
All media were incubated for 3 days for bacteria (Clostridium sporogenes, Pseudomonas aeruginosa, Staphylococcus aureus, and Bacillus subtilis) and 5 days for fungi (Aspegillus niger and Candida albicans) at 35°C and 22°C for TPB and TSPB, respectively. For each microorganism a Gram stain was performed at the end of incubation to confirm microorganism identity as well as a growth promotion test as a positive control.
Validation of mycoplasma detection test
Demonstration of specificity requires the use of a suitable range of bacterial species other than mycoplasmas. Bacterial genera with close phylogenetic relation to mycoplasmas are most appropriate for this validation, for example, Lactobacillus [18]. To determine the LOD for mycoplasma assay, the performance and reproducibility of the analytical procedure was demonstrated with three reference strains known as Mycoplasma orale (NCTC 10112), Acholeplasma laidlawii (NCTC 10116), and Mycoplasma fermentans (NCTC 10117) [26]. The LOD studied was 10 CFU/mL for each strain [18]. Each sample (5×105 cells by 500 μL of supernatant) was inoculated by M. orale, A. laidlawii, and M. fermentans with a final concentration of 10 CFU/mL. All reference strains were obtained from European Directorate for the Quality of Medicines (EDQM). Briefly, in the inoculated samples, the mycoplasma assay was carried out eight times for each sample in the same PCR assay. This procedure was repeated three times on different days to analyze the variability and robustness [27].
Validation of endotoxin assay
To validate the analytical procedure used for endotoxin assay, two studies were conducted: confirmation of the labeled lysate sensitivity and test for interfering factors.
Specificity was demonstrated by the absence of interfering factors; for determination of LOD, a visual evaluation of the gelation was performed with a lysate sensitivity of 0.03 EU/mL and four replicates of each concentrations equivalent to 2λ, λ, 0.5λ, and 0.25λ by diluting the standard endotoxin with test solution and the robustness was studied by the reproducibility [20].
Confirmation of the labeled lysate sensitivity: This study was performed in three different batches of LAL (I, II, and III) to confirm the labeled lysate sensitivity (λ), which must be 0.03 EU/mL [17]. λ was the antilog10 of this mean log endpoint (the last positive result in the series of endotoxin decreasing concentration).
For each batch, four endotoxin standard dilutions were used (2λ, 1λ, 0.5λ, and 0.25λ). In a pyrogen-free tube with 100 μL of LAL, 100 μL of each standard dilution was added. Each dilution was assayed in quadruplicate. All tubes were incubated at 37°C for 1 h in water bath. Then, the presence/absence of gelation in each tube was observed, and the endpoints were determined. Each endpoint was converted to log10 and the mean and the antilog10 were calculated.
Test for interfering factors: This study was performed in three samples of intermediate product (phase of MCB, WCB, and final cellular medicine on three different cultures) to check the presence of interfering factors. Test solution was used at a dilution 1:10 (less than the MVD). Four solutions were necessary, namely, A, B, C, and D. Solution A (four replicates of test solution) was the diluted sample in water for BET (1:10). Solution B (B consisted of four replicates of each concentrations equivalent to 2λ, λ, 0.5λ, and 0.25λ by diluting the standard endotoxin with test solution) was prepared concentrations of 2λ, 1λ, 0.5λ, and 0.25λ. 2λ was elaborated with 100 μL standard solution of 20λ and 900 μL of diluted sample. 1λ was manufactured with 100 μL solution of 2λ and 100 μL of diluted sample. 0.5λ was made up with 100 μL solution of λ and 100 μL of diluted sample. 0.25λ was produced with 100 μL solution of 0.5λ and 100 μL of diluted sample. Solution C (2 replicates of each concentrations equivalent to 2λ, λ, 0.5λ, and 0.25λ by diluting the standard endotoxin with water for BET) was standard concentrations of 2λ, 1λ, 0.5λ, and 0.25λ. Solution D (two replicates of water for BET) was the water for BET.
In a pyrogen-free tube, 100 μL of each solution (A, B, C, and D) was added with 100 μL of LAL. All tubes were incubated at 37°C for 1 h in water bath. Solutions A and B were assayed in quadruplicate and solutions C and D in duplicate. Then, the presence/absence of gelation was observed.
Results
Isolation and culture of MSCs
Three different cultures (named 1, 2, and 3) of MSCs from human adipose tissue were analyzed according to the QCP proposed (as shown in Fig. 1) for the manufacturing of a cellular medicine under GMP conditions. Three samples of adipose tissue were processed to carry out the ex vivo expansion. The donor of the biological sample both autologous and allogeneic should be studied from the point of view serological, to know whether there is any infectious diseases that can cause quality problems and conservation for ex vivo expansion [28]. For each biological tissue sample, a serological report was required. The results of the three samples were negative for immunoglobulin M (IgM) A hepatitis, surface B antigen, IgG C hepatitis, and HIV, and positive for anti-HBc and anti-HBs. All materials and reagents necessary for the elaboration of the hMSCs were controlled, their sterility was analyzed, and all results were negative; no microbiological growth was shown in any media (TSB and TSPB). Also, the endotoxin assay was performed and the results demonstrated that all materials and reagents were endotoxin free.
For ex vivo expansion, different critical points have been defined: MCB, WCB, and final cellular medicine; in each point, the analytic techniques required to ensure the microbiological quality of the culture were proposed. Throughout the process a qualitative and quantitative environmental analyses of microbiological quality of air and surfaces were carried out; the results were within the standards required by the GMP guidelines as previously described [22].
Sterility test and Gram stain
Before carrying out the sterility test of samples, the effectiveness of two isolation media (TSB and TSPB) was demonstrated by growth promotion test with reference strains (C. sporogenes, P. aeruginosa, S. aureus, A. niger, B. subtilis, and C. albicans). Turbidity was observed in all media. On the other hand, sterility test of each medium was also satisfactory.
After verification of the culture media, they were inoculated with the test sample of the different phases and, after 14 days of incubation, none showed turbidity, so there was no evidence of microbial growth and the sterility of the cells was accepted for the three cultures. All negative control samples with NaCl were negative after the required incubation period.
From the results of Gram stains, no CFU was observed, in none of the three final cellular medicines studied.
Detection of mycoplasma
The presence of mycoplasma in each sample was analyzed by DNA amplification and the presence of 267-bp amplicon; none of the samples of the different phases showed presence of this band (data not shown). A successful PCR was performed without polymerase inhibition. Internal control DNA was demonstrated by a 191-bp band in the gel, which verified extraction, reverse transcription, amplification, and detection that were carried out since the internal control was added to the sample before isolating the nucleic acid.
Endotoxin assay
The threshold of endotoxin was calculated using the formula endotoxin limit (EL)=K/M [20]. Where K is the threshold pyrogenic dose of endotoxin per kilogram of body mass in 1-h period and M is the maximum recommended dose of the product per kilogram of body mass in 1-h period. For intravenous administration, the suggested value of K by the EP is 5.0 EU/kg·h and M was 1 mL/kg·h for the final cellular medicine manufactured in our laboratory, thus
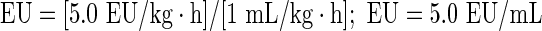 |
The threshold of endotoxin was defined for each sample as 5.0 EU/mL. LAL used for the endotoxin assay had a sensitivity of 0.03 EU/mL, which is lower than EL calculated in-house, 5.0 EU/mL, thereby, preventing false negatives. For the preparation of sample, MVD was calculated according to the formula:
 |
The sample could be diluted 166.7 times; in our method, the selected dilution was 1:10, far below the permitted MVD. In each phase for ex vivo expansion, diluted samples were tested and including negative control no sample showed gelation except the two positive controls.
In addition, a study of storage stability was performed; the concentration selected for this study (0.08 EU/mL) was based on the preparation of a concentration similar to positive control. The positive control was calculated according to the formula
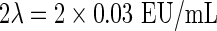 |
The positive control was 0.06 EU/mL. All samples showed gelation except the negative control samples for the duration of the study (6 months) at 4°C and −20°C. The gelation demonstrated the presence of endotoxin from E. coli. And therefore, the storage conditions of a sample in a QCP defined for endotoxin assay were at 4°C or −20°C for a maximum of 6 months.
Microbiological monitoring in process
Results were interpreted according to the annex I-GMP (Grade A: <1 CFU/4 h and <1 CFU/glove, for environmental and staff monitoring, respectively) [29].
The effectiveness of the aseptic procedures has to be continuously evaluated in order to guarantee the safety of the medicine and to identify foci of contamination risk. The results obtained for environmental and staff monitoring during the ex vivo expansion of the three cultures were within the recommended specifications for microbial contamination in TSA and SDC.
Validation of the analytical method
Analytical methods described in pharmacopoeia monographs are considered validated; however, the laboratory must also confirm the in-house procedure with the samples for study in each technique initially, ensuring that there is no interference.
Validation of sterility test
No antimicrobial activity was observed in the three cultures studied of any of the microorganisms tested. The media inoculated (with the supernatant and the final cellular medicine) and the microorganisms presented a similar growth as in the promotion test, proving that the antibiotics present in the intermediate product or the excipients of the final suspension do not interfere with the results of the sterility test. Gram stains of these media inoculated evidenced the microorganisms added.
In the study of cell culture sterility, the presence of antibiotic in the expansion medium as microbial growth inhibitory substance must be taken into account. The antibiotic concentration in 1 mL of supernatant was 10,000 IU/mL of penicillin and 10 mg/mL of streptomycin, and was inactivated with 103 IU/mL of penicillinase contained in the medium (TPB and TSPB).
Absence of false positive results confirmed the specificity of the test. The LOD was shown because all the microorganisms were grown at a concentration of 30 CFU/125 mL. The robustness was demonstrated satisfactorily by three replicate testing.
Validation of mycoplasma detection
The studied specificity by the kit manufacturer did not detect any of the phylogenetically related microorganisms: Clostridium, Lactobacillus, and Streptococcus.
To evaluate LOD, M. orale was selected in terms of the most possible source of contamination along with A. laidlawii and M. fermentans. The Fig. 2 shows the results to M. orale (Fig. 2). The required LOD of 10 CFU/mL was reached in all conditions (n=24 results: eight test replicates for each of the three samples with each strain) using different combinations of different reagents and reagent lots at different working dates by different analysts. On different days and with different samples, the same results were obtained; the three strains of tested mycoplasma were detected with equal certainty. The results presented demonstrate the robustness of mycoplasma PCR-based test.
FIG. 2.

Detection of PCR products by gel electrophoresis of 1 mL sample with 10 CFU Mycoplasma orale. Lane 1: negative control; lanes 3–10: replicates 1–8 (10 CFU/mL); Lane 11: positive control; lane 13: size marker 50 bp. The presence of mycoplasma in the sample was indicated by an amplification product at ∼267 bp. A successfully performed PCR without polymerase inhibition was indicated by a 191-bp band (internal control DNA). Three individual sample batches were tested with eight replicates for each one. On different days the same results were obtained; M. orale was detected with equal certainty. The detection limit (10 CFU/mL) was confirmed.
Validation of endotoxin assay
Confirmation of labeled lysate sensitivity: Three batches of Pyrogent Ultra Gel Clot with labeled sensitivity of 0.03 EU/mL were evaluated with standard solutions. The standard solution potency had been previously established using the current FDA Reference Standard Endotoxin. The results of these studies are described in Table 2. The results from this study confirmed the λ. The sensitivities measured were not <0.5λ (0.015 EU/mL) and not more than 2λ (0.06 EU/mL).
Table 2.
Results of Calculated Lysate Sensitivity (LAL)
| Endotoxin standard dilution (EU/mL) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Batch of LAL | 0.06 | 0.03 | 0.015 | 0.0075 | Endpoint (EU/mL) | Log10 endpoint | Mean | Antilog10 mean |
| I | + | + | − | − | 0.03 | −1.52 | −1.29 | 0.05 |
| + | − | − | − | 0.06 | −1.22 | |||
| + | − | − | − | 0.06 | −1.22 | |||
| + | − | − | − | 0.06 | −1.22 | |||
| II | + | + | − | − | 0.03 | −1.52 | −1.37 | 0.04 |
| + | + | − | − | 0.03 | −1.52 | |||
| + | − | − | − | 0.06 | −1.22 | |||
| + | − | − | − | 0.06 | −1.22 | |||
| III | + | − | − | − | 0.06 | −1.22 | −1.29 | 0.05 |
| + | + | − | − | 0.03 | −1.52 | |||
| + | − | − | − | 0.06 | −1.22 | |||
| + | − | − | − | 0.06 | −1.22 | |||
Test for interfering factors: The results of the assay are shown in Table 3. Solution A was negative for three samples, which indicated no detectable endotoxin. The sensitivity was determined with solution B in the three cultures, which was not <0.5λ (0.015 EU/mL) and not >2λ (0.06 EU/mL). The test solution did not contain interfering factors under the experimental conditions used. The sensitivity of the LAL was confirmed with solution C, and the negative control (solution D) was confirmed.
Table 3.
Test Results of Interfering Factors
| Endotoxin dilution (EU/mL) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Studied cultures (phase MCB) | Solutions | 0.06 | 0.03 | 0.015 | 0.0075 | Endpoint (EU/mL) | Log10 endpoint | Mean | Antilog10 |
| 1 | B | + | + | − | − | 0.03 | −1.52 | −1.37 | 0.04 |
| + | + | − | − | 0.03 | −1.52 | ||||
| + | − | − | − | 0.06 | −1.22 | ||||
| + | − | − | − | 0.06 | −1.22 | ||||
| C | + | + | − | − | 0.03 | −1.52 | −1.37 | 0.04 | |
| + | − | − | − | 0.06 | −1.22 | ||||
| 2 | B | + | + | − | − | 0.03 | −1.52 | −1.29 | 0.05 |
| + | − | − | − | 0.06 | −1.22 | ||||
| + | − | − | − | 0.06 | −1.22 | ||||
| + | − | − | − | 0.06 | −1.22 | ||||
| C | + | + | − | − | 0.03 | −1.52 | −1.52 | 0.03 | |
| + | + | − | − | 0.03 | −1.52 | ||||
| 3 | B | + | + | − | − | 0.03 | −1.52 | −1.29 | 0.05 |
| + | − | − | − | 0.06 | −1.22 | ||||
| + | − | − | − | 0.06 | −1.22 | ||||
| + | − | − | − | 0.06 | −1.22 | ||||
| C | + | + | − | − | 0.03 | −1.52 | −1.37 | 0.04 | |
| + | − | − | − | 0.06 | −1.22 | ||||
Solution A, solution of the preparation being examined that is free of detectable endotoxins; solution B, test for interference; solution C, control of the labeled lysate sensitivity; solution D, negative control (water for bacterial endotoxin test).
For endotoxin test absence of interfering factors validated the specificity. The LOD was confirmed for an amebocyte lysate sensitivity of 0.03 EU/mL and the robustness was demonstrated by three times satisfactory testing. The three analytical procedures (sterility, mycoplasma, and endotoxin) satisfied the required characteristics for a limit test: specificity, LOD, and robustness.
Discussion
The quality of any pharmaceutical product, including biological medicines as a CTMP, is established by the European Medicine Agency guidelines in conjunction with GMP guidelines and general chapters of the EP [30–32]. In addition, each laboratory should establish a quality system tailored to its own process and the characteristic of the cellular medicine it is producing.
Medicines are generally subjected to end-product batch testing as a means of quality control; in the case of a biologic medicine using stem cell, for clinical use, this control must be amplified due to limitations inherent in the cells, such as viability, genetic stability related to extended culture time, and microbiological contamination [33,34]. Microbiological contamination is one of the major risks associated with the administration of a CTMP. Therefore, it will be necessary to establish minimum standards of microbiological quality during the manufacture of these medicines through a QCP as proposed in this article, encompassing microbiological analysis of biological sample, materials, reagents, intermediate, and final products, as well as the environmental microbiological quality of air, surfaces, and staff [35]. Regarding all the reagents used for clinical-scale manufacturing of MSCs, they should be clinical grade. The use of reagents of animal origin must be justified and approved by the appropriate regulatory agencies. In the case of FBS, it is one of the most widely used reagents in the expansion of MSCs as cell culture supplement but the FBS presents a high variability between different batches as well as risk of contamination. As alternatives to this supplement, human factors from serum, plasma, or platelet derivatives have been suggested [36,37]. All of them must be included in the QCP, with stringent criteria of donor eligibility as well as sensitive viral NAT testing and serology, ensuring safety of the starting material for MSC manufacturing [37].
In this study, our results showed no microbiological contamination in any of the phases of cultures studied according to the QCP. In each of the three cultures studied, quality of adipose tissue sample was analyzed based on its serology and the existence of cellular pathogens and exogenous contaminants after the process of ex vivo expansion. Further, all starting materials and reagents involved in the expansion process were tested for sterility and endotoxin and their traceability was followed [34, 38].
A CTMP must be viable for its administration, besides being sterile [39, 40]. So that the manufacture of these medicines should be carried out under aseptic conditions, in a clean room and under GMP conditions, preventing contamination of the medicine and ensuring its quality, which requires identifying and controlling the critical aspects of ex vivo expansion [41].
A sterility test may be defined as a critical; the traditional method described in EP [15, 41] for the sterility test takes 14 days, although other rapid methods have been published and supported in the last annex II of the GMP [42], which entail 7 days [43], the “shelf-lives” of cells do not exceed 48 h. Therefore, the final cell medicine must be released parametrically [44], being necessary to provide all additional quality controls described in QCP to ensure that the medicine is free of contaminating microorganisms at the time of release [45]. Through sterility tests prior to release, in the intermediate phases (MCB and WCB) and on the starting material and reagents, besides endotoxin and mycoplasma assays, Gram stain and management of the monitorization process in the final cellular medicine, the absence of viable and active microorganisms can be ensured [6].
That sterility validation must demonstrate that the presence of antibiotics does not interfere with the results of sterility test. On the other hand, the use of antibiotics should be controlled to avoid resistances in patients after infusion of final cellular medicine, so the cellular pool should be washed with PBS three or four times, eliminating any trace of antibiotics.
Regarding the contamination risk of a biologic medicine, mycoplasma contamination is a major problem [46,47]. It can produce a myriad of different effects with a dramatic alteration of biological characteristics of the contaminated cells: alteration of proliferation characteristics, immune reactions, viruses' proliferation, chromosomal aberrations, and more besides. These organisms are resistant to most antibiotics commonly employed in cell cultures [16]; therefore, it is essential to analyze both the intermediate products and the final medicine in the development of a CTMP to control disturbances not only to the cells, but also to the inhibition of cell growth and hence the difficulty to obtain the final dose. Although the incidence of mycoplasma infection in the cultures is low [48], in most cases it has a human origin, making staff the major source of contamination, particularly M. orale. Accordingly, staff should receive regular training for the aseptic manipulation of therapeutics [22]. Also, both animal serum products and environment are possible sources of contamination [49].
Endotoxin assay is another requirement for parenteral medicines. The endotoxin effects are different depending on the cell types. It can induce contractile dysfunction, and increased production of immunoglobulin light chains. Equally, the absence of bacterial endotoxins in a product implies the absence of pyrogenic components.
We performed three successful validation runs to demonstrate the sensitivity, LOD, and robustness of the method through the analytical technique validation [50]. The validation parameters of different assays involved in the proposed QCP (sterility, mycoplasma, and endotoxin) demonstrated that the reagents used in each analytical technique were free of inhibitory factors and verified the suitability of each assay [51], based on the presence or absence of a detected analyte [52] by turbidity appearance, gelation after incubation, and/or detection of mycoplasma as band from PCR amplification.
In conclusion, this work proposes a control plan to analyze the safety of hMSCs as cellular medicine for clinical use, based on the evaluation of bacteriological agent contamination throughout the manufacturing process.
The efficiency of the proposed QCP in this article, carried out during the ex vivo expansion of three different cultures of hMSCs for the elaboration of autologous cell medicine under GMP conditions, has been premised on the standardization and validation in situ of each analytical technique, to ensure that the products are manufactured in a reliable and safe manner. Minimum controls of microbiological quality of the cells to be administered parenterally to patients have been established, encompassing both control samples and starting, intermediate, and final materials or products. Based on the QCP proposed, it was observed that the contamination risk could be prevented.
Acknowledgments
This study was supported by the Fundación Progreso y Salud, Consejería de Salud, Junta de Andalucía (grant PI-0022/2008); Consejería de Innovación Ciencia y Empresa, Junta de Andalucía (grant CTS-6505; INP-2011-1615-900000); FEDER co-funded grants from Instituto de Salud Carlos III (Red TerCel grant RD06/0010/0025; PI10/00964 and PI10/00871); and the Ministry of Health and Consumer Affairs (Advanced Therapies Program grant TRA-120). Support from FSED and FAID allows access to databanks. CIBERDEM is an initiative of the Instituto de Salud Carlos III.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Galvez P, Clares B, Hmadcha A, Ruiz A. and Soria B. (2012). Development of a cell-based medicinal product: regulatory structures in the European Union. Br Med Bull 105:85–105 [DOI] [PubMed] [Google Scholar]
- 2.Regulation (EC) No 1394/2007 of the European Parliament and of the council of 13 November 2007 on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004. Official J. Eur. Union L 324 Available at http://eurlex.europa.eu/LexUriServ/LexUriServ.do?.uri=OJ:L:2007:324:0121:0137:en:PDF Accessed January07, 2014 [Google Scholar]
- 3.Commission Regulation (EC) No 668/2009 of 24 July 2009 implementing Regulation (EC) No 1394/2007 of the European Parliament and of the Council with regard to the evaluation and certification of quality and non-clinical data relating to advanced therapy medicinal products developed by micro, small and medium-sized enterprises. Official J Eur Union L 194 Available at http://eurlex.europa.eu/LexUriServ/LexUriServ.do?.uri=OJ:L:2009:194:0007:0010:EN:PDF Accessed January07, 2014 [Google Scholar]
- 4.Commission Directive 2009/120/EC of 14 September 2009 amending Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use as regards advanced therapy medicinal products. Official J. Eur. Union L 242 Available at www.biosafety.be/PDF/2009_120_EC.pdf Accessed January07, 2014 [Google Scholar]
- 5.Sensebe L. (2008). Clinical grade production of mesenchymal stem cells. Biomed Mater Eng 18:3–10 [PubMed] [Google Scholar]
- 6.Inamdar MS, Healy L, Sinha A. and Stacey G. (2012). Global solutions to the challenges of setting up and managing a stem cell laboratory. Stem Cell Rev 8:830–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo CJ, Gao Y, Hou D, Shi DY, Tong XM, Shen D, Xi YM. and Wang JF. (2011). Preclinical transplantation and safety of HS/PCs expanded from human umbilical cord blood. World J Stem Cells 3:43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hillyer CD, Josephson CD, Blajchman MA, Vostal JG, Epstein JS. and Goodman JL. (2003). Bacterial contamination of blood components: risks, strategies, and regulation: joint ASH and AABB educational session in transfusion medicine. Hematology Am Soc Hematol Educ Program 2013:575–589 [DOI] [PubMed] [Google Scholar]
- 9.Cobo F, Stacey GN, Cortes JL. and Concha A. (2006). Environmental monitoring in stem cell banks. Appl Microbiol Biotechnol 70:651–662 [DOI] [PubMed] [Google Scholar]
- 10.Menard C, Pacelli L, Bassi G, Dulong J, Bifari F, Bezier I, Zanoncello J, Ricciardi M, Latour M, et al. (2013). Clinical-Grade Mesenchymal Stromal Cells Produced Under Various Good Manufacturing Practice Processes Differ in Their Immunomodulatory Properties: Standardization of Immune Quality Controls. Stem Cells Dev 22:1789–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cobo F, Stacey GN, Hunt C, Cabrera C, Nieto A, Nieto A, Montes R, Cortés JL, Catalina P, Barnie A. and Concha A. (2005). Microbiological control in stem cell banks: approaches to standardisation. Appl Microbiol Biotechnol 68:456–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serabian MA. and Pilaro AM. (1999). Safety assessment of biotechnology-derived pharmaceuticals: ICH and beyond. Toxicol Pathol 7:27–31 [DOI] [PubMed] [Google Scholar]
- 13.WHO Expert Committee on Biological Standardization. (2012). World Health Organ Tech Rep Ser 964:1–228 [PubMed] [Google Scholar]
- 14.Cobo F, Cabrera C, Catalina P. and Concha A. (2006). General safety guidances in stem cell bank installations. Cytotherapy 8:47–56 [DOI] [PubMed] [Google Scholar]
- 15.European Pharmacopoeia.(2008). Section 2.6.1 (Sterility), 7th edn. Maisonneuve SA, Sainte Ruffine, France [Google Scholar]
- 16.PIC/S. Pharmaceutical Inspection Convention, Pharmaceutical Inspection Co-operation Scheme. Recommendation on sterility testing. Available at: www.picscheme.org/ Accessed December8, 2013
- 17.Halebian S, Harris B, Finegold SM. and Rolfe RD. (1981). J Clin Microbiol 13:444–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.European Pharmacopoeia. (2008). Section 2.6.7 (Mycoplasma), 7th edn. Maisonneuve SA, Sainte Ruffine: France [Google Scholar]
- 19.Volokhov DV, Graham LJ, Brorson KA. and Chizhikov VE. (2011). Mycoplasma testing of cell substrates and biologics: Review of alternative non-microbiological techniques. Mol Cell Probes 25:69–77 [DOI] [PubMed] [Google Scholar]
- 20.European Pharmacopoeia. (2008). Section 2.6.14 (Endotoxin), 7th edn. Maisonneuve SA, Sainte Ruffine, France [Google Scholar]
- 21.Ogawa Y. (1994). Application of a bacterial endotoxin test for parenteral drugs. Eisei Shikenjo hokoku 112:209–211 [PubMed] [Google Scholar]
- 22.Martin PG, Gonzalez MB, Martinez AR, Lara VG. and Naveros BC. (2012). Isolation and characterization of the environmental bacterial and fungi contamination in a pharmaceutical unit of mesenchymal stem cell for clinical use. Biologicals 40:330–337 [DOI] [PubMed] [Google Scholar]
- 23.ICH. Harmonised Tripartite Guideline. Validation of analytical procedures: text and methodology Q2 (R1). Available at www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf Accessed December2, 2013
- 24.WHO. Expert Committee on Specifications for Pharmaceutical Preparations. 46th report. Avalilable at www.who.int/medicines/areas/quality_safety/quality_assurance/expert_committee/TRS-970-pdf1.pdf Accessed December2, 2013
- 25.European Pharmacopoeia. (2008). Section 2.6.27 (Microbiological Control of Celular Products), 7th edn. Maisonneuve SA, Sainte Ruffine, France [Google Scholar]
- 26.Dabrazhynetskaya A, Volokhov DV, David SW, Ikonomi P, Brewer A, Chang A, and Chizhikov V. (2011). Preparation of reference strains for validation and comparison of mycoplasma testing methods. J Appl Microbiol 111:904–914 [DOI] [PubMed] [Google Scholar]
- 27.Zhi Y, Mayhew A, Seng N. and Takle GB. (2010). Validation of a PCR method for the detection of mycoplasmas according to European Pharmacopoeia section 2.6.7. Biologicals 38:232–237 [DOI] [PubMed] [Google Scholar]
- 28.Eichler H, Schrezenmeier H, Schallmoser K, Strunk D, Nystedt J, Kaartinen T, Korhonen M, Cappellesso Fleury-S, Sensebé L, et al. (2013). Donor selection and release criteria of cellular therapy products. Vox Sang 104:67–91 [DOI] [PubMed] [Google Scholar]
- 29.EudraLex. Volume 4: EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use. Annex 1: Manufacture of Sterile Medicinal Products (corrected version). Directives 91/356/EEC, as amended by Directive 2003/94/EC, and 91/412/EEC. Available at http://ec.europa.eu/health/files/eudralex/vol-4/2008_11_25_gmp-an1_en.pdf Accessed November21, 2013
- 30.European Medicines Agency (EMA). Guideline on human cell-based medicinal products (EMEA/CHMP/410869/2006). Available at www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003894.pdf Accessed January07, 2014
- 31.European Medicines Agency (EMA). Guideline on safety and efficacy follow-up—risk management of advanced therapy medicinal products. (EMEA/149995/2008). Available at www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500006329.pdf Accessed January07, 2014
- 32.European Medicines Agency (EMA). Guideline on the risk-based approach according to annex I, part IV of directive 2001/83/EC applied to advanced therapy medicinal products (EMA/CAT/CPWP/686637/2011). Available at www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/03/WC500139748.pdf Accessed January07, 2014
- 33.Carmen J, Burger SR, McCaman M. and Rowley JA. (2012). Developing assays to address identity, potency, purity and safety: cell characterization in cell therapy process development. Regen Med 7:85–100 [DOI] [PubMed] [Google Scholar]
- 34.Klug B, Reinhardt J. and Schroder C. (2010). Requirements for long-term follow-up on efficacy and safety of advanced therapy medicinal products. Risk management and traceability. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz 53:58–62 [DOI] [PubMed] [Google Scholar]
- 35.Coecke S, Balls M, Bowe G, Davis J, Gstraunthaler G, Hartung T, Hay R, Merten OW, Price A, et al. (2005). Guidance on good cell culture practice. A report of the second ECVAM task force on good cell culture practice. Altern Lab Anim 33:261–287 [DOI] [PubMed] [Google Scholar]
- 36.Bieback K, Hecker A, Kocaömer A, Lannert H, Schallmoser K, Strunk D. and Klüter H. (2009). Human alternatives to fetal bovine serum for the expansion of mesenchymal stromal cells from bone marrow. Stem Cells 27:2331–2341 [DOI] [PubMed] [Google Scholar]
- 37.Bieback K. (2013). Platelet lysate as replacement for fetal bovine serum in mesenchymal stromal cell cultures. Transfus Med Hemother 40:326–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Isasi R. and Knoppers BM. (2011). From banking to international governance: fostering innovation in stem cell research. Stem Cells Int 2011:498132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rayment EA. and Williams DJ. (2010). Concise review: mind the gap: challenges in characterizing and quantifying cell- and tissue-based therapies for clinical translation. Stem Cells 28:996–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Migliaccio G. and Pintus C. (2012). Role of the EU framework in regulation of stem cell-based products. Adv Biochem Eng Biotechnol: in press; [DOI] [PubMed] [Google Scholar]
- 41.Martin PG, Martinez AR, Lara VG. and Naveros BC. (2012). Regulatory considerations in production of a cell therapy medicinal product in Europe to clinical research. Clin Exp Med 14:25–33 [DOI] [PubMed] [Google Scholar]
- 42.EudraLex. Volume 4: EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use. Annex 2: Manufacture of Biological active substances and Medicinal Products for Human Use. Directives 91/356/EEC, as amended by Directive 2003/94/EC, and 91/412/EEC. Available at http://ec.europa.eu/health/files/eudralex/vol-4/vol4-an2__2012-06_en.pdf Accessed July21, 2013
- 43.Parveen S, Kaur S, David SA, Kenney JL, McCormick WM, Gupta RK. (2011). Evaluation of growth based rapid microbiological methods for sterility testing of vaccines and other biological products. Vaccine 29:8012–8023 [DOI] [PubMed] [Google Scholar]
- 44.EudraLex. Volume 4: EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use. Annex 17: Parametric Release. Directives 91/356/EEC, as amended by Directive 2003/94/EC, and 91/412/EEC. Available at http://ec.europa.eu/health/files/eudralex/vol-4/pdfs-en/v4an17_en.pdf Accessed November21, 2013
- 45.Hock SC, Constance NX. and Wah CL. (2012). Toward higher QA: from parametric release of sterile parenteral products to PAT for other pharmaceutical dosage forms. PDA J Pharm Sci Technol 66:371–391 [DOI] [PubMed] [Google Scholar]
- 46.Xu S, Sharma H. and Mutharasan R. (2010). Sensitive and selective detection of mycoplasma in cell culture samples using cantilever sensors. Biotechnol Bioeng 105:1069–1077 [DOI] [PubMed] [Google Scholar]
- 47.Young L, Sung J, Stacey G. and Masters JR. (2010). Detection of Mycoplasma in cell cultures. Nat Protoc 5:929–934 [DOI] [PubMed] [Google Scholar]
- 48.Barile MF, Grabowski MW, Zoumbos Kapatais-K, Brown B, Hu PC. and Chandler DK. (1993). Experimentally induced Mycoplasma pneumoniae pneumonia in chimpanzees. Microb Pathog 15:243–253 [DOI] [PubMed] [Google Scholar]
- 49.Drexler HG. and Uphoff CC. (2002). Mycoplasma contamination of cell cultures: Incidence, sources, effects, detection, elimination, prevention. Cytotechnology 39:75–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Griesinger C, Hoffmann S, Kinsner A, Coecke S. and Hartung T. (2009). Possible improvement of information sources on hazard and risk. Hum Exp Toxicol 28:157. [DOI] [PubMed] [Google Scholar]
- 51.Stacey G. (2004). Validation of cell culture media components. Hum Fertil 7:113–118 [DOI] [PubMed] [Google Scholar]
- 52.Stacey G. (2012). Banking stem cells for research and clinical applications. Prog Brain Res 200:41–58 [DOI] [PubMed] [Google Scholar]