

Abstract
Some mutations of the insulin gene cause hyperinsulinemia or hyperproinsulinemia. Replacement of biologically important amino acid leads to defective receptor binding, longer half‐life and hyperinsulinemia. Three mutant insulins have been identified: (i) insulin Chicago (F49L or PheB25Leu); (ii) insulin Los Angeles (F48S or PheB24Ser); (iii) and insulin Wakayama (V92L or ValA3Leu). Replacement of amino acid is necessary for proinsulin processing results in hyperproinsulinemia. Four types have been identified: (i) proinsulin Providence (H34D); (ii) proinsulin Tokyo (R89H); (iii) proinsulin Kyoto (R89L); and (iv) proinsulin Oxford (R89P). Three of these are processing site mutations. The mutation of proinsulin Providence, in contrast, is thought to cause sorting abnormality. Compared with normal proinsulin, a significant amount of proinsulin Providence enters the constitutive pathway where processing does not occur. These insulin gene mutations with hyper(pro)insulinemia were very rare, showed only mild diabetes or glucose intolerance, and hyper(pro)insulinemia was the key for their diagnosis. However, this situation changed dramatically after the identification of insulin gene mutations as a cause of neonatal diabetes. This class of insulin gene mutations does not show hyper(pro)insulinemia. Mutations at the cysteine residue or creating a new cysteine will disturb the correct disulfide bonding and proper conformation, and finally will lead to misfolded proinsulin accumulation, endoplasmic reticulum stress and apoptosis of pancreatic β‐cells. Maturity‐onset diabetes of the young (MODY) or an autoantibody‐negative type 1‐like phenotype has also been reported. Very recently, recessive mutations with reduced insulin biosynthesis have been reported. The importance of insulin gene mutation in the pathogenesis of diabetes will increase a great deal and give us a new understanding of β‐cell biology and diabetes. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2011.00100.x, 2011)
Keywords: Insulin gene mutation, Endoplasmic reticulum stress, Neonatal diabetes
Introduction
Diabetes mellitus is not a single disease, but a heterogeneous group of metabolic diseases characterized by chronic hyperglycemia resulting from the shortage of insulin action, which is caused by a decrease of insulin secretion and/or increased insulin resistance. Although the underlying etiology of the most common forms of diabetes has not been fully clarified, some forms of the disease are characterized by their specific etiology or pathogenesis. Biologically inactive or structurally abnormal insulin or its precursor, proinsulin, has been thought to be one of the possible causes of diabetes mellitus. The first mutant human insulin protein was identified by Tager et al. in 19791. Until now, three types of mutant insulins with hyperinsulinemia2–8 and four types of mutant proinsulins with hyperproinsulinemia9–17 have been identified. Those are very rare and have shown only mild diabetes or glucose intolerance. Hyperinsulinemia or hyperproinsulinemia were the key for their diagnosis. The possibility of insulin gene mutations, which reduce insulin biosynthesis and therefore do not show hyperinsulinemia, has been speculated. The first example of this class of insulin gene mutations was identified in mice, but not in humans. The mouse models of hypoinsulinemic insulin gene mutations are Akita mice (C96Y mutation in Insulin gene 2)18–20 and Munich mice (C95S mutation in Insulin gene 2)21. Both of these mouse models show early‐onset diabetes and β‐cell loss. Mutation at the cysteine residue causes the disturbance of correct disulfide bonding and proper conformation of the proinsulin molecule, which leads to misfolded protein accumulation in the endoplasmic reticulum (ER), increased ER stress and, finally, apoptosis of pancreatic β‐cells. The same class of insulin gene mutations in humans has been identified as the cause of neonatal diabetes, and the cause of MODY, autoantibody‐negative type 1‐like diabetes and early‐onset type 2‐like diabetes22–31. These mutations are heterozygous and work in a dominant manner. However, very recently, recessive mutations with reduced insulin biosynthesis have been identified in neonatal diabetes32. The spectrum of insulin gene mutations is spreading wider and wider. And its importance in the pathogenesis of diabetes is growing. In the present review, we summarize these various aspects of insulin gene mutations.
Gene structure, biosynthesis and processing of insulin
The human insulin gene cloned by Bell et al.33 consists of three exons. Exon 1 (42 bp) contains only the 5′‐untranslated region and exon 2 (204 bp) encodes the signal peptide, the B‐chain and part of the connecting peptide (C‐peptide), and exon 3 (219 bp) encodes the rest of the C‐peptide and the A‐chain. Humans have a single insulin gene, although rats, mice and at least several fish species have two insulin genes34. The single human insulin gene corresponds to rat or mouse insulin gene 2. All of the three genes have two introns at corresponding positions, although rat and mouse insulin gene 1 lost one of two introns.
Insulin is synthesized almost specifically in pancreatic β‐cells of the islets of Langerhans. Preproinsulin, the first translational product from the insulin gene, is a 110 amino acid polypeptide with the 24 amino acid signal peptide. The signal peptide is bound by the signal‐recognition particle (SRP), and through interaction of the SRP receptor in the ER membrane, the penetration of preproinsulin into ER lumen occurs, and the proteolytic cleavage of signal peptide from preproinsulin yields proinsulin. Thereafter, proinsulin undergoes appropriate folding, forming disulfide bonds. Appropriately folded proinsulin is then transferred to the Golgi apparatus and sorted to constitutive or regulated secretory pathways. The constitutive pathway, as its name implies, is not regulated by secretagogues. In contrast, the regulated pathway, where almost all of the proinsulin molecules are sorted, includes the packaging of prohormones into secretory granules and following exocytosis in response to secretagogues. Conversion of proinsulin to insulin and C‐peptide occurs within secretory granules. Proinsulin, the precursor of insulin, which was discovered by Steiner et al.35,36, is converted to insulin by enzymatic removal of C‐peptide at the sites of dibasic amino acids. Prohormone convertase 1/3, 2 and carboxypeptidase E are involved in this process. The initial event of this processing is endoproteolytic cleavage on the carboxy‐terminal side of one of the basic residues linking the insulin chains to the C‐peptide. Two prohormone convertases, PC1/3 and PC2, involved in this process have preferences for specific sites. PC1/3 preferentially cleaves on the connecting site between the insulin β‐chain and the C‐peptide, whereas PC2 cleaves on the connecting site between the insulin α‐chain and the C‐peptide. The residual basic amino acids left are removed by carboxypeptidase E. In humans, the initial cleavage will occur predominantly between the insulin β‐chain and the C‐peptide; therefore, in two of the proinsulin conversion intermediates, a much larger amount of des‐64,65 split proinsulin exists in the circulation than that of des‐31,32 split proinsulin. A further round of cleavage and trimming of another site generates insulin and C‐peptide at the equimolar ratio. Biosynthesis and processing of insulin has also been reviewed elsewhere37–40.
Insulin gene mutations with hyperinsulinemia
Missense mutations in the insulin gene leading to the production of structurally abnormal insulins with reduced biological activity and receptor binding can cause diabetes mellitus. To date, three distinct mutant insulins have been identified (Figure 1)1–8,41,42. In 1979, Tager et al.1 reported the first case, insulin Chicago (F49L or PheB25Leu). The family with the second mutant insulin, insulin Los Angeles (F48S or PheB24Ser), was reported subsequently3–5. The third mutant insulin, insulin Wakayama (V92L or ValA3Leu), was found in Japan and three families of this mutation have been reported from Japan5–8. The biological activity and receptor binding of these three mutant insulins are dramatically reduced, and therefore their half‐lives are prolonged (Table 1). Among the three mutant insulins, insulin Wakayama showed the lowest receptor binding and biological activity, and the longest half‐life. There is a negative correlation between receptor binding and half‐life. Not all individuals with mutant insulin develop diabetes. Because affected subjects have both normal and mutant alleles, overproduction of normal insulin will compensate. The assessment of endogenous insulin secretory capacity by serum CPR response to the 75‐g oral glucose tolerance test (Table 2) clearly showed that affected subjects secrete approximately twice the amount of insulin compared with control subjects with corresponding glucose tolerance. If other factors, such as aging, causing insulin resistance or pancreatic β‐cell secretory dysfunction, are added in the affected subjects, they might easily develop diabetes or glucose intolerance.
Figure 1.
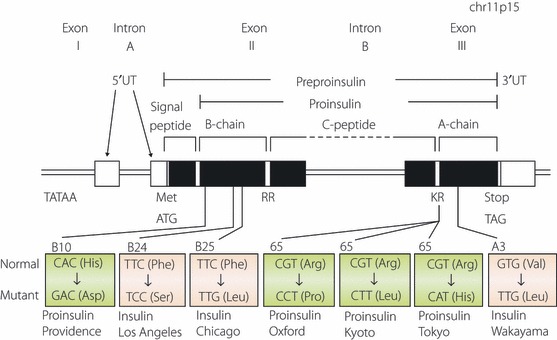
Schematic representation of human insulin gene structure and mutations causing hyperinsulinemia or hyperproinsulinemia. Exons are shown as boxes. Protein coding regions are shown as closed boxes, whereas non‐coding regions are shown as open boxes. Mutations resulting in hyperinsulinemia and hyperproinsulinemia are colored in blue and in pink, respectively. Translation initiation codon (ATG), termination or stop codon (TAG) or dibasic cleavage sites (RR and KR) are shown.
Table 1. Receptor binding, biological activity and serum half‐life of three abnormal insulins.
| Normal Human | Chicago LeuB25 | Los Angeles SerB24 | Wakayama LeuA3 | |
|---|---|---|---|---|
| In vitro receptor binding (%) | 100 | 2.0–2.8 | 0.5–1.8 | 0.3–0.7 |
| Biological activity (%) | 100 | 1.9–2.5 | 0.6–1.5 | 0.4–0.8 |
| In vivo serum half‐life in dog (min) | 3.6 | 15.0 | 24.5 | 35.0 |
Human Insulin served as standard and was defined as 100% receptor binding and biological activity. Receptor binding was determined by using rat adipocytes, and biological activity was assessed by the stimulation of glucose uptake or oxidation in rat adipocytes. In vivo half‐life was determined by using somatostatin infused dog.
Table 2. Endogenous insulin secretion capacity in patients with abnormal insulin, Insulin Wakayama.
| NGT | IGT | DM | |
|---|---|---|---|
| Insulin Wakayama | 7.43 ± 0.22 (n = 3) | 3.56 ± 0.56 (n = 3) | 1.37 ± 0.21 (n = 3) |
| Controls | 3.56 ± 0.29 (n = 15) | 2.07 ± 0.22 (n = 10) | 0.72 ± 0.16 (n = 9) |
△CPR(30′) at 75 g oral glucose tolerance test was shown.
△CPR(30′) = CPR(30′) – CPR(0′) (ng/mL)
DM, diabetes mellitus; IGT, impaired glucose tolerance; NGT, normal glucose tolerance.
Reduced urinary excretion of mutant insulin, insulin Wakayama, compared with normal insulin has been reported43,44. Because arginine (inhibitor of renal tubular reabsorption) load increases urinary excretion of insulin Wakayama44, there might exist a receptor‐mediated secretion system in the kidney.
Insulin gene mutations with hyperproinsulinemia
Insulin gene mutations affecting the conversion of proinsulin to insulin cause hyperproinsulinemia. Four different mutations have been identified thus far (Figure 1)9–17,41. The R89 at the C‐peptide–α‐chain junction is mutated in three of the mutations (proinsulin Tokyo, R89H; proinsulin Kyoto, R89L; and proinsulin Oxford, R89P). Another mutation is at the β‐chain (proinsulin Providence, H34D). The degree of glucose intolerance varies from normal to mild diabetes. The mutations at the position of R89 disturb the processing of des‐31,32 split proinsulin to insulin and C‐peptide by PC2. Therefore, these patients have a high level of the circulating des‐31,32 split proinsulin, which cleaves only the β‐chain–C‐peptide junction by PC1/3. Another mechanism is involved in the cause of hyperproinsulinemia by H34D insulin gene mutation. Transfection of the mutant insulin gene or transgenic mice both showed that a significant amount of mutant H34D proinsulin is secreted from the constitutive pathway where processing does not occur45,46. The reason why H34D proinsulin missorted to the constitutive pathway is unclear. However, H34D insulin has approximately fivefold enhanced receptor binding compared with normal insulin47. Therefore, the mutant H34D proinsulin might bind to the recycling insulin receptor much more strongly than normal proinsulin. Since recycling insulin receptors are transferred to cell surface through the constitutive pathway, mutant proinsulins binding with receptors might be sorted to this pathway.
Insulin gene mutations without hyperinsulinemia or hyperproinsulinemia (animal model)
In contrast to insulin gene mutations causing hyper(pro)insulinemia, the possibility of insulin gene mutations, which reduce insulin biosynthesis and do not show hyperinsulinemia, had been speculated. The Akita mouse is a spontaneous diabetic mouse with reduced β‐cell mass and without insulitis or obesity18,19. Genetic analysis showed that a mutation of C96Y in the insulin 2 gene is responsible for the diabetic phenotype in this mouse20. Because mice have two insulin genes, neither knockout mice of insulin 1 or insulin 248 develop severe diabetes as Akita mice do. The C96Y mutation disrupts a disulfide bond between the α‐ and β‐chains of insulin and therefore are thought to induce a drastic conformational change of the molecule. In the pancreas of Akita mice, mRNAs for ER‐stress induced genes were increased and the targeted disruption of the C/EBP homologous protein (CHOP) gene, ER‐stress associated apoptosis factor, delayed the onset of diabetes in this mouse49. Overexpression of mutant C96Y insulin in MIN6 insulinoma cells results in apoptosis49. Therefore, diabetes in Akita mice is a result of β‐cell apoptosis induced by increased ER stress through the production of misfolded protein, rather than a simple decrease of insulin production49,50. Another example of this class of insulin gene mutation is the Munich (C95S mutation in Ins2 gene) mouse21. This mouse has been identified as the novel non‐obese diabetic mouse by the screening of N‐ethyl‐N‐nitrosourea (ENU) mouse mutagenesis project. The C95S mutation will disrupt the intrachain disulfide bond of the insulin α‐chain, and causes hypoinsulinemic hyperglycemia by 1 month‐of‐age. Electron microscopic observation showed a decreased number and size of insulin granules, enlarged ER and swollen mitochondria consistent with increased ER stress by abnormally folded mutant insulin molecules21.
ER stress and β‐cells
The ER is a membrane‐bound organelle that supports the biosynthesis of approximately one‐third of the cellular proteins. The ER is a place for protein folding and post‐translational modification of polypeptide destined for the plasma membrane, intracellular organelles or extracellular secretion. Protein folding is facilitated or assisted by molecular chaperones and folding enzymes including binding Ig protein (BiP) or protein disulfide isomerase (PDI). Successfully folded proteins are sent to the Golgi apparatus. Proteins improperly folded are, however, subjected to retention in the ER lumen and eventually destroyed. ER stress, which is caused by the accumulation of misfolded proteins in the ER, elicits stress signaling pathways termed the unfolded protein response (UPR). The presence of unfolded proteins is sensed by dsRNA‐activated protein kinase (PKR)‐like ER kinase (PERK), also known as eukaryotic translation initiation factor (eIF) 2‐a kinase 3 (EIF2aK3), inositol requiring protein 1a (IRE1a) and activating transcription factor 6 (ATR6). When the unfolded protein load exceeds the capacity, PERK phosphorylates eIF2a and attenuates mRNA translation initiation of new proteins. However, several mRNA species need eIF2a phosphorylation for their translation initiation. One example is ATF4, a transcriptional factor inducing the expression of a subset of genes including ER chaperones. IRE1a, when activated, showed RNase activity and degradated mRNAs to reduce the ER stress. IRE1a also cleaves X‐box binding protein 1 (XBP1) mRNA, which is necessary for the splicing reaction required for the translation of active XBP1 isoform (XBP1s). XBP1s activates the transcription of genes, including ER chaperones and genes for the ER‐associated degradation (ERAD) system51. ATF6 is another transcription factor that binds to the ER stress response elements in the promoters of UPR‐responsive genes. PERK, IRE1a and ATF6 are maintained in an inactive state through interaction with the chaperone protein, BiP. Accumulation of unfolded proteins in the ER results in the release of BiP from these sensor proteins. And the dimerization of PERK or IRE1a will cause their activation. ATF6 is activated after transfer to the Golgi apparatus and after proteolytic processing. If the ER stress is too much and UPR cannot handle it, ER stress‐mediated apoptosis might occur. Apoptotic signals from the ER includes: (i) PERK/eIF2a‐dependent induction of the proapoptotic transcription factor CHOP; (ii) BAK/BAX‐regulated Ca2+ release from the ER; (iii) IRE1a‐mediated activation of apoptotic signal‐regulating kinase 1(ASK1)/c‐Jun amino terminal kinase (JNK); and (iv) activation of procaspase 12.
Because pancreatic β‐cells have to produce a huge amount of insulins, almost 1 million molecules per minute per cell, conditions impair protein folding and processing would easily cause β‐cell failure and diabetes. In fact, UPR gene induction was reported both in the islets of diabetic patients and diabetic model mice, showing the involvement of ER stress in the pathogenesis of diabetes. The importance of ER stress in β‐cells has been summarized in other review articles52–57.
Neonatal diabetes
Neonatal diabetes mellitus (NDM) is defined as diabetes before 6 months‐of‐age and is divided into two categories: (i) permanent neonatal diabetes mellitus (PNDM); and (ii) transient neonatal diabetes mellitus (TNDM)58,59. The former requires continuous treatment for diabetes. In contrast, the latter is characterized by spontaneous remission within the first few months of life, although frequent relapse of the disease might occur later in life. NDM is rare, estimated approximately one case per 300,000–500,000 live births, and over half of NDM cases are TNDM. Recent studies have shown the genetic causes of both TNDM and PNDM. In TNDM, the major cause is chromosome 6q24 abnormalities. Overexpression of genes in this region, which is normally imprinted (silencing of the maternal allele), by paternal uniparental isodisomy (UPD), by paternal duplication of this region or by loss of DNA methylation is thought to be a cause of TNDM60. The 6q24 abnormalities explain approximately 70% of TNDM, followed by adenosine triphosphate (ATP)‐binding cassette, subfamily C, member 8 (ABCC8) mutations, and potassium channel, inwardly rectifying, subfamily J, member 11 (KCNJ11) mutations, both of which are components of pancreatic β‐cell ATP‐sensitive potassium channel58,59,61. Mutations of hepatocyte nuclear factor‐1β, well‐known as the cause of maturity‐onset diabetes of the young (MODY) 5, have been also reported as a cause of TNDM62,63. In PNDM, the major cause is defects of the ATP‐sensitive potassium channel (ABCC8 or KCNJ11 mutations)58,59,61. Glucokinase and pancreas/duodenum homeobox protein (PDX) 1, both of which have been known to cause MODY in heterozygous situations, cause PNDM in homozygous or compound heterozygous states64–68. Rare genetic disorders with syndromic features also cause PNDM. The genes responsible include forkhead box P3 (FOXP3) in immunodysregulation polyendocrinopathy, enteropathy, X‐linked (IPEX)69,70, eukaryotic translation initiation factor 2‐α kinase 3 (EIF2AK3) in Wolcott–Rallison syndrome71, pancreas transcription factor (PTF) 1a in PNDM with cerebellar agenesis72, regulatory factor X 6 (RFX6) in PNDM with pancreatic hypoplasia, intestinal atresia and gallbladder aplasia73, and glioma‐associated oncogene‐similar family zinc finger 3 (GLIS3) in PNDM with congenital hypothyroidism74. Among these genes, PERK is an important component in the UPR, and therefore PNDM, as a result of PERK dysfunction, was originally thought to be caused by a failure to control ER stress. However, recent studies have also claimed this notion. The decrease of β‐cell mass was a result of decreased proliferation rather than increased apoptosis. PERK also regulates proinsulin trafficking75,76. Further study will be necessary to gain a better understanding of the physiological roles of PERK or ER stress in β‐cells.
Insulin gene mutations in neonatal diabetes
In 2007, Stoy et al.22 reported that mutations of the insulin gene are a novel cause of PNDM. Ten heterozygous mutations in 16 families were identified and this discovery stimulated further screening and led to the identification of more mutations (Figure 2)23–31. Furthermore, the clinical entity of insulin gene mutations is not restricted in ND. Insulin gene mutations have been identified in MODY, type 1b (autoantibody‐negative) like patients and early‐onset type 2‐like patients. To date, insulin gene mutations in the heterozygous state have been reported in 66 probands. Among them, 18 out of 66 (27%) were inherited from an affected parent, whereas 48 out of 66 (73%) were originated from de novo mutations. The heterozygous insulin gene mutation could explain approximately 14% of PNDM patients born to non‐consanguineous parents. The incidence of heterozygous insulin gene mutation in diabetes other than PNDM is supposed to be much lower and is estimated to be less than 2%77.
Figure 2.
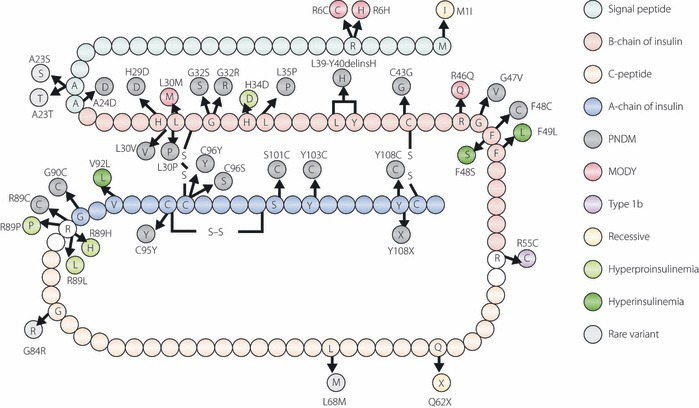
Schematic representation of the amino acid sequence of human preproinsulin showing the positions of mutations. Signal peptide, β‐chain or α‐chain of insulin, and C‐peptide are shown in different colors. Mutations identified in patients with permanent neonatal diabetes mellitus, maturity‐onset diabetes of the young, type 1b‐like, hyperproinsulinemia, hyperinsulinemia or recessive mutation are shown in different colors. Modified from Stoy J, Steiner DF, Park S‐Y et al. Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev Endocr Metab Disord 2010; 11: 205–215.
Recently, recessive (homozygous or compound heterozygous) mutations of the insulin gene have been reported. Ten mutations in 15 families were identified (Figure 3)32. These mutations lead to decreased insulin biosynthesis through different mechanisms: gene deletion, mutated start codon, mutated polyadenylation signal that would affect mRNA stability, and promoter mutations. Compared to patients with heterozygous insulin gene mutations, these recessive patients showed lower birth weight (−3.2SD vs−2.0SD) and earlier diagnosis (1 week vs 10 weeks), showing the severity of β‐cell defects in these patients. TNDM is only seen in patients with recessive mutations (26%vs 0%). TNDM is only found in patients with non‐coding mutations, and they have a higher birth weight and are diagnosed later, which implies mutations causing TNDM are less severe than those with PNDM.
Figure 3.
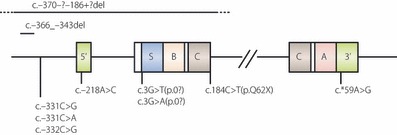
Schematic representation of the structure of the human insulin gene showing the sites of recessive mutations. Exons are boxed. Coding regions for signal peptide, α‐ and β‐chains of insulin, C‐peptide are shown as S, A, B and C, respectively, and 5′‐ and 3′‐untranslated regions are indicated as 5′ and 3′, respectively. Recessive insulin gene mutations are shown. Modified from Garin I, Edghill EL, Akerman I et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci USA 2010; 107: 3105–3110.
Studies of the function of each insulin gene mutation have been carried out. Mutations affecting processing or folding do indeed accumulate in the ER, poorly secreted, and cause the induction of UPR and apoptosis78–81. This class of mutant insulin exerts a dominant‐negative effect on the synthesis and secretion of normal insulin as a result of UPR and the consequent attenuation of translation82,83.
Recessive mutations in the insulin gene, in contrast, results in reduced insulin biosynthesis through various mechanisms including gene deletion, mutated start codon, mutated polyadenylation signal that would affect mRNA stability, and promoter mutations32.
The insulin variable number of tandem repeats and type 1a diabetes
Insulin gene variable number of tandem repeats (VNTR) is located approximately 0.5 kb upstream of the insulin gene. This polymorphic repeat consists of a 14–15 bp unit of consensus sequence (ACAGGGGTCTGGGG) with a slight variation of the repeat sequence84, and is classified as class I (small, frequency approximately 70% in Caucasians, but more than 90% in Japanese), class II (intermediate, rare) or class III (large, frequency approximately 30% in Caucasians). The insulin VNTR was found to be associated with type 1 diabetes and is now referred to as the IDDM2 susceptibility locus85–88. The shorter class I allele predisposes to type 1 diabetes, whereas the class III allele showed resistance to type 1 diabetes. Not only seen in Caucasians, this correlation has also been proved in Japanese89. The expression of the insulin gene is highly restricted in pancreatic β‐cells with very few exceptions. The thymus is one of the exceptions. Genes encoding for self molecules have been found to be expressed in the thymus, including insulin90,91. And the thymic expression of self antigens might be crucial for the development of self tolerance or negative selection. Insulin mRNA levels in the thymus were found to correlate with the VNTR allele92,93. The class III VNTR alleles are transcribed at much higher levels in the thymus than class I alleles. Because thymic expression of self antigens and their levels of expression affect the development of self tolerance or negative selection of autoreactive T‐lymphocytes, the insulin gene VNTR allele might modulate tolerance to insulin by affecting its expression in the thymus. Supporting this hypothesis, transgenic expression of proinsulin in the thymus of non‐obese diabetic (NOD) mice prevents insulitis and diabetes94.
Acknowledgements
The authors thank current and former colleagues of the First Department of Medicine, Wakayama Medical University, and University of Chicago, especially Donald F Steiner, Arthur H Rubenstein and Graeme I Bell. MN belongs to an endowed department (Novo‐Nordisk Japan). With the exception of that point, the authors have no conflict of interest.
References
- 1.Tager H, Given B, Baldwin B, et al. A structurally abnormal insulin causing human diabetes. Nature 1979; 281: 122–125 [DOI] [PubMed] [Google Scholar]
- 2.Given BD, Mako ME, Tager HS, et al. Diabetes due to secretion of an abnormal insulin. N Engl J Med 1980; 302: 129–135 [DOI] [PubMed] [Google Scholar]
- 3.Haneda M, Chan SJ, Kwok SCM, et al. Studies on mutant human insulin gene: identification and sequence analysis of a gene encoding Ser B24 insulin. Proc Natl Acad Sci USA 1983; 80: 6366–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shoelson S, Fickova M, Haneda M, et al. Identification of a mutant human insulin predicted to contain a serine‐for‐phenylalanine substitution. Proc Natl Acad Sci USA 1983; 80: 7390–7394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shoelson S, Haneda M, Blix P, et al. Three mutant insulin in man. Nature 1983; 302: 540–543 [DOI] [PubMed] [Google Scholar]
- 6.Nanjo K, Sanke T, Miyano M, et al. Diabetes due to secretion of a structurally abnormal insulin (insulin Wakayama). J Clin Invest 1986; 77: 514–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakura H, Iwamoto Y, Sakamoto Y, et al. Structurally abnormal insulin in a diabetic patient: patient characterization of the mutant insulin A3(val‐to‐leu) isolated from the pancreas. J Clin Invest 1986; 78: 1666–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nanjo K, Miyano M, Kondo M, et al. Insulin Wakayama: familial mutant insulin syndrome in Japan. Diabetologia 1987; 30: 87–92 [DOI] [PubMed] [Google Scholar]
- 9.Gabby KH, Bergenstal RM, Wilff J, et al. Familial hyperproinsulinemia: partial characterization of circulating proinsulin‐like material. Proc Natl Acad Sci USA 1979; 76: 2881–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robbins DC, Blix PM, Rubenstein AH, et al. A human proinsulin variant at arginine 65. Nature 1981; 291: 679–681 [DOI] [PubMed] [Google Scholar]
- 11.Robbins DC, Shoelson SE, Rubenstein AH, et al. Familial hyperproinsulinemia: two cohorts secreting indistinguishable type II intermediates of proinsulin conversion. J Clin Invest 1984; 73: 714–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gruppuso PA, Gordon P, Kahn CR, et al. Familial hyperproinsulinemia due to a proposed defects in conversion of proinsulin to insulin. N Engl J Med 1984; 311: 629–634 [DOI] [PubMed] [Google Scholar]
- 13.Shibasaki Y, Kawakami T, Kanazawa Y, et al. Posttranslational cleavage of proinsulin is blocked by a point mutation in familial hyperproinsulinemia. J Clin Invest 1985; 76: 378–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yano H, Kitano N, Morimoto M, et al. A novel point mutation in the human insulin gene giving rise to hyperproinsulinemia (proinsulin Kyoto). J Clin Invest 1992; 89: 1902–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oohashi H, Ohgawara H, Nanjo K, et al. Familial hyperproinsulinemia associated with NIDDM. Diabetes Care 1993; 16: 1340–1346 [DOI] [PubMed] [Google Scholar]
- 16.Roder ME, Vissing H, Nauck MA. Hyperproinsulinemia in a three‐generation Caucasian family due to mutant proinsulin (Arg65‐>His) not associated with impaired glucose tolerance: the contribution of mutant proinsulin to insulin bioactivity. J Clin Endocrinol Metab 1996; 81: 1634–1640 [DOI] [PubMed] [Google Scholar]
- 17.Warren‐Perry MG, Manley SE, Ostrega D, et al. A novel point mutation in the insulin gene giving rise to hyperproinsulinemia. J Clin Endocrinol Metab 1997; 82: 1629–1631 [DOI] [PubMed] [Google Scholar]
- 18.Yoshioka M, Kayo T, Ikeda T, et al. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early‐onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 1997; 46: 887–894 [DOI] [PubMed] [Google Scholar]
- 19.Kayo T, Koizumi A. Mapping of murine diabetogenic gene Mody on chromosome 7 at D7Mit258 and its involvement in pancreatic islet and b cell development during the perinatal period. J Clin Invest 1998; 101: 2112–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Takeuchi T, Tanaka S, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic b‐cell dysfunction in the Mody mouse. J Clin Invest 1999; 103: 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herbach N, Rathkolb B, Kemter E, et al. Dominant‐negative effects of a novel mutated Ins2 allele causes early‐onset diabetes and severe b‐cell loss in Munich Ins2C95S mutant mice. Diabetes 2007; 56: 1268–1276 [DOI] [PubMed] [Google Scholar]
- 22.Stoy J, Edghill EL, Flanagan SE, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci USA 2007; 104: 15040–15044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edghill EL, Flanagan SE, Patch A‐M, et al. Insulin mutation screening in 1044 patients with diabetes. Diabetes 2008; 57: 1034–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polak M, Decaume A, Cave H, et al. Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy. Diabetes 2008; 57: 1115–1119 [DOI] [PubMed] [Google Scholar]
- 25.Molven A, Ringdal M, Nordbo AM, et al. Mutations in the insulin gene can cause MODY and autoantibody‐negative type 1 diabetes. Diabetes 2008; 57: 1131–1135 [DOI] [PubMed] [Google Scholar]
- 26.Colombo C, Porzio O, Liu M, et al. Seven mutations in the human insulin gene linked to permanent neonatal/infancy‐onset diabetes mellitus. J Clin Invest 2008; 118: 2148–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonfanti R, Columbo C, Nocerino V, et al. Insulin gene mutations as cause of diabetes in children negative for five type 1 diabetes autoantibodies. Diabetes Care 2009; 32: 123–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rubio‐Cabezas O, Edghill EL, Argente J, et al. Testing for monogenic diabetes among children and adolescents with antibody‐negative clinically defined type 1 diabetes. Diabet Med 2009; 26: 1070–1074 [DOI] [PubMed] [Google Scholar]
- 29.Ahamed A, Unnikrishnan AG, Pendsey SS, et al. Permanent neonatal diabetes mellitus due to a C96Y heterozygous mutation in the insulin gene. A case report. J Pancreas 2008; 9: 715–718 [PubMed] [Google Scholar]
- 30.Meur G, Simon A, Harun N, et al. Insulin gene mutations resulting in early‐onset diabetes: marked difference in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes 2010; 59: 653–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boesgaard TW, Pruhova S, Andersson EA, et al. Further evidence that mutations in INS can be a rare cause of Maturity‐Onset Diabetes of the Young (MODY). BMC Med Genet 2010; 11: 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garin I, Edghill EL, Akerman I, et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci USA 2010; 107: 3105–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bell Gi, Pictet RL, Rutter WJ, et al. Sequence of the human insulin gene. Nature 1980; 284: 26–32 [DOI] [PubMed] [Google Scholar]
- 34.Lomedico P, Rosenthal N, Efstratiadis A, et al. The structure and evolution of the two non‐allelic rat preproinsulin genes. Cell 1979; 18: 545–558 [DOI] [PubMed] [Google Scholar]
- 35.Steiner DF, Oyer PE. The biosynthesis of insulin and probable precursor of insulin by a human islet cell adenoma. Proc Natl Acad Sci USA 1967; 57: 473–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steiner DF, Cunningham D, Spigelman L, et al. Insulin biosynthesis: evidence for a precursor. Science 1967; 157: 697–700 [DOI] [PubMed] [Google Scholar]
- 37.Steiner DF, Bell GI, Rubenstein AH, et al. Chemistry and biosynthesis of the islet hormones: insulin, islet amyloid polypeptide (amylin), glucagon, somatostatin, and pancreatic polypeptide In: DeGroot LG (ed.). Endocrinology, 14th edn vol. 1. WB Sannders, Philadelphia, 2001; 667–696 [Google Scholar]
- 38.Rhodes CJ, Shoelson S, Halban PA. Insulin biosynthesis, processing, and chemistry In: Kahn CR (ed.). Joslin’s Diabetes Mellitus, 14th edn Lippincott Williams & Wilkins, Philadelphia. 2005:65–82 [Google Scholar]
- 39.Steiner DF, Park S‐Y, Stoy J, et al. A brief perspective on insulin production. Diabetes Obes Metab 2009; 11(Suppl 4): 189–196 [DOI] [PubMed] [Google Scholar]
- 40.Weiss MA. Proinsulin and the genetics of diabetes mellitus. J Biol Chem 2009; 284: 19159–19163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steiner DF, Tager HS, Chan SJ, et al. Lessons learned from molecular biology of insulin‐gene mutations. Diabetes Care 1990; 13: 600–609 [DOI] [PubMed] [Google Scholar]
- 42.Nanjo K, Kondo M, Sanke T, et al. Abnormal insulinemia. Diabetes Res Clin Pract 1994; 24(Suppl.): S135–S141 [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto R, Iwamoto Y, Sakura H, et al. Reduced urinary insulin clearance in patients with abnormal insulinemia. Diabetes 1987; 36: 602–606 [DOI] [PubMed] [Google Scholar]
- 44.Hanabusa T, Oki C, Nakano Y, et al. The renal metabolism of insulin: urinary excretion in patients with mutant insulin syndrome (Insulin Wakayama). Metabolism 2001; 50: 863–867 [DOI] [PubMed] [Google Scholar]
- 45.Carroll RJ, Hammer RE, Chan SJ, et al. A mutant human proinsulin is secreted from islets of Langerhans in increased amounts via an unregulated pathway. Proc Natl Acd Sci USA 1988; 85: 8943–8947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gross DJ, Halban PA, Kahn RC, et al. Partial diversion of a mutant proinsulin (B10 aspartic acid) from the regulated to the constitutive secretory pathways in transfected AtT‐20 cells. Proc Natl Acad Sci USA 1989; 86: 4107–4111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwartz GP, Burke SGT, Katsoyannis PG. A superactive insulin:[B10‐aspartic acid] insulin (human). Proc Natl Acad Sci USA 1987; 84: 6408–6411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leroux L, Desbois P, Lamotte L, et al. Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes 2001; 50(Suppl 1): S150–S153 [DOI] [PubMed] [Google Scholar]
- 49.Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress‐mediated diabetes. J Clin Invest 2002; 109: 525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Izumi T, Yokota‐Hashimoto H, Zhao S, et al. Dominant negative pathogenesis by mutant proinsulin in the Akita diabetic mouse. Diabetes 2003; 52: 409–416 [DOI] [PubMed] [Google Scholar]
- 51.Allen JR, Nguyen LX, Sargent KEG, et al. High ER stress in b‐cells stimulates intracellular degradation of misfolded insulin. Biochem Biophys Res Commun 2004; 324: 166–170 [DOI] [PubMed] [Google Scholar]
- 52.Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress‐mediated apoptosis in pancreatic b‐cells. Apoptosis 2002; 7: 335–342 [DOI] [PubMed] [Google Scholar]
- 53.Araki E, Oyadomari S, Mori M. Impact of endoplasmic reticulum stress pathway on pancreatic b‐cells and diabetes mellitus. Exp Biol Med 2003; 228: 1213–1217 [DOI] [PubMed] [Google Scholar]
- 54.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 2008; 29: 42–61 [DOI] [PubMed] [Google Scholar]
- 55.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with b‐cell failure and diabetes. Endocr Rev 2008; 29: 317–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fonseca SG, Burcin M, Gromada J, et al. Endoplasmic reticulum stress in b‐cells and development of diabetes. Curr Opin Pharmacol 2009; 9: 763–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thomas SE, Dalton LE, Daly M‐L, et al. Diabetes as a disease of endoplasmic reticulum stress. Diabetes Metab Res Rev 2010; 26: 611–621 [DOI] [PubMed] [Google Scholar]
- 58.Aguilar‐Bryan L, Bryan J. Neonatal diabetes. Endocr Rev 2008; 29: 265–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Greeley SAW, Tucker SE, Naylor RN, et al. Neonatal diabetes mellitus: A model for personalized medicine. Trends Endocrinol Metab 2010; 21: 464–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Temple IK, Shield JP. Transient neonatal diabetes, a disorder of imprinting. J Med Genet 2002; 39: 872–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Glaser G. Insulin mutations in diabetes. The clinical spectrum. Diabetes 2008; 57: 799–800 [DOI] [PubMed] [Google Scholar]
- 62.Yorifuji T, Kurokawa K, Mamada M, et al. Neonatal diabetes and neonatal polycystic, dysplastic kidneys: phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor‐1b gene due to germline mozaicism. J Clin Endocrinol Metab 2004; 89: 2905–2908 [DOI] [PubMed] [Google Scholar]
- 63.Edghill EL, Bingham C, Slingerland AS, et al. Hepatocyte nuclear factor‐1b mutations cause neonatal diabetes and intrauterine growth retardation: support for a critical role of HNF‐1b in human pancreatic development. Diabet Med 2006; 23: 1301–1306 [DOI] [PubMed] [Google Scholar]
- 64.Njolstad PR, Sovik O, Cuesta‐Munoz A, et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med 2001; 344: 1588–1592 [DOI] [PubMed] [Google Scholar]
- 65.Njolstad PR, Sagen JV, Bjorkhaug L, et al. Permanent neonatal diabetes caused by glucokinase deficiency: in born error of the glucose‐insulin signaling pathway. Diabetes 2003; 52: 2854–2860 [DOI] [PubMed] [Google Scholar]
- 66.Porter JR, Shaw NJ, Barrett TG, et al. Permanent neonatal diabetes in an Asian infant. J Pediatr 2005; 146: 131–133 [DOI] [PubMed] [Google Scholar]
- 67.Stoffers DA, Zinkin NT, Stanojevic V, et al. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet 1997; 15: 106–110 [DOI] [PubMed] [Google Scholar]
- 68.Schwitzgebel VM, Mamin A, Brun T, et al. Agenesis of human pancreas due to decreased half‐life of insulin promoter factor‐1. J Clin Endocrinol Metab 2003; 88: 4398–4406 [DOI] [PubMed] [Google Scholar]
- 69.Chatila TA, Blaeser F, Ho N, et al. JM2, encoding a fork head‐related protein, is mutated in X‐linked autoimmunity‐allergic disregulation syndrome. J Clin Invest 2000; 106: R75–R81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wildin RS, Ramsdell F, Peake J, et al. X‐linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 2001; 27: 18–20 [DOI] [PubMed] [Google Scholar]
- 71.Delepine M, Nicolino M, Barrett T, et al. EIF2AK3, encoding translation initiation factor 2‐a kinase 3, is mutated in patients with Wolcott‐Rallison syndrome. Nat Genet 2000; 25: 406–409 [DOI] [PubMed] [Google Scholar]
- 72.Sellick GS, Barker KT, Stolte‐Dijkstra I, et al. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet 2004; 36: 1301–1305 [DOI] [PubMed] [Google Scholar]
- 73.Smith SB, Qu HQ, Taleb N, et al. Rfx6 directs islet formation and insulin production in mice and humans. Nature 2010; 463: 775–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Senee V, Chelala C, Duchatelet S, et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet 2006; 38: 682–687 [DOI] [PubMed] [Google Scholar]
- 75.Gupta S, McGrath B, Cavener DR. PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes 2010; 59: 1937–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cavener DR, Gupta S, McGrath BC. PERK in beta cell biology and insulin biogenesis. Trends Endocrinol Metab 2010; 21: 714–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stoy J, Steiner DF, Park S‐Y, et al. Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev Endocr Metab Disord 2010; 11: 205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu M, Hodish I, Haataja L, et al. Proinsulin misfolding and diabetes: mutant INS gene‐induced diabetes of youth. Trends Endocrinol Metab 2010; 21: 652–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rajan S, Eames SC, Park S‐Y, et al. In vitro processing and secretion of mutant insulin proteins that cause permanent neonatal diabetes. Am J Physiol Endocrinol Metab 2010; 298: E403–E410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Park S‐Y, Ye H, Steiner DF, et al. Mutant proinsulins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted. Biochem Biophys Res Commun 2010; 391: 1449–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hartley T, Siva M, Lai E, et al. Endoplasmic reticulum stress response in an INS‐1 pancreatic b‐cell line with inducible expression of a folding‐deficient proinsulin. BMC Cell Biol 2010; 11: 59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hodish I, Liu M, Rajpal G, et al. Misfolded proinsulin affects bystander proinsulin in neonatal diabetes. J Biol Chem 2010; 285: 685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu M, Haataja L, Wright J, et al. Mutant INS‐Gene induced diabetes of youth:proinsulin cysteine residues impose dominant‐negative inhibition on wild‐type proinsulin transport. PLoS ONE 2010; 5: e13333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bell GI, Selby MJ, Rutter WJ. The highly polymorphic region near the insulin gene is composed of simple tandemly repeating sequences. Nature 1982; 295: 31–35 [DOI] [PubMed] [Google Scholar]
- 85.Bell GI, Horita S, Karam JH. A polymorphic locus near the human insulin gene is associated with insulin‐dependent diabetes mellitus. Diabetes 1984; 33: 176–183 [DOI] [PubMed] [Google Scholar]
- 86.Julier C, Hyer RN, Davies J, et al. Insulin‐IGF2 region on chromosome 11p encodes a gene impricated in HLA‐DR4‐dependent diabetes susceptibility. Nature 1991; 354: 155–159 [DOI] [PubMed] [Google Scholar]
- 87.Lucassen AM, Julier C, Beressi JP, et al. Susceptibility to insulin dependent diabetes mellitus maps to a 4.1kbsegement of DNA spanning the insulin gene and associated with VNTR. Nat Genet 1993; 4: 305–310 [DOI] [PubMed] [Google Scholar]
- 88.Bennett ST, Luccassen AM, Gough SC, et al. Susceptibility to type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat Genet 1995; 9: 284–292 [DOI] [PubMed] [Google Scholar]
- 89.Awata T, Kawasaki E, Ikegami H, et al. Insulin gene/IDDM2 locus in Japanese type 1 diabetes: contribution of class I alleles and influence of class I subdivision in susceptibility to type 1 diabetes. J Clin Endocrinol Metab 2007; 92: 1791–1795 [DOI] [PubMed] [Google Scholar]
- 90.Heath VL, Moore NC, Parnell SM, et al. Intrathymic expression of genes involved in organ specific autoimmune disease. J Autoimmun 1998; 11: 309–318 [DOI] [PubMed] [Google Scholar]
- 91.Derbinski J, Schulte A, Kyewski B, et al. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol 2001; 2: 1031–1039 [DOI] [PubMed] [Google Scholar]
- 92.Vafiadis P, Bennett ST, Todd JA, et al. Insulin expression in the thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet 1997; 15: 289–292 [DOI] [PubMed] [Google Scholar]
- 93.Pugliese A, Zeller M, Fernandez AJ, et al. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR‐IDDM2 susceptibility locus for type 1 diabetes. Nat Genet 1997; 15: 293–297 [DOI] [PubMed] [Google Scholar]
- 94.French MB, Allison J, Cram AS, et al. Transgenic expression of mouse proinsulin II prevents diabetes in nonobese diabetic mice. Diabetes 1997; 46: 34–39 [DOI] [PubMed] [Google Scholar]