

Abstract
Type 2 diabetic patients are insulin resistant as a result of obesity and a sedentary lifestyle. Nevertheless, it has been known for the past five decades that insulin response to nutrients is markedly diminished in type 2 diabetes. There is now a consensus that impaired glucose regulation cannot develop without insulin deficiency. First‐phase insulin response to glucose is lost very early in the development of type 2 diabetes. Several prospective studies have shown that impaired insulin response to glucose is a predictor of future impaired glucose tolerance (IGT) and type 2 diabetes. Recently discovered type 2 diabetes‐risk gene variants influence β‐cell function, and might represent the molecular basis for the low insulin secretion that predicts future type 2 diabetes. We believe type 2 diabetes develops on the basis of normal but ‘weak’β‐cells unable to cope with excessive functional demands imposed by overnutrition and insulin resistance. Several laboratories have shown a reduction in β‐cell mass in type 2 diabetes and IGT, whereas others have found modest reductions and most importantly, a large overlap between β‐cell masses of diabetic and normoglycemic subjects. Therefore, at least initially, the β‐cell dysfunction of type 2 diabetes seems more functional than structural. However, type 2 diabetes is a progressive disorder, and animal models of diabetes show β‐cell apoptosis with prolonged hyperglycemia/hyperlipemia (glucolipotoxicity). β‐Cells exposed in vitro to glucolipotoxic conditions show endoplasmic reticulum (ER) and oxidative stress. ER stress mechanisms might participate in the adaptation of β‐cells to hyperglycemia, unless excessive. β‐Cells are not deficient in anti‐oxidant defense, thioredoxin playing a major role. Its inhibitor, thioredoxin‐interacting protein (TXNIP), might be important in leading to β‐cell apoptosis and type 2 diabetes. These topics are intensively investigated and might lead to novel therapeutic approaches. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2010.00094.x, 2011)
Keywords: β‐Cell failure, Insulin secretion, Insulin resistance
Introduction
To present day readers of this journal, it might seem strange that until the 1970s1 it was not known that type 1 diabetes is an autoimmune disease entirely distinct from type 2 diabetes. Diabetes specialists who have reached a certain age today, including the senior author of this review, were trained to view the differences between various types of diabetes as a matter of degree, not of kind; some patients required insulin treatment to survive, whereas others managed their metabolic state with dietary means or pharmacological agents. As insulin became more widely used, it also appeared that in many patients whom today we characterize as typical type 2 diabetic patients, achieving metabolic control with insulin treatment was difficult if not impossible. Thus, half a century before the insulin receptor and its signaling pathways were identified, the British clinician HP Himsworth2,3 wrote ‘…I would suggest the possibility of the existence of a type of diabetes due not to diminished secretion of insulin by the pancreas, but to a greater or less impairment of the organism’s susceptibility to insulin’. In other words, the idea that diabetes might be the result of insulin resistance was aired already in the 1930s. Indeed, the presence of obesity in the vast majority of patients with type 2 diabetes makes it a reasonable assumption that insulin resistance must exist in this disorder.
Strikingly different is the approach that has dominated the last decades of the 20th century, viewing insulin resistance as the main, often sole, etiological factor in type 2 diabetes, negating any role of deranged insulin secretion. Summarizing this view, the Journal of Clinical Investigation as recently as 2000 published a ‘Perspective’ series entitled ‘On diabetes: insulin resistance’4. It is difficult to understand how insulin deficiency could so widely be ignored; already in the early 1960s it was shown by several investigators that the insulin responses to glucose challenge is markedly reduced in type 2 diabetes, including in normoglycemic subjects with glucose intolerance only (IGT)5–9. Fortunately, over the years, in the face of dogmatic‐monolithic positions, balanced views have also been presented10–12 that point to the fact that the biology of type 2 diabetes is not simple; pure β‐cell deficiency or exclusive insulin resistance are rare events, because in reality, insulin secretion and insulin action are interconnected, as would be expected in any feedback regulatory loop.
Also, clinical evidence points to the fact that in many type 2 diabetic patients the metabolic state cannot be ascribed to insuperable insulin resistance. Indeed, although conventional insulin therapy in type 2 diabetes often fails or gives suboptimal results, intensified insulin treatment [multiple dose administration or continuous subcutaneous infusion of insulin (CSII)] is able to near‐normalize blood glucose in many patients. Thus, in pilot studies in 23 type 2 diabetic patients, we could achieve fasting and postprandial normoglycemia with a mean daily CSII insulin dose of 0.55 U/kg bodyweight13,14; this is shown in Figure 1. Recently, Retnakaran et al.15 achieved similar results with a multiple injection protocol administering a mean daily insulin dose of 0.65 U/kg. Finally, in contrast to the aforementioned studies that were carried out mainly in Caucasian patients, Weng et al.16 achieved near‐normoglycemia with CSII in a large group of Chinese type 2 diabetic patients with a daily insulin dose of 0.68 U/kg. The interest of these studies is not only that excellent metabolic control could be achieved in type 2 diabetic patients with exogenous insulin, but that the daily insulin requirement was not substantially different from that used as replacement therapy in insulin‐deficient type 1 diabetic patients. We certainly recognize the important role of insulin resistance in the pathophysiology of type 2 diabetes, but conclude nevertheless that type 2 diabetes is first of all a disorder of insulin deficit; the input of insulin resistance to its pathogenesis increases with the severity of obesity, acting as a magnifier of insulin deficiency.
Figure 1.
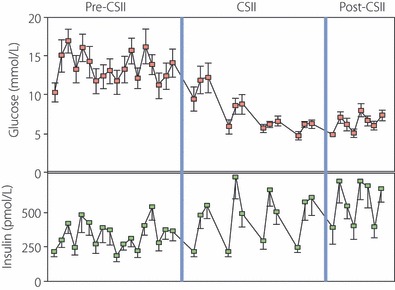
Induction of near‐normoglycemia in type 2 diabetes patients by continuous subcutaneous infusion of insulin (CSII). A total of 12 patients were treated with CSII for 2 weeks; towards the end of the period, daily blood glucose excursions were markedly reduced and approached normal values. The data shown are part of a larger study, presented by Ilkova et al.14
Plasma insulin in type 2 diabetes
It is important to remember that both insulin response to glucose and peripheral sensitivity to insulin show a remarkably wide range of variation in non‐diabetic lean subjects as well as obese subjects, with a continuous distribution of the parameters; that is, without evidence of population segregation17–20. This is shown in Figure 2 with data selected from two studies as examples. In obese subjects, although mean insulin sensitivity is reduced and insulin response augmented, the wide variation of these parameters leads to a major overlap with the levels of lean subjects. Although less marked, the variability of insulin sensitivity is also important in type 2 diabetic patients (Figure 2b). In substantial numbers of subjects with either markedly low insulin response or low sensitivity to insulin, normal glucose tolerance is retained; furthermore, clinical experience shows that more than two out of three obese subjects never develop impaired glucose tolerance (IGT), despite insulin resistance. In type 2 diabetic patients, except in its very severe forms, fasting and postprandial plasma insulin levels are normal or higher than normal. This observation provides the rationale for insulin resistance in diabetes; it is, however, a static view of a highly dynamic regulatory system, confusing cause and effect: what degree of hyperinsulinemia is adequate for a given degree of hyperglycemia? An example of the fallacy of the argument is provided by our study in 15 mildly obese type 2 diabetic patients treated with a sulfonylurea for 6 months in whom, with the normalization of blood glucose, the initially high fasting plasma insulin levels fell to the normal range, despite the use of the β‐cell stimulating sulfonylurea13. Thus, fasting insulin is also under the control of blood glucose. Obviously, similar arguments might (and should) be applied to postprandial insulin levels in type 2 diabetes. For example, the bell‐shaped insulin curve used to describe changes in β‐cell function during the transition from normal to IGT and type 2 diabetes is an artefact as a result of the use of 120‐min plasma insulin values in the oral glucose tolerance test (OGTT); the higher glucose levels in patients with IGT amplify the secretion of insulin, resulting in a typical late insulin peak. When earlier (e.g. 30‐min) time‐points are chosen, the insulin response to OGTT shows a continuous fall from normal over IGT to type 2 diabetes21–23. We wish to reiterate with emphasis that, provided plasma insulin data are interpreted with full reference to the physiology of regulated insulin secretion, β‐cell responsiveness to glucose is lower than normal in IGT, and more so in type 2 diabetes. The data presented in Figure 2b show this point; the glucose responsiveness of β‐cells assessed by computer simulation was dramatically lower in mild type 2 diabetic patients than in control subjects.
Figure 2.
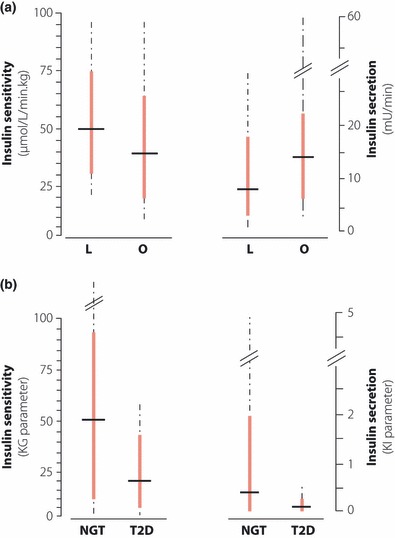
The wide variability of insulin sensitivity and insulin secretion. (a) Whole body insulin‐mediated glucose uptake (left panel) and basal post‐hepatic insulin delivery rate (right panel) were assessed in 608 lean (L) and 538 obese (O) non‐diabetic subjects with euglycemic hyperinsulinemic clamps by Ferrannini et al.20; the data shown here were recalculated from the original publication. The thick vertical bars show the results between the 5th and 95th percentile, whereas the dotted lines show the data range; the short horizontal bars denote the median. (b) Whole body glucose clearance stimulated by endogenous insulin (insulin sensitivity, KG, left panel) and sensitivity of insulin secretion to glucose (insulin secretion, KI, right panel) were assessed by modeling of data generated with a glucose infusion test in 226 lean normal glucose tolerant controls (NGT) and 25 lean type 2 diabetes patients (T2D)17. The results of Efendic et al.17 have been replotted here in a similar manner to those of Figure 2a. Note the markedly skewed distribution of the values; the range of insulin sensitivities in NGT subjects exceeded the limits of the figure (166.0 KG units). Note also the markedly lower insulin response of the type 2 diabetes patients, and their more modestly reduced insulin sensitivity.
When glucose tolerance is impaired, the earliest modifications of the insulin response to glucose to be detected concern the so‐called first‐phase insulin response, which is rapidly reduced and then lost, and the disruption of the oscillatory character of insulin release5,6,8,24,25. Assessment of insulin oscillations is technically demanding and therefore seldom utilized in larger clinical studies. In contrast, the early insulin response to glucose can be measured during oral or i.v. glucose tolerance tests, whereas glucose clamps allow detailed definition of the insulin response kinetics. The first‐phase response is markedly reduced in subjects with IGT, and further diminishes with the advent of fasting hyperglycemia. The later or second‐phase insulin response to glucose is retained in early or moderately advanced type 2 diabetes, but as the disease progresses this phase of insulin secretion also collapses (Figure 3). Some studies suggest that type 2 diabetic patients might also present changes in incretin secretion and action, with consequences for the remaining β‐cell function. For extensive reading, please see a recent review in this journal26.
Figure 3.
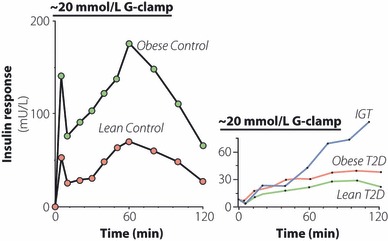
Insulin response to glucose infusion. Typical examples of the insulin response in lean and obese normal glucose tolerant (left panel) and type 2 diabetes subjects at different stages of the disease (right panel). Note the loss of the first‐phase insulin response already at the stage of impaired glucose tolerance (IGT).
Low first‐phase insulin response is also found in a proportion of subjects with normal glucose tolerance5,6 (Figure 2b). Several studies over past years have shown that a low insulin response is a predictor of future glucose intolerance and type 2 diabetes, in lean as well as obese subjects belonging to various ethnic groups27–29. To give an example, we followed a large group of lean and physically active Swedish subjects with normal glucose tolerance for a mean period of 25 years; the initially‐measured first‐phase insulin response corrected for insulin sensitivity (disposition index) was significantly correlated to later glucose tolerance, low values predicting IGT and type 2 diabetes27.
Genetic causes of reduced insulin secretion
Extensive studies over past decades in family members of diabetic patients and control subjects, including monozygotic twin pairs, have shown that many aspects of the plasma insulin response to glucose administration in humans are under strong genetic control30–33. However, it is only recently that data have emerged that allow some insight into the possible cellular mechanisms responsible for the decrease of β‐cell function in subjects at risk of developing diabetes. Indeed, numerous whole genome association studies carried out over the past decade have identified allelic variants of several genes that collectively participate in the risk of type 2 diabetes development. The majority of these genes are involved in β‐cell development, function and survival34. As the number of risk alleles that a subject carries increases, several aspects of β‐cell function deteriorate; most pertinently, the insulin response to oral or i.v. glucose decreases in proportion to the number of risk alleles35,36. By which cellular mechanisms these risk alleles impair insulin secretion is not known. However, recent data suggests that, at least regarding the highest‐risk gene transcription factor 7‐like 2 (TCF7L2), distal steps in exocytosis, including insulin granule connection with voltage‐gated calcium channels in the β‐cell, might be involved, thus reducing the efficiency of the insulin exocytotic machinery, although other mechanisms have also been proposed37–40. It can be expected that within a short time the molecular mechanisms of the low insulin response to glucose, which is a strong risk factor for type 2 diabetes, will be fully clarified at the molecular level, as has been done for the various forms of monogenic diabetes41–43.
Progressive deterioration of β‐cell function in type 2 diabetes
It has become axiomatic that type 2 diabetes is a progressive disease; it is indeed a common clinical experience that with prolonged duration, the severity of diabetes increases and requires augmenting numbers and doses of anti‐diabetic drugs. This experience has been confirmed and extended by the United Kingdom Prospective Diabetes Study (UKPDS) investigators; whatever the treatment modality chosen, the level of hemoglobin A1c (HbA1c) increases with time44. A very important question is whether type 2 diabetes is an inherently progressive disorder as a result of the molecular nature of its pathogenesis, or whether progression is secondary to the metabolic state including hyperglycemia. It is indeed nearly impossible to obtain normoglycemia throughout the day over the life of a diabetic patient, whichever treatment modality is chosen. Notwithstanding its molecular cause, the progressive deterioration of metabolism in type 2 diabetes is accompanied by a progressive decline in β‐cell function44. This decline might be as a result of either a reduction in the function of individual β‐cells or a reduction in the number of β‐cells, that is, β‐cell mass (or both).
Is β‐Cell Mass Reduced in Type 2 Diabetes?
Studies with classical pathology methods carried out more than 30–40 years ago established the existence of structural abnormalities in islets and some reduction in β‐cell mass in diabetic patients45–47. This concept has recently been revived and is strongly advocated by Butler et al.48, whose studies suggest that β‐cell mass is already markedly reduced at the stage of IGT, a further deficit being apparent in overt diabetes, even when treated by diet alone. Contrasting with these dramatic data showing 50–60% reduction in β‐cell mass independent of the severity of diabetes, other studies in Europe and Asia have found considerably less reduction in β‐cell mass in type 2 diabetes49–52. Of special interest is the study by Rahier et al.52, where absoluteβ‐cell mass was estimated (Butler et al.48 measured β‐cell area, which gives only an approximation of β‐cell mass). Most importantly, Rahier et al.52 clearly showed the extraordinarily wide range of β‐cell masses that exist both in the diabetic and non‐diabetic populations, with a major overlap between the hyperglycemic and normoglycemic subjects (Figure 4). These observations make it difficult to ascribe a determining role to reduced β‐cell mass in the etiology of hyperglycemia in type 2 diabetes. Nevertheless, it might be questioned why the high blood glucose of the patients did not stimulate β‐cell mass to increase as a compensatory mechanism. In a physiological situation of insulin resistance, pregnancy, β‐cell mass is indeed augmented by approximately 40%53. To our knowledge, no study has reported increased β‐cell mass in type 2 diabetes.
Figure 4.
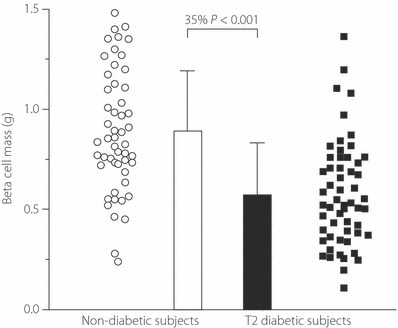
β‐Cell mass in type 2 diabetes. β‐Cell mass in absolute terms (mg/pancreas) was measured in autopsy material from 52 non‐diabetic control cases and 57 typical type 2 diabetes patients. Note that despite a statistically significant reduction of mean β‐cell mass, the very large variation of the results in both groups leads to a major overlap between the diabetic and non‐diabetic pancreases. Minimally modified from Rahier et al.52, with permission.
There exist several problems that must be overcome when ascribing a role to β‐cell mass changes in the development of type 2 diabetes. In addition to the technical problems relating to the quality of the pancreas obtained post‐mortem, the information generated is only cross‐sectional and static, and thus does not reflect the dynamics of β‐cell turnover during the development of type 2 diabetes. Presently, intensive effort is being made to develop non‐invasive β‐cell imaging techniques; however, these are as yet in their infancy54. Therefore, to gain insight into the β‐cell mass dynamics in type 2 diabetes, animal models of diabetes are the only tools presently available that permit detailed longitudinal observations on the pancreas. We studied this in an animal model of nutrition‐dependent type 2 diabetes, the gerbil Psammomys obesus. These animals have an inborn insulin resistance, but retain normal glucose tolerance under caloric restriction; when given a diet with approximately 40% higher calories and low fibre content they rapidly become hyperglycemic55. Figure 5 shows that as the animals develop hyperglycemia, they rapidly lose their pancreatic insulin stores because the β‐cells are forced to discharge all their insulin granules in the face of the unrelenting hyperglycemic stimulation. Nevertheless, β‐cell mass remains normal for a considerable period; it is even slightly increased as a result of increased β‐cell proliferation induced by the high glucose levels55. β‐Cell mass collapses only after prolonged diabetes duration, with severe worsening of hyperglycemia (so‐called end‐stage diabetes). Thus, in this model, possibly in analogy with European and Asian type 2 diabetic patients, physiologically significant β‐cell mass reductions occur only in long‐standing and advanced type 2 diabetes. In earlier stages, the β‐cell deficiency seems to be more of a functional nature. Therefore, the term ‘functional β‐cell mass’ should be preferentially used to denote the globally insufficient insulin delivery state in type 2 diabetes, until we gain access to in vivo imaging techniques with the ability to determine β‐cell mass in situ.
Figure 5.
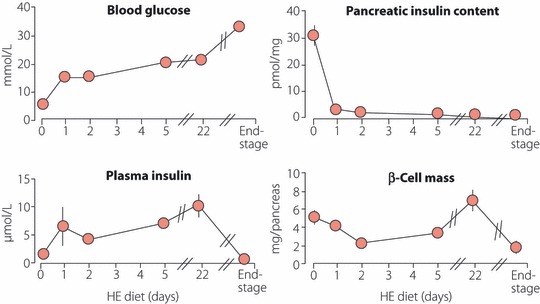
Evolution of metabolic parameters in diabetic Psammomys obesus. Normoglycemic animals were switched from a low‐energy diet to a high‐energy (HE) diet at day 0. After several weeks on a HE diet, animals developed severe diabetes (end‐stage). Vertical bars denote SEM; when not shown, SEM was smaller than the symbol for the mean value.
The β‐cell in chronic hyperglycemia
The close tie between hyperglycemia and β‐cell dysfunction raises the possibility that elevated glucose itself is deleterious to the β‐cell (so‐called glucotoxicity). Near‐normalization of blood glucose by short‐term insulin treatment of newly diagnosed diabetic patients or of patients with secondary failure to oral hypoglycemic agents improved β‐cell function14, and a recent multicenter study showed that intensive insulin treatment in newly‐diagnosed type 2 diabetic patients enhanced β‐cell function, resulting in prolonged diabetes remission in approximately 50% of the patients16. These findings support the hypothesis that glucotoxicity plays an important role in maintaining the hyperglycemic state in type 2 diabetes. In Psammomys obesus, hyperglycemia is associated with a marked depletion of pancreatic insulin content, increased proinsulin/insulin ratio and β‐cell apoptosis55. Strikingly, normalization of blood glucose using the glucosuric drug phlorizin or by changing the diabetogenic diet to a low‐energy diet rapidly reversed these β‐cell abnormalities55. This further emphasizes the importance of glucotoxicity for the β‐cell dysfunction of diabetes.
The mechanisms underlying glucotoxicity are not clear. Gene expression analysis in islet preparations of patients with type 2 diabetes identified multiple changes in the expression of genes known to be important for β‐cell function, including a major decrease in β‐cell transcription factors, the insulin receptor and its downstream effectors, as well as in several genes involved in glucose metabolism56. However, a recent study on β‐cell‐enriched tissue obtained by laser capture microdissection from type 2 diabetic patients failed to confirm these findings57. A limitation of these studies is the considerable heterogeneity in β‐cell purity, and differences in degree of ischemia and stress between tissue preparations. Furthermore, the level of hyperglycemia and the response to metabolic stress can vary substantially between pancreas donors. Several mechanisms have been implicated in glucotoxicity, including inflammation, endoplasmic reticulum (ER) stress and oxidative stress58,59. Intricate interactions exist between the various stress pathways, which culminate in an impairment of β‐cell function and survival. In humans, the β‐cell turnover rate is slow60; however, over the years, small but persistent β‐cell loss might eventually decrease the β‐cell mass48. Cytokines, such as interleukin‐1β (IL‐1β), are probably involved in hyperglycemia‐induced β‐cell dysfunction, as shown in animal models and diabetic patients61,62. Notably, treatment of uncontrolled type 2 diabetic patients with IL‐1β receptor antagonist improved insulin secretion and ameliorated diabetes62. Thus, protection against inflammatory stress might become a therapeutic strategy in diabetes.
ER Stress and β‐Cell Glucotoxicity
The role of ER stress in glucose‐induced β‐cell dysfunction is controversial. Accumulation of misfolded proteins in the ER stimulates a signaling pathway called the unfolded protein response (UPR), which protects the cell by translational attenuation, induction of chaperone synthesis and ER‐associated protein degradation (ERAD). Severe β‐cell ER stress leading to strong activation of the UPR might cause apoptosis, which is mediated by stress kinases and transcription factors, such as Jun N‐terminal kinase (JNK) and C/Ebp‐homologous protein (CHOP). Importantly, disruption of the CHOP gene was shown to inhibit β‐cell apoptosis, expand β‐cell mass and improve glycemic control in mouse models of diabetes, suggesting that the UPR plays an important role in mediating the β‐cell dysfunction of diabetes63. We found that glucose moderately stimulates ER stress; however, high glucose levels synergize with fatty acids to stimulate UPR and JNK with β‐cell apoptosis as a consequence64. Therefore, hyperglycemia‐induced ER stress seems to become apparent mainly under conditions of glucolipotoxicity.
Increased expression of ER stress markers was observed in islets of patients with type 2 diabetes; however, the number of β‐cells expressing stress markers was small65. Furthermore, most studies showing that ER stress is involved in β‐cell dysfunction and apoptosis were carried out in vitro. Intriguingly, transplantation to mice of β‐cell‐enriched tissue derived from pancreases of non‐diabetic subjects, conditions that exposed the human β‐cells to mild to moderate hyperglycemia, increased the expression of UPR genes without stimulating pro‐apoptotic genes66. Thus, UPR stimulation by hyperglycemia might be adaptive, rather than deleterious. Altogether, the conclusion that ER stress plays an important role in the β‐cell dysfunction of type 2 diabetes should be drawn with caution.
Oxidative Stress and β‐Cell Glucotoxicity
Chronic exposure to high glucose is expected to increase the metabolic flux in mitochondria and through the hexosamine pathway, leading to excess production of reactive oxygen species (ROS). The level of oxidative stress exerted on the β‐cell depends on its capacity to scavenge ROS and other free radicals generated under conditions of glucotoxicity and glucolipotoxicity. It is widely believed that β‐cells are particularly vulnerable to oxidative stress as a result of low expression of the main anti‐oxidant enzymes, superoxide dismutase, catalase and glutathione peroxidase67,68. However, a recent report describing an effective adaptive response of diabetic GK rat islets to oxidative stress, with increased expression of anti‐oxidants and high glutathione content, challenges this notion69. Furthermore, in vitro studies showed that ROS production in β‐cells is maximal at low glucose concentrations when nicotinamide adenine dinucleotide phosphate (reduced; NADPH) is low, whereas it is markedly diminished by high glucose, which increases NADPH production70. Thus, it is possible that NADPH‐dependent anti‐oxidant systems operate well in β‐cells and do protect the cells from oxidative stress and apoptosis under conditions of hyperglycemia.
Glutaredoxin and thioredoxin are the main redox acceptor proteins for NADPH electrons71. Located in distinct subcellular domains, they are highly expressed in β‐cells72; their abundance argues against the dogma that β‐cell anti‐oxidant capacity is generally poor. Thioredoxin is emerging as an important anti‐oxidant in the β‐cell defence against oxidative stress. Thioredoxin partners with thioredoxin reductase and thioredoxin peroxidase to reduce oxidized proteins and scavenge free radicals73. In addition to its anti‐oxidative function, thioredoxin inhibits apoptosis through interaction with signaling molecules and transcription factors, such as redox effector protein‐1 (Ref‐1), activator protein 1 (AP‐1), nuclear factor κB (NF‐κB) and apoptosis signal regulating kinase‐1 (ASK1)73.
Thioredoxin‐interacting protein (TXNIP), also known as vitamin D3 upregulated protein‐1 (VDUP‐1) and thioredoxin binding protein‐2 (TBP‐2), is an endogenous inhibitor of thioredoxin, which, by binding to its redox‐active cysteine residues, inhibits the anti‐oxidative function of thioredoxin74. Under conditions of oxidative stress, TXNIP shuttles from the nucleus to the mitochondria, binds and oxidizes thioredoxin, thereby reducing its binding to ASK‐175. This in turn activates ASK‐1 with subsequent induction of mitochondrial cell death. TXNIP expression is robustly induced by glucose in islets and β‐cell lines76–79. Strikingly, islets derived from TXNIP‐deficient mice are fully protected from glucose‐induced β‐cell apoptosis80. Such mice have increased β‐cell mass and are resistant to streptozotocin‐induced β‐cell apoptosis and diabetes81. Furthermore, crossing the HcB‐19 TXNIP‐mutant mice with ob/ob mice protected against diabetes and β‐cell apoptosis81. Similarly, we have shown that partial knockdown of TXNIP in insulinoma‐1E (INS‐1E) cells was sufficient to prevent β‐cell apoptosis in response to high glucose82. This suggests that TXNIP is an important mediator of β‐cell glucotoxicity.
Interestingly, TXNIP was shown to participate in the activation of nucleotide‐binding oligomerization domain (NOD)‐like receptor 3 (NLRP3) inflammasomes, leading to secretion of IL‐1β83. Thus, TXNIP might provide a molecular link between hyperglycemia, oxidative stress and inflammation, and serve as an important mediator of β‐cell damage in type 2 diabetes.
Concluding Remarks
The considerable clinical heterogeneity of type 2 diabetes, and the large number of genes that seem to be involved in this disease (more than 2534), clearly show that the pathogenesis of type 2 diabetes is not simple. That β‐cell defects play a preponderant role and that clinically overt diabetes is not possible in the absence of impaired insulin production seems universally accepted nowadays. Nevertheless, β‐cell deficiency in type 2 diabetic patients is modest compared with that of type 1 diabetics; it is therefore questionable whether type 2 diabetes would reach its present epidemic proportions without the concourse of additional factors; that is, an environment characterized by overfeeding and decreased exercise (an idea already discussed several decades ago84). Thus, type 2 diabetes is a classic example of gene–environment interaction. Indeed, what the numerous diabetes‐related polymorphic alleles seem to do is to somewhat reduce the functional (and perhaps survival) capabilities of the β‐cell; that is, place the cell in the lower‐end of the normal variation in terms of its adaptation and resistance to the stress exerted by overfeeding and insulin resistance. By themselves, these characteristics do not define such a β‐cell as abnormal or sick, nor can the aforementioned polymorphic alleles be strictly described as diabetes‐causing gene variants, because subjects carrying them are free of disease unless exposed for a prolonged time to the inappropriate environment (even then only a fraction of the population at risk develops type 2 diabetes). Indeed, it can be speculated that in a different context, for example, after World War 2 in the undernourished but physically very active populations of Europe and Asia, genome‐wide association studies would probably fail to discover any of the diabetes‐related gene variants hotly debated today. Our view of type 2 diabetes development is that, the greater the caloric intake and the lower the insulin sensitivity of a subject, the greater the need for the β‐cells to increase their insulin output to maintain normal glucose homeostasis. To do so, the β‐cells must have the capacity to continuously augment insulin secretion and proinsulin biosynthesis, and probably also expand the β‐cell mass; failure to fine‐tune the adaptation of means to needs ineluctably leads to impaired glucose homeostasis. It is therefore of utmost importance that the mechanisms that allow the full adaptation of β‐cells to increased functional demands be well understood; enhancing these mechanisms will provide the means to prevent transition from normal glucose tolerance to IGT, that is, prevention of type 2 diabetes. Once IGT appears, glucolipotoxic events impair β‐cell function and well‐being, and accelerate the loss of glucose homeostasis. We are only starting to understand the mechanisms of glucolipotoxicity and to describe the molecular mechanisms involved in β‐cell ER stress as well as oxidative stress. A picture is emerging ascribing to the UPR and to the TXNIP‐thioredoxin couple key roles in determining whether β‐cells exposed to the pressure of extended hyperglycemia succeed to adapt the functional β‐cell mass to demand, or fail and undergo apoptotic cell death. However, we know nothing of gene polymorphisms that might render these stress responses adaptive or deleterious for the β‐cell. This is indeed a major area of future research, with a great impact on our understanding of the pathophysiology of type 2 diabetes and on the discovery of a new generation of diabetes drugs.
As is apparent from the present discussion, most studies on the β‐cell in the context of type 2 diabetes deal with various aspects of glucolipotoxicity. This is obviously relevant to β‐cell fate in the diabetic environment, and thus to diabetes progression. However, it might be questioned whether the glucolipotoxicity mechanisms that are responsible for the deterioration of β‐cell function and induction of β‐cell death, discussed earlier, are also responsible for the initial events in the development of type 2 diabetes; that is, the transition from normal glucose tolerance to IGT. Expressed differently, the question is whether the β‐cell stress mechanisms described earlier are only a secondary consequence of diabetes (glucolipotoxicity), which can be referred to as a complication of the disease, or whether similar mechanisms are also operative early in the pathogenic events that lead to the gradual loss of glucose homeostasis. Future research should answer this important question.
Acknowledgements
Our previous and present work that formed the basis on which we developed the ideas exposed in this review has been supported by too many agencies to be individually cited here; our sincere thanks to all of them. We also thank the Endocrine Laboratories of the Hadassah Hospital and our present students and collaborators for their valuable input. We declare that there is no conflict of interest involved with the present manuscript.
References
- 1.Bottazzo GF, Florin‐Christensen A, Doniach D. Islet‐cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 1974; 304: 1279–1283 [DOI] [PubMed] [Google Scholar]
- 2.Himsworth HP. High carbohydrate diets and insulin efficiency. Br Med J 1934; 2: 57–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Himsworth HP. Diabetes mellitus. Its differentiation into insulin‐sensitive and insulin‐insensitive types. Lancet 1936; 227: 127–130 [DOI] [PubMed] [Google Scholar]
- 4.Saltiel AR. The molecular and physiological basis of insulin resistance: emerging implications for metabolic and cardiovascular disease. J Clin Invest 2000; 106: 163–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cerasi E, Luft R. Plasma insulin response to sustained hyperglycaemia induced by glucose infusion in human subjects. Lancet 1963; 282: 1359–1361 [DOI] [PubMed] [Google Scholar]
- 6.Cerasi E, Luft R. The plasma insulin response to glucose infusion in healthy subjects and in diabetes mellitus. Acta Endocrinol 1967; 55: 278–304 [DOI] [PubMed] [Google Scholar]
- 7.Pyke DA, Taylor KW. Glucose tolerance and serum insulin in unaffected identical twins of diabetics. Br Med J 1967; 4: 21–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seltzer HS, Allen EW, Herron AL, et al. Insulin secretion in response to glycemic stimulus: relation of delayed initial release to carbohydrate intolerance in mild diabetes mellitus. J Clin Invest 1967; 46: 323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soeldner JS, Gleason RE, Williams RF, et al. Diminished serum insulin response to glucose in genetic prediabetic males with normal glucose tolerance. Diabetes 1968; 17: 17–26 [DOI] [PubMed] [Google Scholar]
- 10.Cerasi E. Insulin deficiency and insulin resistance in the pathogenesis of type 2 diabetes: is a divorce possible? Diabetologia 1995; 38: 992–997 [DOI] [PubMed] [Google Scholar]
- 11.Kulkarni RN, Bruning JC, Winnay JN, et al. Tissue‐specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 1999; 96: 329–339 [DOI] [PubMed] [Google Scholar]
- 12.Cavaghan MK, Ehrman DA, Polonsky KS. Interactions between insulin resistance and insulin secretion in the development of glucose intolerance. J Clin Invest 2000; 106: 329–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Della Casa L, Del Rio G, Glaser B, et al. Effect of 6‐month gliclazide treatment on insulin release and sensitivity to endogenous insulin in NIDDM: role of initial continuous subcutaneous insulin infusion‐induced normoglycemia. Am J Med 1991; 90: 37S–45S [DOI] [PubMed] [Google Scholar]
- 14.Ilkova H, Glaser B, Tunçkale A, et al. Induction of long‐term glycemic control in newly diagnosed type 2 diabetic patients by transient intensive insulin treatment. Diabetes Care 1997; 20: 1353–1356 [DOI] [PubMed] [Google Scholar]
- 15.Retnakaran R, Yakubovitch N, Qi Y, et al. The response to short‐term intensive insulin therapy in type 2 diabetes. Diabetes Obes Metab 2010; 12: 65–71 [DOI] [PubMed] [Google Scholar]
- 16.Weng J, Li Y, Wen X, et al. Effect of intensive insulin therapy on ß‐cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: A multicentre randomised parallel‐group trial. Lancet 2008; 371: 1753–1760 [DOI] [PubMed] [Google Scholar]
- 17.Efendic S, Cerasi E, Elander I, et al. Studies on low insulin responders. Acta Endocrinol 1979; 90(Suppl. 224): 5–32 [PubMed] [Google Scholar]
- 18.Hollenbeck CB, Chen N, Chen Y‐DI, et al. Relationship between the plasma insulin response to oral glucose and insulin‐stimulated glucose utilization in normal subjects. Diabetes 1984; 33: 460–463 [DOI] [PubMed] [Google Scholar]
- 19.Lillioja S, Mott DM, Spraul M, et al. Insulin resistance and insulin secretory dysfunction as precursors of non‐insulin‐dependent diabetes mellitus. Prospective studies of Pima Indians. N Engl J Med 1993; 329: 1988–1992 [DOI] [PubMed] [Google Scholar]
- 20.Ferrannini E, Natali A, Bell P, et al. Insulin resistance and hypersecretion in obesity. J Clin Invest 1997; 100: 1166–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cerasi E, Efendic S, Luft R. Dose‐response relationship of plasma insulin and blood glucose levels during oral glucose loads in prediabetic and diabetic subjects. Lancet 1973; 301: 794–797 [DOI] [PubMed] [Google Scholar]
- 22.Hales CN. The pathogenesis of NIDDM. Diabetologia 1994; 37(Suppl. 2): S162–S168 [DOI] [PubMed] [Google Scholar]
- 23.Ferrannini E, Gastaldelli A, Miyazaki Y, et al. β‐cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: a new analysis. J Clin Endocrinol Metab 2005; 90: 493–500 [DOI] [PubMed] [Google Scholar]
- 24.Brunzell JD, Robertson RP, Lerner RL, et al. Relationships between fasting plasma glucose levels and insulin secretion during intravenous glucose tolerance tests. J Clin Endocrinol Metab 1976; 42: 222–229 [DOI] [PubMed] [Google Scholar]
- 25.Pørksen N, Holligdal M, Juhl C, et al. Pulsatile insulin secretion: detection, regulation, and role in diabetes. Diabetes 2002; 51(Suppl. 1): S245–S254 [DOI] [PubMed] [Google Scholar]
- 26.Seino Y, Fukushima N, Yabe D. GIP and GLP‐1, the two incretin hormones: similarities and differences. J Diabetes Invest 2010; 1: 8–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alvarsson M, Wajngot A, Cerasi E, et al. K‐value and low insulin secretion in a non‐obese white population predicted glucose tolerance after 25 years. Diabetologia 2005; 48: 2262–2268 [DOI] [PubMed] [Google Scholar]
- 28.Utzschneider KM, Prigeon RL, Faulenbach MV, et al. Oral disposition index predicts the development of future diabetes above and beyond fasting and 2‐h glucose levels. Diabetes Care 2009; 32: 335–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cali AMG, Dalla Man C, Cobelli C, et al. Primary defects in β‐cell function further exacerbated by worsening of insulin resistance mark the development of impaired glucose tolerance in obese adolescents. Diabetes Care 2009; 32: 456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cerasi E, Luft R. Insulin response to glucose infusion in diabetic and non‐diabetic monozygotic twin pairs. Genetic control of insulin response? Acta Endocrinol 1967; 55: 330–345 [DOI] [PubMed] [Google Scholar]
- 31.Pyke DA, Cassar J, Todd J, et al. Glucose tolerance and serum insulin in identical twins of diabetics. Br Med J 1970; 4: 649–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iselius L, Lindsten J, Morton NE, et al. Genetic regulation of the kinetics of glucose‐induced insulin release in man – studies in families with diabetic and non‐diabetic probands. Clin Genet 1985; 28: 8–15 [DOI] [PubMed] [Google Scholar]
- 33.Simonis‐Bik AMC, Eekhoff EMW, de Moor MHM, et al. Genetic influences on the insulin response of the beta cell to different secretagogues. Diabetologia 2009; 52: 2570–2577 [DOI] [PubMed] [Google Scholar]
- 34.Staiger H, Machicao F, Fritsche A, et al. Pathomechanisms of type 2 diabetes genes. Endocr Rev 2009; 30: 557–585 [DOI] [PubMed] [Google Scholar]
- 35.Pascoe L, Frayling TM, Weedon MN, et al. Beta cell glucose sensitivity is decreased by 39% in non‐diabetic subjects carrying multiple diabetes‐risk alleles compared with those with no risk alleles. Diabetologia 2008; 51: 1989–1992 [DOI] [PubMed] [Google Scholar]
- 36.t’Hart LM, Simonis‐Bik AM, Nijpels G, et al. Combined risk allele score of eight type 2 diabetes genes is associated with reduced first‐phase glucose‐stimulated insulin secretion during hyperglycaemic clamps. Diabetes 2010; 59: 287–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gloyn AL, Braun M, Rorsman P. Type 2 diabetes susceptibility gene TCF7L2 and its role in β‐cell function. Diabetes 2009; 58: 800–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Da Silva Xavier G, Loder MK, MacDonald A, et al. TCF7L2 regulates late events in insulin secretion from pancreatic islet beta‐cells. Diabetes 2009; 58: 894–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shu L, Matveyenko AV, Kerr‐Conte J, et al. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP and GLP‐1 receptors and impaired beta‐cell function. Hum Mol Genet 2009; 18: 2388–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mondal AK, Das SK, Baldini G, et al. Genotype and tissue‐specific effects on alternative splicing of the transcription factor 7‐like 2 gene in humans. J Clin Endocrinol Metab 2010; 95: 1450–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vaxillaire M, Bonnefond A, Froguel P. Breakthroughs in monogenic diabetes genetics: from pediatric forms to young adulthood diabetes. Pediatr Endocrinol Rev 2009; 6: 405–417 [PubMed] [Google Scholar]
- 42.Hattersley A, Bruining J, Shield J, et al. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes 2009; 10(Suppl. 12): 33–42 [DOI] [PubMed] [Google Scholar]
- 43.Støy J, Steiner DF, Park SYet al.Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev Endocr Metab Disord 2010; 11: 205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holman RR. Long‐term efficacy of sulfonylureas: a United Kingdom Prospective Diabetes Study perspective. Metabolism 2006; 55(Suppl. 1): S2–S5 [DOI] [PubMed] [Google Scholar]
- 45.Maclean N, Ogilvie RF. Quantitative estimation of the pancreatic islet tissue in diabetic subjects. Diabetes 1955; 4: 367–376 [DOI] [PubMed] [Google Scholar]
- 46.Gepts W. Die histopathologischen Veränderungen der Langerhansschen Inseln und ihre Bedeutung in der Frage des Pathogenese des menschlichen Diabetes. Endokrinologie 1958; 36: 185–211 [PubMed] [Google Scholar]
- 47.Saito K, Takahashi T, Yaginuma N, et al. Islet morphometry in the diabetic pancreas of man. Tohoku J Exp Med 1978; 125: 185–197 [DOI] [PubMed] [Google Scholar]
- 48.Butler AE, Janson J, Bonner‐Weir S, et al. Beta‐cell deficit and increased beta‐cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52: 102–110 [DOI] [PubMed] [Google Scholar]
- 49.Clark A, Jones LC, de Koning E, et al. Decreased insulin secretion in type 2 diabetes: a problem of cellular mass or function? Diabetes 2001; 50(Suppl. 1): S169–S171 [DOI] [PubMed] [Google Scholar]
- 50.Sakuraba H, Mizukami H, Yagihashi N, et al. Reduced beta‐cell mass and expression of oxidative stress‐related DNA damage in the islet of Japanese type II diabetic patients. Diabetologia 2002; 45: 85–96 [DOI] [PubMed] [Google Scholar]
- 51.Yoon KH, Ko SH, Cho JH, et al. Selective β‐cell loss and α‐cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocr Metab 2003; 88: 2300–2308 [DOI] [PubMed] [Google Scholar]
- 52.Rahier J, Guiot Y, Goebbels RM, et al. Pancreatic beta‐cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 2008; 10(Suppl. 4): 32–42 [DOI] [PubMed] [Google Scholar]
- 53.Butler AE, Cao‐Minh L, Galasso R, et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010; 53: 2167–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ichise M, Harris PE. Imaging of beta‐cell mass and function. J Nucl Med 2010; 51: 1001–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaiser N, Yuli M, Üçkaya G, et al. Dynamic changes in beta‐cell mass and pancreatic insulin during the evolution of nutrition‐dependent diabetes in Psammomys obesus. Impact of glycemic control. Diabetes 2005; 54: 138–145 [DOI] [PubMed] [Google Scholar]
- 56.Gunton JE, Kulkarni RN, Yim S, et al. Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic‐islet dysfunction in human type 2 diabetes. Cell 2005; 122: 337–349 [DOI] [PubMed] [Google Scholar]
- 57.Marselli L, Thorne J, Dahiya S, et al. Gene expression profiles of beta‐cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PLoS ONE 2010; 5: e11499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest 2006; 116: 1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaiser N, Leibowitz G. Failure of beta‐cell adaptation in type 2 diabetes: lessons from animal models. Front Biosci 2009; 14: 1099–1115 [DOI] [PubMed] [Google Scholar]
- 60.Cnop M, Hughes SJ, Igoillo‐Esteve M, et al. The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia 2010; 53: 321–330 [DOI] [PubMed] [Google Scholar]
- 61.Maedler K, Sergeev P, Ris F, et al. Glucose‐induced beta cell production of IL‐1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 2002; 110: 851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larsen CM, Faulenbach M, Vaag A, et al. Interleukin‐1‐receptor antagonist in type 2 diabetes mellitus. N Engl J Med 2007; 356: 1517–1526 [DOI] [PubMed] [Google Scholar]
- 63.Song B, Scheuner D, Ron D, et al. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 2008; 118: 3378–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bachar E, Ariav Y, Ketzinel‐Gilad M, et al. Glucose amplifies fatty acid‐induced endoplasmic reticulum stress in pancreatic beta‐cells via activation of mTORC1. PLoS ONE 2009; 4: e4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang CJ, Lin CY, Haataja L, et al. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta‐cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007; 56: 2016–2027 [DOI] [PubMed] [Google Scholar]
- 66.Kennedy J, Katsuta H, Jung MH, et al. Protective unfolded protein response in human pancreatic beta cells transplanted into mice. PLoS ONE 2010; 5: e11211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med 1996; 20: 463–466 [DOI] [PubMed] [Google Scholar]
- 68.Tiedge M, Lortz S, Drinkgern J, et al. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin‐producing cells. Diabetes 1997; 46: 1733–1742 [DOI] [PubMed] [Google Scholar]
- 69.Lacraz G, Figeac F, Movassat J, et al. Diabetic beta‐cells can achieve self‐protection against oxidative stress through an adaptive up‐regulation of their antioxidant defenses. PLoS ONE 2009; 4: e6500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martens GA, Cai Y, Hinke S, et al. Glucose suppresses superoxide generation in metabolically responsive pancreatic beta cells. J Biol Chem 2005; 280: 20389–20396 [DOI] [PubMed] [Google Scholar]
- 71.Holmgren A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid Redox Signal 2000; 2: 811–820 [DOI] [PubMed] [Google Scholar]
- 72.Ivarsson R, Quintens R, Dejonghe S, et al. Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 2005; 54: 2132–2142 [DOI] [PubMed] [Google Scholar]
- 73.Yoshioka J, Schreiter ER, Lee RT. Role of thioredoxin in cell growth through interactions with signaling molecules. Antioxid Redox Signal 2006; 8: 2143–2151 [DOI] [PubMed] [Google Scholar]
- 74.Patwari P, Higgins LJ, Chutkow WA, et al. The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange. J Biol Chem 2006; 281: 21884–21891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saxena G, Chen J, Shalev A. Intracellular shuttling and mitochondrial function of thioredoxin‐interacting protein. J Biol Chem 2010; 285: 3997–4005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Minn AH, Hafele C, Shalev A. Thioredoxin‐interacting protein is stimulated by glucose through a carbohydrate response element and induces beta‐cell apoptosis. Endocrinology 2005; 146: 2397–2495 [DOI] [PubMed] [Google Scholar]
- 77.Schulze PC, Yoshioka J, Takahashi T, et al. Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin‐interacting protein. J Biol Chem 2004; 279: 30369–30374 [DOI] [PubMed] [Google Scholar]
- 78.Qi W, Chen X, Gilbert RE, et al. High glucose‐induced thioredoxin‐interacting protein in renal proximal tubule cells is independent of transforming growth factor‐beta1. Am J Pathol 2007; 171: 744–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Turturro F, Friday E, Welbourne T. Hyperglycemia regulates thioredoxin‐ROS activity through induction of thioredoxin‐interacting protein (TXNIP) in metastatic breast cancer‐derived cells MDA‐MB‐231. BMC Cancer 2007; 7: 96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen J, Saxena G, Mungrue IN, et al. Thioredoxin‐interacting protein: a critical link between glucose toxicity and beta‐cell apoptosis. Diabetes 2008; 57: 938–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen J, Hui ST, Couto FM, et al. Thioredoxin‐interacting protein deficiency induces Akt/Bcl‐xL signaling and pancreatic beta‐cell mass and protects against diabetes. FASEB J 2008; 22: 3581–3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shaked M, Ketzinel‐Gilad M, Ariav Y, et al. Insulin counteracts glucotoxic effects by suppressing thioredoxin‐interacting protein production in INS‐1E beta cells and in Psammomys obesus pancreatic islets. Diabetologia 2009; 52: 636–644 [DOI] [PubMed] [Google Scholar]
- 83.Zhou R, Tardivel A, Thorens B, et al. Thioredoxin‐interacting protein links oxidative stress to inflammasome activation. Nat Immunol 2010; 11: 136–140 [DOI] [PubMed] [Google Scholar]
- 84.Cerasi E, Luft R. “What is inherited ‐ what is added” hypothesis for the pathogenesis of diabetes mellitus. Diabetes 1967; 16: 615–627 [DOI] [PubMed] [Google Scholar]