

Abstract
Microvascular loss may be an unappreciated root cause of chronic rejection for all solid organ transplants. As the only solid organ transplant that does not undergo primary systemic arterial revascularization at the time of surgery, lung transplants rely on the establishment of a microcirculation and are especially vulnerable to the effects of microvascular loss. Microangiopathy, with its attendant ischemia, can lead to tissue infarction and airway fibrosis. Maintaining healthy vasculature in lung allografts may be critical for preventing terminal airway fibrosis, also known as the bronchiolitis obliterans syndrome (BOS). BOS is the major obstacle to lung transplant success and affects up to 60% of patients surviving 5 years. The role of complement in causing acute microvascular loss and ischemia during rejection has recently been examined using the mouse orthotopic tracheal transplantation; this is an ideal model for parsing the role of airway vasculature in rejection. Prior to the development of airway fibrosis in rejecting tracheal allografts, C3 deposits on the vascular endothelium just as tissue hypoxia is first detected. With the eventual destruction of vessels, microvascular blood flow to the graft stops altogether for several days. Complement deficiency and complement inhibition lead to markedly improved tissue oxygenation in transplants, diminished airway remodeling, and accelerated vascular repair. CD4+ T cells and antibody-dependent complement activity independently mediate vascular destruction and sustained tissue ischemia during acute rejection. Consequently, interceding against complement-mediated microvascular injury with adjunctive therapy during acute rejection episodes, in addition to standard immunosuppression which targets CD4+ T cells, may help prevent the subsequent development of chronic rejection.
16.1 Introduction
Chronic rejection after transplantation is the primary cause of long-term morbidity and mortality in solid organ transplant recipients (Libby and Pober 2001). Although not widely studied at this time, emerging clues from preclinical models and clinical studies suggest that the maintenance of a functional microvasculature is required for immunosuppression to be effective (Ozdemir et al. 2004; Babu et al. 2007). Chronic rejection of solid organ transplants develops in close association with microvascular attrition (Luckraz et al. 2004, 2006; Bishop et al. 1989; Matsumoto et al. 1993). In lung transplantation, chronic rejection is manifested by BOS (Trulock et al. 2006; Yousem et al. 1985). Microvascular loss results in local tissue ischemia and may be an important cause of fibrotic wound healing (Babu et al. 2007; Luckraz et al. 2004, 2006; Minami et al. 2006; Platt et al. 1991). While ischemia-reperfusion injury due to the sudden recirculation of devitalized tissue following transplantation surgery is well recognized, microvascular-injury-associated ischemia, which occurs because of acute rejection, was only recently described by our group (Babu et al. 2007; Jiang et al. 2011). Therapeutics targeting critical pathways involved in microvascular injury are expected to improve clinical outcomes in transplantation (Contreras and Briscoe 2007), but information is lacking about what immune factors are directly responsible for tissue ischemia during acute rejection. This chapter mainly focuses on the role of complement in vascular destruction in transplanted lungs, a phenomenon that is presumably at play in other solid organ transplants.
To study this issue in a model relevant to lung transplantation, our group has utilized mouse orthotopic tracheal transplants (OTTs) (Babu et al. 2007; Jiang et al. 2011; Khan et al. 2011). Grafted trachea is functional transplants through which mice breathe and, in rejection, the airways pathologically replicate lymphocytic bronchiolitis (a large airway precursor of BOS) (Sato and Keshavjee 2008). Findings about fibrosis development in large airways from OTT research can, with appropriate caveats, be extrapolated to fibrogenesis in terminal bronchioles (Babu et al. 2007; Jiang et al. 2011; Murakawa et al. 2005; Kuo et al. 2006). The OTT model is useful because the well-organized planar anatomy of airway microvasculature supports the study of relatively long segments of microvessels (Babu et al. 2007). Recently, the Papworth autopsy study demonstrated a marked loss of microvessels in preobliterative bronchiolitis (OB) foci of human lung transplants which suggested that a loss of microcirculation and airway ischemia precede the onset of OB (Luckraz et al. 2004, 2006) (Fig. 16.1). These preclinical and clinical studies cumulatively suggest that preserving normal airway circulation is of likely benefit to the overall health (and patency) of the respiratory tree.
Fig. 16.1. Microvascular dropout prior to BOS development.

An autopsy study (Luckraz et al. 2004, 2006) has shown that lung transplant patients who die without BOS (A) have normal numbers of blood vessels around airways. In those patients dying with BOS, otherwise normal airways adjacent to BOS airways (i.e., pre-BOS) exhibit diminished microvasculature (B), whereas in adjacent lung with BOS, there are increased numbers of small-caliber blood vessels (C)
During acute rejection, profound physiologic events are occurring in the transplanted tissue beyond inflammation: most notably significant tissue hypoxia due to microvascular injury (Babu et al. 2007; Jiang et al. 2011). Typical histological techniques do not capture this information, and subsequently, the dynamic changes of microvascular blood flow and tissue oxygenation remain unknown for solid organ transplants undergoing acute rejection. Endothelial cells are a well-established target for both adaptive and innate immune responses in allograft rejection (Al-Lamki et al. 2008). Prior studies have determined that effector CD4+ T cells and complement appear to be directly injurious to allogeneic endothelial cells (Minami et al. 2006; Platt et al. 1991; Shiao et al. 2007; Baldwin et al. 2000; Choi et al. 2007). As described below, we recently determined that antibody-dependent complement activity, working independently of CD4+ cells, is sufficient to induce microvascular injury and tissue ischemia observed during acute rejection. This chapter will review the special role that complement-mediated vascular injury plays in airway transplant damage and chronic rejection.
16.2 Lung Transplantation and the Vasculature
BOS is characterized by a decline in ventilatory function and fibro-obliteration of small airways and is the major obstacle to survival following lung transplantation (Trulock et al. 2006). Despite identification of risk factors for the development of BOS, such as acute rejection and cytomegalovirus infection, the etiology of the fibroproliferative changes associated with BOS remains unknown. Recent autopsy studies from Luckraz and colleagues at Papworth Hospital, Cambridge, demonstrated a significant loss of microvasculature in nonobliterated small airways from BOS lungs, suggesting that airway ischemia is a preceding condition to airway fibrosis (Luckraz et al. 2004, 2006). In this study, it was shown that lung transplant patients who die without BOS have normal numbers of blood vessels around their airways. In those patients dying with BOS, otherwise normal airways adjacent to BOS airways (i.e., pre-BOS airways) exhibit significantly diminished microvasculature, whereas in adjacent lung with BOS, there are increased numbers of small-caliber blood vessels (Fig. 16.1).
Lung transplants are the only solid organ allografts that do not routinely undergo direct systemic arterial reconnection at the time of surgery. Airways are normally supplied by a dual circulation derived from the bronchial arteries and the pulmonary artery, and the source of microvessels around airways following transplantation is not fully known. Only the pulmonary artery circulation is restored at the time of transplantation rejection. Because the bronchial anastomosis heals well with the current surgical procedure, reconnecting the bronchial arteries has been deemed unnecessary (Patterson 1993). Therefore, following lung transplantation, the low-O2 pulmonary artery circulation is the major source of blood and microvasculature for transplanted lungs. It is important to note that, prior to transplantation, about 50% of blood flow to airways normally comes from the bronchial arteries and 50% from the poorly oxygenated pulmonary artery circulation (Barman et al. 1988).
Bronchial artery revascularization at the time of lung transplantation is feasible and was originally performed in several hundred patients (mainly in Copenhagen) in the early 1990s. However, this procedure was no longer utilized when it was noted that tracheal anastomoses healed well without bronchial artery reconnection. For this reason, the highly oxygenated bronchial artery circulation is now sacrificed in all lung transplant recipients. Omitting this rearterialization step may have more distant effects not evident in the early postoperative clinical course. Preclinical and preliminary clinical studies demonstrate that bronchial artery revascularization is durable (Norgaard et al. 1997), improves tissue perfusion with more highly oxygenated blood (Sundset et al. 1997; Kamler et al. 2004), is associated with less epithelial metaplasia (Norgaard et al. 1999), is protective of pulmonary endothelium and type II pneumocytes (Nowak et al. 2002), and may delay the development of BOS while improving patient survival (Norgaard et al. 1998). Without this native circulation, lung transplants rely on the establishment of a microcirculation to provide perfusion to the airways. However, even if bronchial artery revascularization were routinely performed, donor vasculature expressing foreign MHC antigens will always remain a target for alloimmune rejection responses.
16.3 Hypoxia, Ischemia, and Fibrosis
In airway transplantation, there is evidence that the constellation of hypoxia, alloimmune in flammation, and ultimately ischemia culminates in irreversible fibrosis (Babu et al. 2007). The mechanisms by which hypoxia and ischemia contribute to postinflammatory fibrosis are not established but are observed in several clinical scenarios such as normal skin wound healing (Gurtner et al. 2008) and chronic kidney diseases (Fine and Norman 2008). In pulmonary fibrosis studies in humans and rodents, microarray data sets demonstrate that hypoxia signaling is a prominently dysregulated pathway (Cosgrove et al. 2004; Kaminski and Rosas 2006; Zuo et al. 2002). In vitro studies have shown profibrotic phenotypic change of fibroblasts in response to hypoxia (Cool et al. 2006; Karakiulakis et al. 2007). Epithelial and endothelial cells can undergo mesenchymal transition (EMT) under ischemia to become another source of activated fibroblasts (Manotham et al. 2004). Hypoxia likely directly contributes to the progression of fibrosis by increasing the release of major extracellular matrix proteins (Distler et al. 2007). Transforming growth factor-β2-induced fibrosis is associated with remarkable vasoconstriction and tissue hypoxia (Ledbetter et al. 2000). Which of the above phenomena (i.e., activated fibroblasts, EMT, release of matrix proteins) contributes the most to airway fibrosis is not established, but it is clear that in flamed tissue subject to low pO2 and ischemia is at considerable risk for fibrotic remodeling.
16.4 Experimental Models of Lung Transplantation
There are currently no completely satisfactory models of experimental lung transplantation which replicate the fibrotic BOS lesion in terminal bronchioles. While orthotopic lung transplantation in mice is a recently developed technique (Okazaki et al. 2007), the procedure itself is technically challenging with three surgical anastomoses and has neither the throughput nor the planar anatomy to be conducive to large-scale studies of the microvasculature. Additionally, this model does not develop airway fibrosis. Heterotopic tracheal transplantation has been employed in mice and does develop an occlusive airways disease akin to BOS (Hertz et al. 1993). While this has been used to dissect out certain mechanisms of fibrotic airway occlusion, its nonnative and nonaerated position also makes it somewhat removed from clinical BOS. The use of OTT as a model for aloimmune airway rejection is now well established (Murakawa et al. 2005; Genden et al. 2002; Minamoto and Pinsky 2002). Airway remodeling as a result of chronic injury is manifested by subepithelial fibrosis, akin to the large airways in human lung transplantation (Paradis et al. 1993). The OTT model is not an obstructive airways disease model, like heterotopic transplants, but rather reproduces the fibrosis that follows lymphocytic bronchitis (i.e., large airway inflammation that can precede BOS) (Ross et al. 1997; Glanville et al. 2008). In clinical BOS, which is characterized by terminal bronchiole luminal fibrosis, large airways correspondingly show subepithelial collagen deposition (Zheng et al. 1997) similar to chronically rejecting OTTs. Therefore, the OTT model is useful for studying airway fibrosis but only allows inferences about the fibrotic process that takes place in terminal bronchioles. The benefit over heterotopic transplants is that the OTT model is functional and has, as its microenvironment, normal superior mediastinal structures. Therefore, dendritic cell clearance of shed antigens to draining lymph nodes for antigen presentation to T cells is reproduced in its native position. Transplanting airways into their normal anatomic sites may preserve the regular dendritic cell and lymphocyte trafficking pathways critical for the study of normal airway immunity. OTTs are exposed to ambient air, and so the unique respiratory interface between host and environment is maintained. Further, the inferior pole of the trachea is normally supplied by the bronchial artery circulation and is interrupted at the time of surgery. A significant advantage of the OTT model is that the microvasculature is densely arrayed in a single tissue plane allowing relatively easy quantitation and description. This is much less readily studied in lung parenchyma or terminal bronchioles where the anatomy is not so concentrated in a defined area. These favorable features have led our group to favor the OTT model for airway vascular research.
16.5 Complement, Antibodies, and Vascular Endothelium
As an important component of the innate immune system, the complement system represents a biochemical cascade consisting of a number of small proteins that circulate as inactive zymogens. When stimulated by one of several triggers such as antibody deposition on vascular endothelium, proteases in the system cleave specific proteins to release cytokines and initiate an amplifying cascade of further cleavages. The complement system is subdivided into three cascades: the classical, alternate, and lectin pathways. All three pathways converge on the C3 protein. This process culminates in the activation of the cell-killing membrane attack complex (MAC). Since the 1990s, the major laboratories examining complement and the microvasculature in transplantation and ischemia-reperfusion injury have been under the direction of Jeffrey Platt, George Tsokos, Stephen Tomlinson, Michael Carroll, Greg Stahl, and Michael Holers. Platt's group has long focused on the deleterious role that complement plays and the protective role of complement regulatory proteins (CRPs) on the vascular endothelium of rejecting xenografts (Dalmasso et al. 1992; Byrne et al. 1995; Bustos et al. 1997; Daggett et al. 1997; Gaca et al. 2006). Endothelial injury in Platt's lung xenotransplantation studies leads to vascular leakiness, macrovascular congestion and thrombus formation, pulmonary edema, and neutrophilic invasion of the microvasculature (Gaca et al. 2006; Yeatman et al. 1999). Like all lung transplantation studies to date, this pioneering work did not assess tissue perfusion or oxygenation but rather globally assesses lung tissue for injury. George Tsokos, Michael Carroll, Mike Holers, and Stephen Tomlinson's groups have primarily focused on ischemia-reperfusion injury. These elegant studies have variously demonstrated the ameliorating impact of targeted complement inhibition and the role of natural antibody in complement-mediated injury. A critical distinction in these ischemia-reperfusion studies is that ischemia happens first by manual manipulation and that complement-mediated injury ensues. The OTT model suggests that the converse process occurs in vivo; complement activity leads to microvascular destruction and ischemia, and tissue injury follows.
Figure 16.2 illustrates the fate of microvessels following rejection in OTT (Babu et al. 2007; Jiang et al. 2011; Khan et al. 2011). Following transplantation, donor and recipient microvessels reconnect through angiogenesis and fusion at the surgical anastomosis site (Babu et al. 2007). Alloimmune injury from antibody-dependent complement activity and CD4+ T cells leads to a loss of connection between the vascular systems of the recipient and the donor; the graft becomes ischemic and not rescuable with immunosuppression (Babu et al. 2007; Khan et al. 2011). Thus, the health of the transplant is closely tied to a functional microvasculature in the transplant. Figures 16.3 and 16.4 illustrate what happens to airway perfusion and oxygenation with rejection and a loss of the functional microvasculature. Figure 16.4 is the whole-mount appearance of animals treated with fluorescein isothiocyanate conjugated to lectin (FITC-lectin) i.v. just prior to sacrifice. All vessels that are connected to the systemic circulation will fluoresce, and when the connection is lost, FITC-lectin staining is absent. Following the loss of microvessels, there is a late return of perfusion as recipient-derived vasculature pierces the ischemic transplant. We have shown that this return of microvessels (vascular repair) occurs through a hypoxia-inducible factor-1 a (HIF-1a)-mediated process (Jiang et al. 2011); augmenting the HIF-1 a response decreases the hypoxic burden of rejection by promoting microvessel integrity through the recruitment of recipient-derived endothelial cells. Thus, a major idea evolving from these animal studies is that rejection is associated with significant tissue hypoxia and limiting the hypoxic burden reduces the extent of fibrotic wound healing and chronic rejection.
Fig. 16.2. Model of microvasculature destruction in airway allografts.
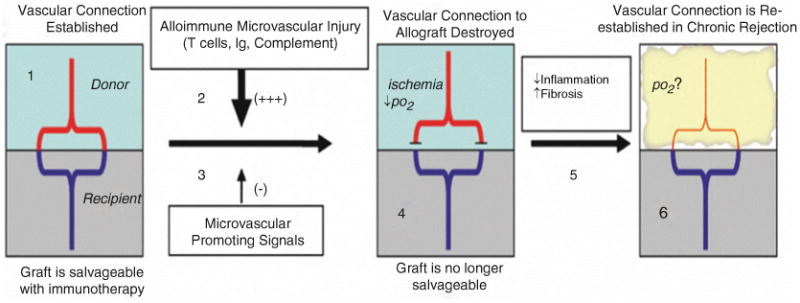
(1) Within days of transplantation, recipient vessels (blue) fuse with donor blood vessels (red) at the anastomosis site, and blood flow to the graft is transiently restored (Luckraz et al. 2006). (2) With uncontrolled rejection, the tissue then becomes progressively hypoxic, and microvasculature is increasingly injured by alloimmune attack; notably T cells, Ig, and complement. (3) Hypoxia induces signals that promote microvascular integrity and growth. HIF-1 a is a “master regulator” of this hypoxic response. However, despite multiple proangiogenic signals, the microvascular connection is eventually destroyed resulting in ischemia, decreased tissue pO2, and a loss of paracrine signaling between the vessels and neighboring tissues. (In diagram, this is indicated by alloimmune injury represented by a larger arrow than the microvascular promoting signals). (4) At this point, immunotherapy will not rescue the graft tissue from chronic rejection (Luckraz et al. 2006). (5) Endogenous anti-in flammatory molecules subsequently promote the clearance of invading leukocytes and early fibrosis occurs. (6) With fibroproliferation, the vascular connection is reestablished, but the fibrotic graft can no longer be restored to normal cellular architecture with immunotherapy. Therefore, the key to understanding and treating chronic rejection may require increased understanding of factors causing microvascular injury and, conversely, the pathways promoting microvascular stabilization
Fig. 16.3. Graphic representation of allograft tissue pO2 and blood vascular perfusion trends in syngeneic and allogeneic transplants during course of acute rejection in mouse OTT model.

Fig. 16.4. FITC-lectin perfusion shows microvascular flow detected in rejecting allograft at different time period during rejection in mouse orthotopic tracheal transplantation model.

16.6 Complement and Rejection in Lung Transplantation
While established in heart and kidney transplantation, complement-mediated injury is less well understood in lung transplantation. In the last several years, there have been diverging reports about the extent and implication of complement deposition in lung transplant recipients (Magro et al. 2003a, b, 2006; Wallace et al. 2005; Ionescu et al. 2005). Baldwin and colleagues demonstrated that rat orthotopic lung transplants performed at different academic centers indeed demonstrated complement deposition on vascular endothelium similar to rejecting heart and kidney transplants (Murata et al. 2008). In heterotopic tracheal transplantation, blockade of complement with soluble complement receptor 1 (sCR1) inhibits airway occlusive disease (Kallio et al. 2000). While Baldwin's group has shown that complement deficiency results in less vascular injury to lung allografts (Nakashima et al. 2002), no study prior to our recent OTT study (Khan et al. 2011) had previously explored how complement-mediated injury to the vascular endothelium affected the functionality of the vessels and whether complement-mediated injury to the vasculature could lead to progressive tissue hypoxia and frank ischemia. A randomized, placebo-controlled clinical study demonstrated that complement inhibition led to improved early outcomes following lung transplantation (as re flected in earlier extubations) (Keshavjee et al. 2005). This therapy was targeting the ischemia-reperfusion injury which occurs at the time of lung transplantation and may have protected the airway microvasculature. In summary, the study of complement in lung transplant rejection is relatively new and, although contentious, shows significant promise for providing insights into disease pathobiology and therapeutic approaches. While generalized complement activation as a result of ischemia perfusion injury at the time of lung transplantation has been investigated, no study in lung transplantation (or any other form of transplantation) has systematically investigated the impact that complement activation has on vascular flow during rejection.
16.7 Complement and Lymphocytes Synergize to Destroy Microvessels in Rejecting Airways
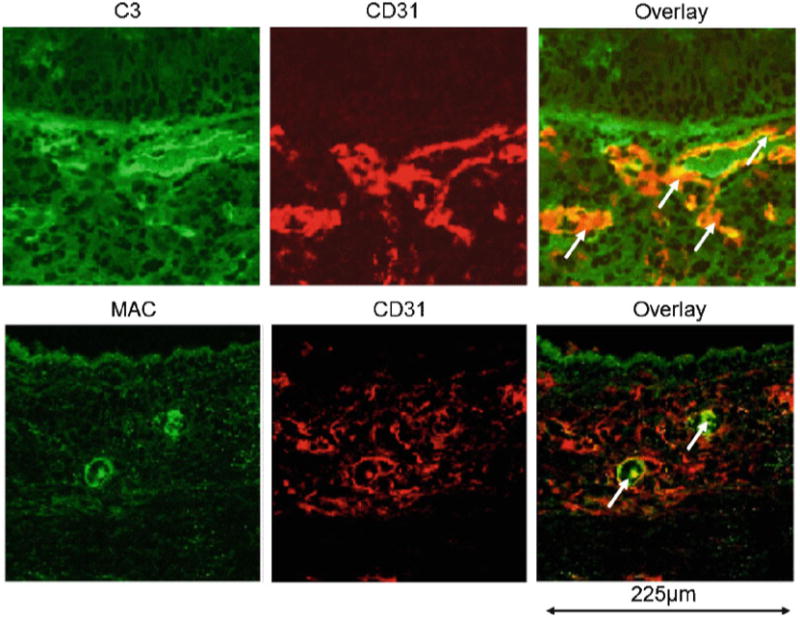
As noted, microvascular loss has been strongly associated with chronic rejection (Ozdemir et al. 2004; Babu et al. 2007; Luckraz et al. 2004, 2006; Jiang et al. 2011; Ishii et al. 2005; Labarrere et al. 2001). An important task was to try to attribute physiologic derangements to individual immune components. By focusing on the functionality of graft microvasculature, we were recently able to demonstrate that CD4+ T cells and antibody-dependent complement activity independently mediated graft ischemia during acute rejection (Khan et al. 2011). C3 deposition is first noted in microvessels approximately after 1 week of rejection, several days prior to the loss of microvessels (Fig. 16.5). C3-mediated vascular injury appears to be mediated by the formation of MAC in airway microvessels (unpublished observations, Fig. 16.6). CD8+ T cells, in isolation, do not induce microvascular-injury-associated ischemia. Airway ischemia during acute rejection was closely associated with the presence of subepi-thelial fibrosis. The duration of airway ischemia closely correlated with the appearance of the overlying epithelium with a brief cessation of airway perfusion during rejection still allowing the preservation of an intact overlying columnar lining but prolonged ischemia being tightly associated with a flattened dysplastic-appearing overlying epithelium. Thus, as our other studies have recently demonstrated (Babu et al. 2007; Jiang et al. 2011), microvascular-injury-associated ischemia in airways, during acute rejection episodes, appears to be causally linked to the airway remodeling observed in chronic rejection.
Fig. 16.5. Schematic representation of complement-mediated microvascular loss and a novel approach to optimize microvascular flow by specific complement inhibitors in lung transplantation.

Fig. 16.6. Activated complement (C3) and membrane attack complex (MAC) deposition during acute rejection. Arrows indicate colocalization of C3 and MAC on CD31+ vascular endothelial cells.
In our recent study (Khan et al. 2011), the following sequence of key results (gleaned from multiple experimental groups) subsequently led to the discovery of two independent effector pathways for microvascular-injury-associated ischemia: (1) Allografts in unreconstituted complement-replete RAG1−/− mice (which lack B and T cells) do not become ischemic. (2) CD4-reconstitution of RAG1−/−recipients results in transplant ischemia. (3) CD4-replete/C3-inhibited recipients demonstrate graft ischemia. (4) Adoptive transfer of donor-specific MHC class II antibodies restores graft ischemia in B and T cell-deficient/complement-replete RAG1−/− recipients. (5) C3-deficient/antibody-replete/CD4-depleted WT recipients do not develop ischemia. The latter two results indicated that in the absence of CD4 cells, C3 and antibody are both required for graft ischemia to occur. The cumulative results indicate that CD4+ T cells and antibody-dependent complement activation independently mediate microvascular-injury-associated ischemia. These results are summarized in Table 16.1. Because C3-deficient/CD4-depleted WT recipients did not become ischemic in this OTT study, additional effector pathways were not required to explain the loss of perfusion in airway transplants. Thus, the possibility was raised that increased complement activity during acute rejection episodes could cause allograft damage that is not addressed with the standard approach of escalating steroids. An unexpected finding in this study, pointing to how adjunctive complement inhibitors may be helpful, was that an administered complement blocker (CR2-Crry) limited graft ischemia by promoting enhanced recipient-derived angiogenesis (i.e., treatment with a complement inhibitor leads to a rapid restoration of graft vascularity by promoting recipient vessels to invade the transplant).
Table 16.1. Role of complement, antibodies and lymphocytes in maintaining microvascular flow in tracheal transplants.
| Experimental groups | Recipient immune characteristics | Allograft characteristics | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||||
| OTT donor | OTT recipient | Treatment | C3 | B cells/Ig | CD4 cells | CD8 cells | Acute rejectiona | Graft hypoxiab | Graft ischemiac | Chronic rejection |
| B6 | B6 | None | Present | Present | Present | Present | No | No | Absent | No |
| BALB/c | B6 | None | Present | Present | Present | Present | Yes | Yes | Moderate | Yes |
| BALB/c | B6 RAGl−/− | None | Present | Absent | Absent | Absent | No | No | Absent | No |
| BALB/c | B6 RAGl−/− | CD4-reconstitution | Present | Absent | Present | Absent | Yes | Yes | Severe | Yes |
| BALB/c | B6 | Anti-CD8 | Present | Present | Present | Absent | Yes | Yes | Severe | Yes |
| BALB/c | B6 RAGl−/− | CD8-reconstitution | Present | Absent | Absent | Present | Yes | No | Absent | (ND)d |
| BALB/c | B6 | Anti-CD4 | Present | Present | Absent | Present | Yes | Yes | Mild | No |
| BALB/c | B6 | Anti-CD4+anti-CD8 | Present | Present | Absent | Absent | No | Yes | Mild | No |
| BALB/c | B6 RAGl−/− | None | Present | Absent | Absent | Absent | Yes | No | Absent | No |
| BALB/c | B6 RAGl−/− | B4 IgM | Present | Anti-Annexin IV IgM | Absent | Absent | No | No | Absent | (ND) |
| BALB/c | B6 RAGl−/− | Anti-IAd MHC II IgG | Present | Donor-Specific IgG | Absent | Absent | No | Yes | Presente | (ND) |
| BALB/c | B6 C3−/− | Anti-CD4 | Absent | Present | Absent | Present | No | No | Absent | No |
| BALB/c | B6 C3−/− | Anti-CD8 | Absent | Present | Present | Absent | Yes | Yes | Severe | (ND) |
| BALB/c | B6 C3−/− | Anti-CD4+ anti-CD8 | Absent | Present | Absent | Absent | No | No | Absent | No |
| BALB/c | B6 | CR2-Crry | Inhibited | Present | Present | Present | Yes | Yes | Mild | No |
Adapted from Khan et al. (2011)
Acute rejection is defined mononuclear infiltration of the transplant
Graft hypoxia defined as significantly different that syngraft oxygenation at any respective time point
Graft ischemia defined as “absent” when none detected, “mild” when the ischemic period was <2 days, “moderate” when perfusion took >2 and <18 days to be restored, and “severe” when reperfusion was not detected during a 28 day period
ND not determined)
Only 1 time point assessed (d10) and, therefore, period of ischemia is not described
16.8 Complement Activation and Dysregulated Angiogenesis
Complement activation causes a dysregulation of normal angiogenesis required for healthy fetal development, and C3 inhibition with Crry-Ig blocks the pathological increase in soluble VEGFR-1, a potent inhibitor of VEGF activity, and rescues pregnancies in mice (Girardi et al. 2006). As just noted, in our recent study, CR2-Crry treatment promoted enhanced neovascularization of rejecting transplants in associated with upregulated proangiogenic factors within the transplant; these factors may have caused the rapid reinvestment of recipient-derived microvessels into these rejected airways (Khan et al. 2011). C3−/− recipients had a similar profile of upregulated proangiogenic factors associated with a faster revascularization period. The loss of donor-derived microvasculature is evident in C3−/− recipients; their rapid replacement with nontransgenic vessels is consistent with recipient vessels growing into the transplant as an accelerated response to increased graft-derived proangiogenic factors. So, while antibody-dependent complement activity was implicated in the destruction of microvessels during rejection, antagonism of C3 activity (when unopposed CD4+ cells are still present and capable of independently destroying the vessels) promoted rapid vascular repair and limited graft ischemia. Thus, there appears to be a dual rationale for complement inhibitor treatment during rejection episodes: (1) preventing vascular destruction and (2) promoting vascular repair.
16.9 Complement-Mediated Microvascular Injury in Transplanted Airways and the Development of Chronic Rejection
Our recent studies have strongly suggested that the duration of hypoxia and ischemia in allograft rejection is relevant to the subsequent development of significant airway remodeling as manifested by a loss of normal epithelium and increased subepithelial fibrosis (Jiang et al. 2011; Khan et al. 2011). Lung transplant recipients are particularly vulnerable to the deleterious effects of airway hypoxia and ischemia. The lung is unique among solid organ transplants because of the lack of the surgical restoration of a vascular connection to the systemic circulation. Blood supply to the airways in lung transplant recipients, in contrast to the normal dual circulation, presumably comes from the deoxygenated pulmonary artery circulation (Nicolls and Zamora 2010). Therefore, from the outset, lung transplant airways have an impaired microcirculation due to the lack of bronchial artery restoration, which, we have recently demonstrated, results in relative airway tissue hypoxia in lung transplant patients (Dhillon et al. 2010). We have hypothesized that this baseline airway hypoxia may be a diathesis for chronic rejection in lung transplant recipients (Dhillon et al. 2010) and therapies which preserve microvascular integrity may be especially relevant.
Profound tissue ischemia, undetectable by histology, may “silently” occur during acute rejection in transplant recipients. Understanding how complement activation can cause microvascular-injury-associated ischemia should help to more logically target therapeutics designed to preserve microves-sel integrity. Recently obtained physiologic information suggests that targeted complement inhibitors could safely synergize with conventional therapy during rejection episodes to prevent sustained tissue ischemia and limit the development of chronic rejection.
References
- Al-Lamki RS, Bradley JR, Pober JS. Endothelial cells in allograft rejection. Transplantation. 2008;86(10):1340–1348. doi: 10.1097/TP.0b013e3181891d8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu AN, Murakawa T, Thurman JM, Miller EJ, Henson PM, Zamora MR, et al. Microvascular destruction identifies murine allografts that cannot be rescued from airway fibrosis. J Clin Invest. 2007;117(12):3774–3785. doi: 10.1172/JCI32311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin WM, III, Qian Z, Ota H, Samaniego M, Wasowska B, Sanfilippo F, et al. Complement as a mediator of vascular inflammation and activation in allografts. J Heart Lung Transplant. 2000;19(8):723–730. doi: 10.1016/s1053-2498(00)00137-6. [DOI] [PubMed] [Google Scholar]
- Barman SA, Ardell JL, Parker JC, Perry ML, Taylor AE. Pulmonary and systemic blood flow contributions to upper airways in canine lung. Am J Physiol. 1988;255(5 Pt 2):H1130–H1135. doi: 10.1152/ajpheart.1988.255.5.H1130. [DOI] [PubMed] [Google Scholar]
- Bishop GA, Waugh JA, Landers DV, Krensky AM, Hall BM. Microvascular destruction in renal transplant rejection. Transplantation. 1989;48(3):408–414. doi: 10.1097/00007890-198909000-00011. [DOI] [PubMed] [Google Scholar]
- Bustos M, Coffman TM, Saadi S, Platt JL. Modulation of eicosanoid metabolism in endothelial cells in a xenograft model. Role of cyclooxygenase-2. J Clin Invest. 1997;100(5):1150–1158. doi: 10.1172/JCI119626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne GW, McCurry KR, Kagan D, Quinn C, Martin MJ, Platt JL, et al. Protection of xenogeneic cardiac endothelium from human complement by expression of CD59 or DAF in transgenic mice. Transplantation. 1995;60(10):1149–1156. doi: 10.1097/00007890-199511270-00016. [DOI] [PubMed] [Google Scholar]
- Choi J, Cho YM, Yang WS, Park TJ, Chang JW, Park SK. Peritubular capillary C4d deposition and renal outcome in post-transplant IgA nephropathy. Clin Transplant. 2007;21(2):159–165. doi: 10.1111/j.1399-0012.2007.00487.x. [DOI] [PubMed] [Google Scholar]
- Contreras AG, Briscoe DM. Every allograft needs a silver lining. J Clin Invest. 2007;117(12):3645–3648. doi: 10.1172/JCI34238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool CD, Groshong SD, Rai PR, Henson PM, Stewart JS, Brown KK. Fibroblast foci are not discrete sites of lung injury or repair: the fibroblast reticulum. Am J Respir Crit Care Med. 2006;174(6):654–658. doi: 10.1164/rccm.200602-205OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove GP, Brown KK, Schiemann WP, Serls AE, Parr JE, Geraci MW, et al. Pigment epithelium-derived factor in idiopathic pulmonary fibrosis: a role in aberrant angiogenesis. Am J Respir Crit Care Med. 2004;170(3):242–251. doi: 10.1164/rccm.200308-1151OC. [DOI] [PubMed] [Google Scholar]
- Daggett CW, Yeatman M, Lodge AJ, Chen EP, Van Trigt P, Byrne GW, et al. Swine lungs expressing human complement-regulatory proteins are protected against acute pulmonary dysfunction in a human plasma perfusion model. J Thorac Cardiovasc Surg. 1997;113(2):390–398. doi: 10.1016/S0022-5223(97)70337-4. [DOI] [PubMed] [Google Scholar]
- Dalmasso AP, Vercellotti GM, Fischel RJ, Bolman RM, Bach FH, Platt JL. Mechanism of complement activation in the hyperacute rejection of porcine organs transplanted into primate recipients. Am J Pathol. 1992;140(5):1157–1166. [PMC free article] [PubMed] [Google Scholar]
- Dhillon GS, Zamora MR, Roos JE, Sheahan D, Sista RR, Van der Starre P, et al. Lung transplant airway hypoxia: a diathesis to fibrosis? Am J Respir Crit Care Med. 2010;182(2):230–236. doi: 10.1164/rccm.200910-1573OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distler JH, Jungel A, Pileckyte M, Zwerina J, Michel BA, Gay RE, et al. Hypoxia-induced increase in the production of extracellular matrix proteins in systemic sclerosis. Arthritis Rheum. 2007;56(12):4203–4215. doi: 10.1002/art.23074. [DOI] [PubMed] [Google Scholar]
- Fine LG, Norman JT. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int. 2008;74(7):867–872. doi: 10.1038/ki.2008.350. [DOI] [PubMed] [Google Scholar]
- Gaca JG, Appel JZ, III, Lukes JG, Gonzalez-Stawinski GV, Lesher A, Palestrant D, et al. Effect of an anti-C5a monoclonal antibody indicates a prominent role for anaphylatoxin in pulmonary xenograft dysfunction. Transplantation. 2006;81(12):1686–1694. doi: 10.1097/01.tp.0000226063.36325.02. [DOI] [PubMed] [Google Scholar]
- Genden EM, Boros P, Liu J, Bromberg JS, Mayer L. Orthotopic tracheal transplantation in the murine model. Transplantation. 2002;73(9):1420–1425. doi: 10.1097/00007890-200205150-00010. [DOI] [PubMed] [Google Scholar]
- Girardi G, Yarilin D, Thurman JM, Holers VM, Salmon JE. Complement activation induces dysregulation of angiogenic factors and causes fetal rejection and growth restriction. J Exp Med. 2006;203(9):2165–2175. doi: 10.1084/jem.20061022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanville AR, Aboyoun CL, Havryk A, Plit M, Rainer S, Malouf MA. Severity of lymphocytic bronchiolitis predicts long-term outcome after lung transplantation. Am J Respir Crit Care Med. 2008;177(9):1033–1040. doi: 10.1164/rccm.200706-951OC. [DOI] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453(7193):314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Hertz MI, Jessurun J, King MB, Savik SK, Murray JJ. Reproduction of the obliterative bronchiolitis lesion after heterotopic transplantation of mouse airways. Am J Pathol. 1993;142(6):1945–1951. [PMC free article] [PubMed] [Google Scholar]
- Ionescu DN, Girnita AL, Zeevi A, Duquesnoy R, Pilewski J, Johnson B, et al. C4d deposition in lung allografts is associated with circulating anti-HLA alloantibody. Transpl Immunol. 2005;15(1):63–68. doi: 10.1016/j.trim.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Ishii Y, Sawada T, Kubota K, Fuchinoue S, Teraoka S, Shimizu A. Injury and progressive loss of peritubular capillaries in the development of chronic allograft nephropathy. Kidney Int. 2005;67(1):321–332. doi: 10.1111/j.1523-1755.2005.00085.x. [DOI] [PubMed] [Google Scholar]
- Jiang X, Khan MA, Tian W, Beilke J, Natarajan R, Kosek J, et al. Adenovirus-mediated HIF-1alpha gene transfer promotes repair of mouse airway allograft microvasculature and attenuates chronic rejection. J Clin Invest. 2011;121(6):2336–2349. doi: 10.1172/JCI46192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio EA, Lemstrom KB, Hayry PJ, Ryan US, Koskinen PK. Blockade of complement inhibits obliterative bronchiolitis in rat tracheal allografts. Am J Respir Crit Care Med. 2000;161(4 Pt 1):1332–1339. doi: 10.1164/ajrccm.161.4.9901114. [DOI] [PubMed] [Google Scholar]
- Kaminski N, Rosas IO. Gene expression profiling as a window into idiopathic pulmonary fibrosis pathogenesis: can we identify the right target genes? Proc Am Thorac Soc. 2006;3(4):339–344. doi: 10.1513/pats.200601-011TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamler M, Nowak K, Bock M, Herold U, Motsch J, Hagl S, et al. Bronchial artery revascularization restores peribronchial tissue oxygenation after lung transplantation. J Heart Lung Transplant. 2004;23(6):763–766. doi: 10.1016/j.healun.2003.07.016. [DOI] [PubMed] [Google Scholar]
- Karakiulakis G, Papakonstantinou E, Aletras AJ, Tamm M, Roth M. Cell type-specific effect of hypoxia and platelet-derived growth factor-BB on extracellular matrix turnover and its consequences for lung remodeling. J Biol Chem. 2007;282(2):908–915. doi: 10.1074/jbc.M602178200. [DOI] [PubMed] [Google Scholar]
- Keshavjee S, Davis RD, Zamora MR, de Perrot M, Patterson GA. A randomized, placebo-controlled trial of complement inhibition in ischemia-reperfusion injury after lung transplantation in human beings. J Thorac Cardiovasc Surg. 2005;129(2):423–428. doi: 10.1016/j.jtcvs.2004.06.048. [DOI] [PubMed] [Google Scholar]
- Khan MA, Jiang X, Dhillon G, Beilke J, Holers VM, Atkinson C, et al. CD4+ T cells and complement independently mediate graft ischemia in the rejection of mouse orthotopic tracheal transplants. Circ Res. 2011;109(11):1290–1301. doi: 10.1161/CIRCRESAHA.111.250167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo E, Bharat A, Goers T, Chapman W, Yan L, Street T, et al. Respiratory viral infection in obliterative airway disease after orthotopic tracheal transplantation. Ann Thorac Surg. 2006;82(3):1043–1050. doi: 10.1016/j.athoracsur.2006.03.120. [DOI] [PubMed] [Google Scholar]
- Labarrere CA, Nelson DR, Park JW. Pathologic markers of allograft arteriopathy: insight into the pathophysiology of cardiac allograft chronic rejection. Curr Opin Cardiol. 2001;16(2):110–117. doi: 10.1097/00001573-200103000-00006. [DOI] [PubMed] [Google Scholar]
- Ledbetter S, Kurtzberg L, Doyle S, Pratt BM. Renal fibrosis in mice treated with human recombinant transforming growth factor-beta2. Kidney Int. 2000;58(6):2367–2376. doi: 10.1046/j.1523-1755.2000.00420.x. [DOI] [PubMed] [Google Scholar]
- Libby P, Pober JS. Chronic rejection. Immunity. 2001;14(4):387–397. doi: 10.1016/s1074-7613(01)00119-4. [DOI] [PubMed] [Google Scholar]
- Luckraz H, Goddard M, McNeil K, Atkinson C, Charman SC, Stewart S, et al. Microvascular changes in small airways predispose to obliterative bronchiolitis after lung transplantation. J Heart Lung Transplant. 2004;23(5):527–531. doi: 10.1016/j.healun.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Luckraz H, Goddard M, McNeil K, Atkinson C, Sharples LD, Wallwork J. Is obliterative bronchiolitis in lung transplantation associated with microvascular damage to small airways? Ann Thorac Surg. 2006;82(4):1212–1218. doi: 10.1016/j.athoracsur.2006.03.070. [DOI] [PubMed] [Google Scholar]
- Magro CM, Klinger DM, Adams PW, Orosz CG, Pope-Harman AL, Waldman WJ, et al. Evidence that humoral allograft rejection in lung transplant patients is not histocompatibility antigen-related. Am J Transplant. 2003a;3(10):1264–1272. doi: 10.1046/j.1600-6143.2003.00229.x. [DOI] [PubMed] [Google Scholar]
- Magro CM, Ross P, Jr, Kelsey M, Waldman WJ, Pope-Harman A. Association of humoral immunity and bronchiolitis obliterans syndrome. Am J Transplant. 2003b;3(9):1155–1166. doi: 10.1034/j.1600-6143.2003.00168.x. [DOI] [PubMed] [Google Scholar]
- Magro CM, Abbas AE, Seilstad K, Pope-Harman AL, Nadasdy T, Ross P., Jr C3d and the septal microvasculature as a predictor of chronic lung allograft dysfunction. Hum Immunol. 2006;67(4–5):274–283. doi: 10.1016/j.humimm.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Manotham K, Tanaka T, Matsumoto M, Ohse T, Inagi R, Miyata T, et al. Transdifferentiation of cultured tubular cells induced by hypoxia. Kidney Int. 2004;65(3):871–880. doi: 10.1111/j.1523-1755.2004.00461.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, McCaughan GW, Painter DM, Bishop GA. Evidence that portal tract microvascular destruction precedes bile duct loss in human liver allograft rejection. Transplantation. 1993;56(1):69–75. doi: 10.1097/00007890-199307000-00012. [DOI] [PubMed] [Google Scholar]
- Minami K, Murata K, Lee CY, Fox-Talbot K, Wasowska BA, Pescovitz MD, et al. C4d deposition and clearance in cardiac transplants correlates with alloantibody levels and rejection in rats. Am J Transplant. 2006;6(5 Pt 1):923–932. doi: 10.1111/j.1600-6143.2006.01281.x. [DOI] [PubMed] [Google Scholar]
- Minamoto K, Pinsky DJ. Recipient iNOS but not eNOS deficiency reduces luminal narrowing in tracheal allografts. J Exp Med. 2002;196(10):1321–1333. doi: 10.1084/jem.20012135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakawa T, Kerklo MM, Zamora MR, Wei Y, Gill RG, Henson PM, et al. Simultaneous LFA-1 and CD40 ligand antagonism prevents airway remodeling in orthotopic airway transplantation: implications for the role of respiratory epithelium as a modulator of fibrosis. J Immunol. 2005;174(7):3869–3879. doi: 10.4049/jimmunol.174.7.3869. [DOI] [PubMed] [Google Scholar]
- Murata K, Iwata T, Nakashima S, Fox-Talbot K, Qian Z, Wilkes DS, et al. C4d deposition and cellular infiltrates as markers of acute rejection in rat models of orthotopic lung transplantation. Transplantation. 2008;86(1):123–129. doi: 10.1097/TP.0b013e31817b0b57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima S, Qian Z, Rahimi S, Wasowska BA, Baldwin WM., III Membrane attack complex contributes to destruction of vascular integrity in acute lung allograft rejection. J Immunol. 2002;169(8):4620–4627. doi: 10.4049/jimmunol.169.8.4620. [DOI] [PubMed] [Google Scholar]
- Nicolls MR, Zamora MR. Bronchial blood supply after lung transplantation without bronchial artery revascularization. Curr Opin Organ Transplant. 2010;15(5):563–567. doi: 10.1097/MOT.0b013e32833deca9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgaard MA, Efsen F, Andersen CB, Svendsen UG, Pettersson G. Medium-term patency and anatomic changes after direct bronchial artery revascularization in lung and heart-lung transplantation with the internal thoracic artery conduit. J Thorac Cardiovasc Surg. 1997;114(3):326–331. doi: 10.1016/S0022-5223(97)70176-4. [DOI] [PubMed] [Google Scholar]
- Norgaard MA, Andersen CB, Pettersson G. Does bronchial artery revascularization in fluence results concerning bronchiolitis obliterans syndrome and/or obliterative bronchiolitis after lung transplantation? Eur J Cardiothorac Surg. 1998;14(3):311–318. doi: 10.1016/s1010-7940(98)00182-1. [DOI] [PubMed] [Google Scholar]
- Norgaard MA, Andersen CB, Pettersson G. Airway epithelium of transplanted lungs with and without direct bronchial artery revascularization. Eur J Cardiothorac Surg. 1999;15(1):37–44. doi: 10.1016/s1010-7940(98)00292-9. [DOI] [PubMed] [Google Scholar]
- Nowak K, Kamler M, Bock M, Motsch J, Hagl S, Jakob H, et al. Bronchial artery revascularization affects graft recovery after lung transplantation. Am J Respir Crit Care Med. 2002;165(2):216–220. doi: 10.1164/ajrccm.165.2.2012101. [DOI] [PubMed] [Google Scholar]
- Okazaki M, Krupnick AS, Kornfeld CG, Lai JM, Ritter JH, Richardson SB, et al. A mouse model of orthotopic vascularized aerated lung transplantation. Am J Transplant. 2007;7(6):1672–1679. doi: 10.1111/j.1600-6143.2007.01819.x. [DOI] [PubMed] [Google Scholar]
- Ozdemir BH, Demirhan B, Ozdemir FN, Dalgic A, Haberal M. The role of microvascular injury on steroid and OKT3 response in renal allograft rejection. Transplantation. 2004;78(5):734–740. doi: 10.1097/01.tp.0000130453.79906.62. [DOI] [PubMed] [Google Scholar]
- Paradis I, Yousem S, Griffith B. Airway obstruction and bronchiolitis obliterans after lung transplantation. Clin Chest Med. 1993;14(4):751–763. [PubMed] [Google Scholar]
- Patterson GA. Airway revascularization: is it necessary? Ann Thorac Surg. 1993;56(4):807–808. doi: 10.1016/0003-4975(93)90335-f. [DOI] [PubMed] [Google Scholar]
- Platt JL, Fischel RJ, Matas AJ, Reif SA, Bolman RM, Bach FH. Immunopathology of hyperacute xenograft rejection in a swine-to-primate model. Transplantation. 1991;52(2):214–220. doi: 10.1097/00007890-199108000-00006. [DOI] [PubMed] [Google Scholar]
- Ross DJ, Marchevsky A, Kramer M, Kass RM. “Refractoriness” of air flow obstruction associated with isolated lymphocytic bronchiolitis/bronchitis in pulmonary allografts. J Heart Lung Transplant. 1997;16(8):832–838. [PubMed] [Google Scholar]
- Sato M, Keshavjee S. Bronchiolitis obliterans syndrome: alloimmune-dependent and -independent injury with aberrant tissue remodeling. Semin Thorac Cardiovasc Surg. 2008 Summer;20(2):173–182. doi: 10.1053/j.semtcvs.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Shiao SL, Kirkiles-Smith NC, Shepherd BR, McNiff JM, Carr EJ, Pober JS. Human effector memory CD4+ T cells directly recognize allogeneic endothelial cells in vitro and in vivo. J Immunol. 2007;179(7):4397–4404. doi: 10.4049/jimmunol.179.7.4397. [DOI] [PubMed] [Google Scholar]
- Sundset A, Tadjkarimi S, Khaghani A, Kvernebo K, Yacoub MH. Human en bloc double-lung transplantation: bronchial artery revascularization improves airway perfusion. Ann Thorac Surg. 1997;63(3):790–795. doi: 10.1016/s0003-4975(96)01273-8. [DOI] [PubMed] [Google Scholar]
- Trulock EP, Edwards LB, Taylor DO, Boucek MM, Keck BM, Hertz MI. Registry of the International Society for Heart and Lung Transplantation: twenty-third official adult lung and heart-lung transplantation report–2006. J Heart Lung Transplant. 2006;25(8):880–892. doi: 10.1016/j.healun.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Wallace WD, Reed EF, Ross D, Lassman CR, Fishbein MC. C4d staining of pulmonary allograft biopsies: an immunoperoxidase study. J Heart Lung Transplant. 2005;24(10):1565–1570. doi: 10.1016/j.healun.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Yeatman M, Daggett CW, Lau CL, Byrne GW, Logan JS, Platt JL, et al. Human complement regulatory proteins protect swine lungs from xenogeneic injury. Ann Thorac Surg. 1999;67(3):769–775. doi: 10.1016/s0003-4975(99)00049-1. [DOI] [PubMed] [Google Scholar]
- Yousem SA, Burke CM, Billingham ME. Pathologic pulmonary alterations in long-term human heart-lung transplantation. Hum Pathol. 1985;16(9):911–923. doi: 10.1016/s0046-8177(85)80130-1. [DOI] [PubMed] [Google Scholar]
- Zheng L, Ward C, Snell GI, Orsida BE, Li X, Wilson JW, et al. Scar collagen deposition in the airways of allografts of lung transplant recipients. Am J Respir Crit Care Med. 1997;155(6):2072–2077. doi: 10.1164/ajrccm.155.6.9196117. [DOI] [PubMed] [Google Scholar]
- Zuo F, Kaminski N, Eugui E, Allard J, Yakhini Z, Ben-Dor A, et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci USA. 2002;99(9):6292–6297. doi: 10.1073/pnas.092134099. [DOI] [PMC free article] [PubMed] [Google Scholar]