

Abstract
Present on archaeal and eukaryotic translation elongation factor 2, diphthamide represents one of the most intriguing post-translational modifications on proteins. The biosynthesis of diphthamide was proposed to occur in three steps requiring seven proteins, Dph1–7, in eukaryotes. The functional assignments of Dph1–5 in the first and second step have been well established. Recent studies suggest that Dph6 (yeast YLR143W or human ATPBD4) and Dph7 (yeast YBR246W or human WDR85) are involved in the last amidation step, with Dph6 being the actual diphthamide synthetase catalyzing the ATP-dependent amidation reaction. However, the exact molecular role of Dph7 is unclear. Here we demonstrate that Dph7 is an enzyme catalyzing a previously unknown step in the diphthamide biosynthesis pathway. This step is between the Dph5- and Dph6-catalyzed reactions. We demonstrate that the Dph5-catalyzed reaction generates methylated diphthine, a previously overlooked intermediate, and Dph7 is a methylesterase that hydrolyzes methylated diphthine to produce diphthine and allows the Dph6-catalyzed amidation reaction to occur. Thus, our study characterizes the molecular function of Dph7 for the first time and provides a revised diphthamide biosynthesis pathway.
Diphthamide is a post-translationally modified histidine residue present on archaeal and eukaryotic elongation factor 2 (eEF-2), a GTPase involved in the translocation of mRNA and tRNA on the ribosome during translation elongation.1−4 This exceptional modification is targeted by the pathogenic bacterium, Corynebacterium diphtheria, which causes the infectious disease diphtheria in humans. Diphtheria toxin (DT) produced by this bacterium catalyzes the ADP-ribosylation of the diphthamide residue of eEF-2 using nicotinamide adenine dinucleotide (NAD) as the ADP-ribosyl donor.5 Irreversible ADP-ribosylation inactivates eEF-2, which in turn stops translation, leading to cell death.6 Diphthamide is reported to be important for preventing −1 translational frame shift in yeast and mammalian cells.7,8 Intriguingly, this modification is not present in EF-G, the bacterial ortholog of eEF-2.
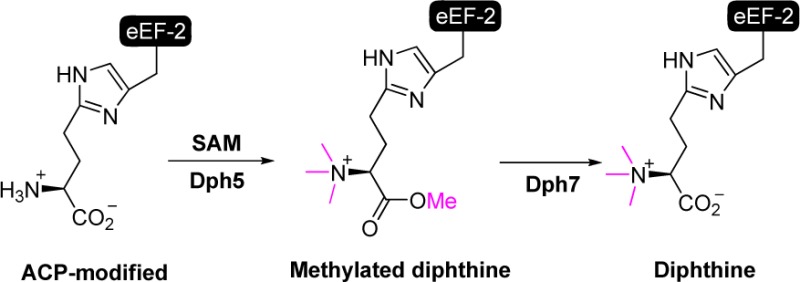
Genetic and biochemical studies in the budding yeast Saccharomyces cerevisiae allowed dissection of the diphthamide biosynthesis pathway. It was initially proposed that the biosynthesis involves three steps (Scheme 1A).9−11 Four proteins, Dph1–4, are required for the first step, which involves the transfer of the 3-amino-3-carboxypropyl (ACP) group from S-adenosyl methionine (SAM) to the C2 carbon of the imidazole ring of His699 of yeast eEF-2 (His715 of mammalian eEF-2). Recent evidence suggests that this step uses a unique [4Fe-4S]-containing enzyme and a radical reaction mechanism.12−14 The second step involves a single methyltransferase, Dph5, which catalyzes the trimethylation of the amino group to form the diphthine intermediate (2, Scheme 1A). The last step is the amidation of the carboxyl group of diphthine (2) to form diphthamide (1), but the proteins required for this step evaded the initial genetic screening11 and remained elusive for a long time. Carette et al. identified human WDR85 (yeast YBR246W) as a new diphthamide biosynthetic protein (later named Dph7) through haploid genetic screening.15 It was initially proposed that Dph7 is involved in the first step of diphthamide biosynthesis, but a later study by our laboratory showed that deletion of Dph7 led to accumulation of 2, suggesting that Dph7 is involved in the last step of diphthamide biosynthesis. However, Dph7 is not the diphthamide synthetase, as it lacks the ATP-binding domain required.16 The actual diphthamide synthetase, Dph6, was identified independently by three groups using comparative genomic analysis,17 yeast cofitness analysis,18 and yeast gene interaction databases.19
Scheme 1. (A) Proposed Diphthamide Biosynthesis Pathway in Current Literature. (B) Revised Diphthamide Biosynthesis Pathway in Eukaryotes.
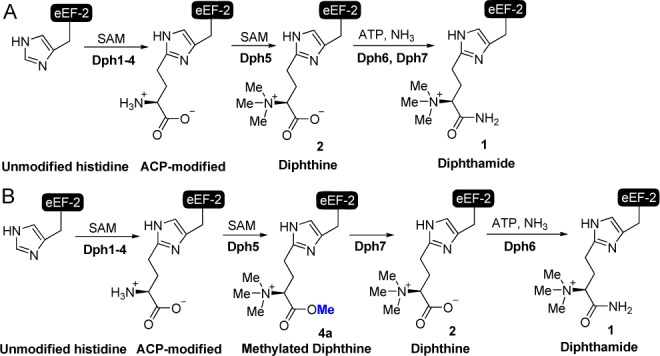
What is the exact role of Dph7 in the pathway then? Initially, we thought that Dph7 could be a scaffold protein for the amidation reaction, as it contains WD40 domains that are known to mediate protein–protein interactions.16 Contrary to this notion, Dph6 and Dph7 were not found to interact with each other by coimmunoprecipitation.19 It was also suggested that Dph7 is required to displace Dph5 after the second step to allow amidation catalyzed by Dph6 to occur, as eEF-2 binds more Dph5 in the absence of Dph7.19 Interestingly, a novel methylated diphthamide (3, Figure 1) was recently reported in a lymphoma cell line with Dph7 gene deletion.20 This methylation was thought to occur on one of the nitrogen atoms of the imidazole ring of the histidine residue (3, Figure 1).20 Here we demonstrate that this modification is actually methylated diphthine with the methyl group on the carboxylate of diphthine (4a, Figure 1) and Dph7 is a methylesterase responsible for the hydrolysis of the methylated diphthine (4a) to generate diphthine (2), which can then be used by Dph6 in the last amidation step (Scheme 1B). Methylated diphthine (4a) is produced by the enzymatic function of Dph5. The present work thus uncovers the molecular function of Dph7 and provides a revised diphthamide biosynthesis pathway (Scheme 1B).
Figure 1.
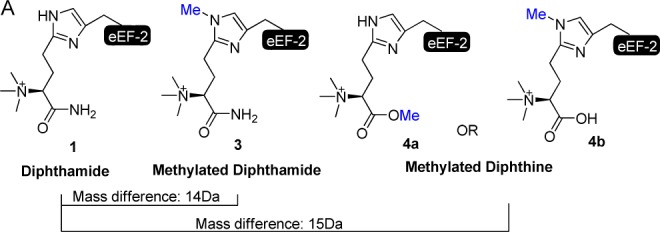
Proposed structures of methylated diphthamide and methylated diphthine. Under the acidic conditions of mass spectrometry analysis, the carboxylic group of 4b is protonated.
To determine if Dph7 had an enzymatic role or merely mediated the interaction of Dph6 with eEF-2, we purified eEF-2 proteins from a yeast strain with Dph6 deletion (Δdph6) and a yeast strain with Dph7 deletion (Δdph7) for in vitro reconstitution of the amidation reaction. The purified eEF-2 proteins were incubated with Dph6, ATP, and ammonium chloride for amidation. Diphthamide formation was detected with fluorescently labeled rhodamine-NAD (Rh-NAD) and a low concentration of DT, as previously described.18 Under these conditions, only diphthamide, but not other intermediate forms, can be labeled by Rh-NAD. Hence, the fluorescence labeling indicates the formation of diphthamide. We found that purified eEF-2 from Δdph7 was not the immediate substrate for the in vitro amidation by Dph6 (Figure 2A, lane 2). In contrast, eEF-2 from Δdph6 was a substrate for the in vitro reaction (Figure 2A, lane 1). Diphthamide was formed on Δdph7 eEF-2 only in the presence of both Dph6 and Dph7 (Figure 2A, lane 3). These results support the notion that Dph7 converts Δdph7 eEF-2 into Δdph6 eEF-2, a form of eEF-2 that can be amidated by Dph6 to generate diphthamide.
Figure 2.
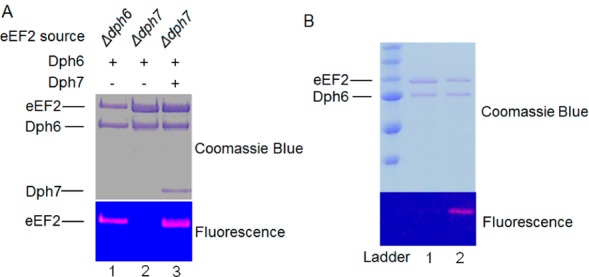
Dph7 converts Δdph7 eEF2 to a substrate for amidation by Dph6. (A) Dph6 and Dph7 are both required for in vitro amidation of Δdph7 eEF2. The fluorescence labels indicate formation of diphthamide by the amidation reaction catalyzed by Dph6. (B) Stepwise in vitro reconstitution of diphthamide formation on Δdph7 eEF2. Lane 1: flag-tagged Δdph7 eEF-2 incubated without Dph7. Lane 2: flag-tagged Δdph7 eEF-2 incubated with Dph7 and then purified to remove Dph7. Both Δdph7 eEF-2 samples were then incubated with Dph6 and labeled with DT and Rh-NAD.
To further demonstrate that Dph7 catalyzes an additional step, we incubated flag-tagged Δdph7 eEF-2 with Dph7 and repurified the Δdph7 eEF-2 to remove Dph7. We found that the repurified Δdph7 eEF-2 was a substrate for the amidation by Dph6 alone (Figure 2B, lane 2). In contrast, flag-tagged Δdph7 eEF-2 incubated without Dph7 did not form diphthamide (Figure 2B, lane 1). Taken together, these findings indicate that there is an additional step before the last amidation step in the diphthamide biosynthesis and Dph7 is the enzyme catalyzing this step.
The conclusion of Dph7 having enzymatic function is seemingly contradictory to previous reports showing that Δdph6 eEF-2 and Δdph7 eEF-2 both contain diphthine (2).16,18,19 Based on these observations, there is no room for any apparent chemical transformation for Dph7’s enzymatic activity. Interestingly, a species with a mass of 15 Da larger than that of diphthamide (1) was reported in a lymphoma cell line with Dph7 gene deletion.20 The proposed structure for this species was methylated diphthamide (3, Figure 1). However, the expected mass difference of 3 and 1 is 14 Da. Therefore, we speculated that the observed species was methylated diphthine instead (4a or 4b, Figure 1). In light of this report, we investigated the presence of methylated diphthine (4a or 4b) in Δdph7 eEF-2 and Δdph6 eEF-2 via liquid chromatography-mass spectrometry (LC-MS) studies. Consistent with previous studies, we found diphthine (2) containing tryptic peptide (686-VNILDVTLHADAIHR-700) from both Δdph7 and Δdph6 eEF-2 samples (Figure S1A). Likewise, we observed a small amount of unmodified peptides, but no diphthamide (1) was detected in either eEF-2 sample.16,19 We also found the presence of methylated diphthine (4a or 4b) in Δdph7 eEF-2 (Figure S1B). Most strikingly, this methylated diphthine (4a or 4b) was not detected in Δdph6 eEF-2. This unexpected form of modification had an m/z larger than those of all the previously known intermediates of diphthamide biosynthesis or diphthamide (1). The results of this investigation suggested a possible enzymatic role for Dph7 as a demethylase. We hypothesized that Dph7 functions to remove the extra methyl group on methylated diphthine (4a or 4b) to form diphthine (2). Furthermore, the fact that 2 was also observed in MS studies of Δdph7 eEF-2 suggested that this methyl group was relatively labile during the sample preparation for MS. Therefore, we proposed that methylated diphthine is a methyl ester which is prone to hydrolysis (4a, Scheme 1B).
To test the hypothesis that methylated diphthine (4a or 4b) is a methyl ester (4a), we examined the nonenzymatic hydrolysis of the methyl ester under mild basic conditions. Purified Δdph7 eEF-2 in Tris-HCl pH 9.0 buffer was incubated at 30 °C for various time intervals. The conversion of 4a to 2 was monitored by the amidation reaction, as Dph6 selectively amidates 2, but not 4a, to form diphthamide (1). The formation of 1 is then detected by a fluorescence label using Rh-NAD and DT. As shown in Figure 3A, an increased incubation time for Δdph7 eEF-2 leads to an increased fluorescence label, indicating the time-dependent conversion of 4a to 2. In contrast, extending the incubation time for Δdph6 eEF-2, which contains 2, has no effect on the fluorescence intensity (Figure 3B). Moreover, the labeling intensities of Δdph7 eEF-2 after incubation at pH 9.0 are considerably lower than those of Δdph6 eEF-2, suggesting incomplete conversion even after 4 h of incubation (Figure 3). This nonenzymatic reaction demonstrated that the extra methylation site is susceptible to hydrolysis. Thus, the most likely configuration of the methylated diphthine is a methyl ester (4a).
Figure 3.
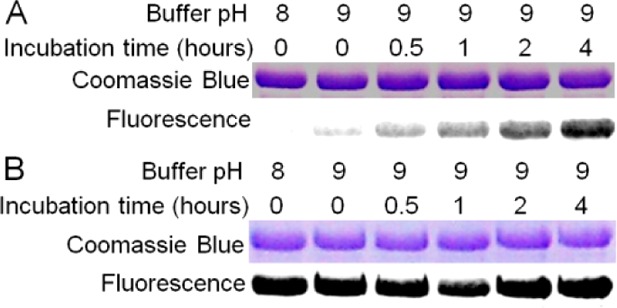
Nonenzymatic hydrolysis of methylated diphthine to diphthine. (A) Δdph7 eEF-2 or (B) Δdph6 eEF-2 were buffer-exchanged into a buffer containing 25 mM Tris-HCl pH 9.0 and 150 mM NaCl and incubated at 30 °C for various time intervals before the amidation reaction by Dph6. Formation of diphthamide was detected by a fluorescence label generated by DT and Rh-NAD.
To further show that Dph7 catalyzes the hydrolysis reaction to form diphthine (2), we incubated Δdph7 eEF-2 with Dph7 protein in Tris-HCl pH 8.0 buffer and examined the levels of methylated diphthine (4a) and 2. Δdph7 eEF-2 incubated with or without Dph7 was subjected to in-solution trypsin digestion and subsequently LC-MS analysis. A tryptic peptide (815-AGEIVLAAR-823) without any post-translational modifications from eEF-2 was used as an internal reference peak for both samples (Figure 4, Figure S2A and S2B). The level of 4a decreases drastically after incubation with Dph7 (Figure 4, Figure S2C–S2F). Correspondingly, the level of 2 increases after treatment with Dph7 (Figure 4, Figure S2G–S2J). Consistent with previous MS analysis, a significant amount of 2 is present in the Δdph7 eEF-2 sample without Dph7, possibly due to nonenzymatic hydrolysis of the methyl ester (4a) during incubation in pH 8.0 buffer and during the sample preparation process for MS analysis.
Figure 4.

Dph7 hydrolyzes methylated diphthine to form diphthine. Relative intensities of tryptic peptides from Δdph7 eEF-2 with (“+Dph7”) or without Dph7 (“–Dph7”) treatment were shown. Intensities of tryptic peptides from Δdph7 eEF-2 without Dph7 treatment were set to 1.
Our finding that Dph7 is a methylesterase converting methylated diphthine (4a) to diphthine (2) suggests that the diphthamide biosynthesis pathway needs to be revised. In the current literature, 2 is proposed as the product of the second step catalyzed by the methyltransferase, Dph5. It was proposed after the fact that acid hydrolysis of eEF-2 with in vitro reconstitution of the second step yields 2.21 However, under such conditions, it was likely that 4a, a methyl ester, was hydrolyzed to 2 and was not detected. To investigate if 4a is produced by Dph5 in the second step, we reconstituted the reaction in vitro using purified eEF-2 from the Δdph5 yeast strain, SAM, and purified Dph5 protein. Δdph5 eEF-2 incubated with SAM but without Dph5 was used as a control. The Δdph5 strain is deficient in the second step of diphthamide biosynthesis, and therefore the eEF2 contains 3-amino-3-carboxypropyl (ACP) modified His699, the product of the first step. Both experimental and control eEF-2 samples were trypsin-digested and subjected to LC-MS analysis. In agreement with previous MS reports, we found ACP-modified peptide and unmodified peptide, but not other intermediates in the Δdph5 eEF-2 sample without Dph5.19 For the Δdph5 eEF-2 sample treated with Dph5 and SAM, the level of ACP-modified peptide was considerably lower than that of the control, indicating that the ACP-modified eEF-2 was consumed (Figure 5, Figure S3C and S3D). In addition, we found three other types of modifications on His699 of the tryptic peptide (686-VNILDVTLHADAIHR-700): monomethylated ACP (Figure S3F), diphthine (Figure S3H), and methylated diphthine (Figure 5, Figure S3J). These three modified forms were not present in the control sample without Dph5 (Figure S3E, S3G, and S3I). The monomethylated ACP-modified eEF-2 was likely an intermediate for the formation of methylated diphthine. Diphthine (2) was again observed, probably due to hydrolysis of 4a. Thus, the MS study demonstrated that 4a is the product of the second step in diphthamide biosynthesis and Dph5 is responsible for the extra methylation.
Figure 5.

Dph5 generates methylated diphthine. Relative intensities of tryptic peptides from Δdph5 eEF-2 with (“+Dph5”) or without Dph5 (“–Dph5”) treatment were shown. Intensities of tryptic peptides from Δdph5 eEF-2 with Dph5 treatment were set to 1.
In summary, our results presented here demonstrate that there is a previously unidentified step in the diphthamide biosynthesis pathway, and we propose a revised scheme of the diphthamide biosynthesis pathway (Scheme 1B). Yeast Dph5 catalyzes the methylation of the amino and the carboxylate groups of ACP, generating methylated diphthine (4a). The molecular function of Dph7 is to convert methylated diphthine (4a) to diphthine (2) so that Dph6 can convert it to diphthamide (1). Although a considerable amount of 2 was observed in both the MS studies of Δdph7 eEF-2 and Δdph5 eEF-2 treated with Dph5, we believe that 4a is the predominant intermediate formed in vivo. This is because, in the yeast Dph7 deletion strain, if both 2 and 4a are formed by Dph5, any 2 formed will be converted to 1 due to the presence of Dph6. Since 1 was not observed in the MS analysis of Δdph7 eEF-2 (as we and others previously reported), we believe that only 4a is formed by yeast Dph5 in vivo. The observation of 2 in Δdph7 eEF-2 during MS analysis is likely due to the hydrolysis of 4a in the sample preparation process.
Previously, we also reported that archaeal Dph5 catalyzes N-trimethylation of the ACP group, which leads to the elimination of the trimethylamino group.22 This elimination reaction was not observed in the yeast Dph5-catalyzed reaction. In contrast, the yeast Dph5 catalyzes tetramethylation of the ACP group. Thus, it seems that archaeal and eukaryotic diphthamide biosyntheses differ in the methylation step. Consistent with this, archaea lack the demethylation enzyme, Dph7. Methyltransferases are known to be able to transfer methyl groups from SAM to diverse acceptor substrates, and methyl transfer to amino and carboxylate groups are known.23 Certain N-methyltransferases and O-methyltransferases are known to be promiscuous.24,25 However, the remarkable promiscuous methylation activity (both N- and O-methylation) of yeast Dph5 has not been observed in other methyltransferases before.
The functional implication of this additional methylation–demethylation step is still unclear at this point. Since archaeal Dph5 does not catalyze the extra methylation, it is hard to believe that this extra methylation is merely a side reaction due to eukaryotic Dph5′s lack of specificity. It is more likely that the extra methylation–demethylation step introduced by the promiscuous methylation activity of eukaryotic Dph5 and the methylesterase Dph7 emerged in the evolution process for a certain purpose. It is possible that this extra methylation introduces a blockage to the pathway, creating a regulatory point for diphthamide biosynthesis. The possibility of a regulation on diphthamide biosynthesis via Dph7 awaits further studies.
To the best of our knowledge, Dph7 is the first WD40 protein to have an enzymatic function. Multiple sequence alignments with Dph7 orthologs reveal conserved serine, aspartic acid, and histidine residues that can potentially form a catalytic Ser-His-Asp triad, commonly found in α/β hydrolases (Figure S4). However, due to the presence of WD40 repeats, Dph7 is predicted to adopt a circularized β-propeller structure,26 lacking the usual α/β hydrolase fold. Pectin methylesterase, which also lacks the α/β hydrolase fold, was demonstrated to adopt a novel esterase active site with two catalytic aspartic residues.27 Thus, it is possible that the catalytic residues of Dph7 differ from the conventional catalytic triad of α/β hydrolases. It will be interesting to investigate the catalytic mechanism of Dph7 in future studies.
Acknowledgments
This work is supported by NIH (R01GM088276). Z.L. is supported by a Singapore Agency for Science, Technology and Research (A*STAR) scholarship. B.C. was supported by the Tsinghua University Xuetang Program.
Supporting Information Available
Experimental materials and methods, supporting figures, and a list of strains used in this study. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ X.S.: Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, New Jersey, 08540. B.C.: Division of Basic Science, The University of Texas Southwestern Medical Center, Dallas, Texas, 75390.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Van Ness B. G.; Howard J. B.; Bodley J. W. J. Biol. Chem. 1980, 255, 10710–10716. [PubMed] [Google Scholar]
- Van Ness B. G.; Howard J. B.; Bodley J. W. J. Biol. Chem. 1980, 255, 10717–10720. [PubMed] [Google Scholar]
- Robinson E. A.; Henriksen O.; Maxwell E. S. J. Biol. Chem. 1974, 249, 5088–5093. [PubMed] [Google Scholar]
- Su X.; Lin Z.; Lin H. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 515–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier R. J. Toxicon 2001, 39, 1793–1803. [DOI] [PubMed] [Google Scholar]
- Honjo T.; Nishizuka Y.; Hayaishi O. J. Biol. Chem. 1968, 243, 3553–5. [PubMed] [Google Scholar]
- Ortiz P. A.; Ulloque R.; Kihara G. K.; Zheng H.; Kinzy T. G. J. Biol. Chem. 2006, 281, 32639–32648. [DOI] [PubMed] [Google Scholar]
- Liu S.; Bachran C.; Gupta P.; Miller-Randolph S.; Wang H.; Crown D.; Zhang Y.; Wein A. N.; Singh R.; Fattah R.; Leppla S. H. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 13817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehring T. J.; Danley D. E.; Moehring J. M. Mol. Cell. Biol. 1984, 4, 642–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. Y.; Bodley J. W.; Livingston D. M. Mol. Cell. Biol. 1985, 5, 3357–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Milne G. T.; Kuremsky J. G.; Fink G. R.; Leppla S. H. Mol. Cell. Biol. 2004, 24, 9487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Zhu X.; Torelli A. T.; Lee M.; Dzikovski B.; Koralewski R. M.; Wang E.; Freed J.; Krebs C.; Ealick S. E.; Lin H. Nature 2010, 465, 891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Dzikovski B.; Su X.; Torelli A. T.; Zhang Y.; Ealick S. E.; Freed J. H.; Lin H. Mol. BioSyst. 2011, 7, 74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong M.; Su X.; Dzikovski B. G.; Dando E. E.; Zhu X.; Du J.; Freed J. H.; Lin H. J. Am. Chem. Soc. 2014, 136, 1754–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette J. E.; Guimaraes C. P.; Varadarajan M.; Park A. S.; Wuethrich I.; Godarova A.; Kotecki M.; Cochran B. H.; Spooner E.; Ploegh H. L.; Brummelkamp T. R. Science 2009, 326, 1231–1235. [DOI] [PubMed] [Google Scholar]
- Su X.; Chen W.; Lee W.; Jiang H.; Zhang S.; Lin H. J. Am. Chem. Soc. 2012, 134, 773–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Crecy-Lagard V.; Forouhar F.; Brochier-Armanet C.; Tong L.; Hunt J. F. Biol. Direct 2012, 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X.; Lin Z.; Chen W.; Jiang H.; Zhang S.; Lin H. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 19983–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uthman S.; Bar C.; Scheidt V.; Liu S.; ten Have S.; Giorgini F.; Stark M. J.; Schaffrath R. PLoS Genet. 2013, 9, e1003334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.; Bera T. K.; Wayne A. S.; Xiang L.; Colantonio S.; Chertov O.; Pastan I. J. Biol. Chem. 2013, 288, 12305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. Y.; Bodley J. W. J. Biol. Chem. 1988, 263, 11692–6. [PubMed] [Google Scholar]
- Zhu X.; Kim J.; Su X.; Lin H. Biochemistry 2010, 49, 9649–9657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert H. L.; Blumenthal R. M.; Cheng X. Trends Biochem. Sci. 2003, 28, 329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; van der Donk W. A. FEBS Lett. 2012, 586, 3391–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopycki J. G.; Rauh D.; Chumanevich A. A.; Neumann P.; Vogt T.; Stubbs M. T. J. Mol. Biol. 2008, 378, 154–64. [DOI] [PubMed] [Google Scholar]
- Xu C.; Min J. Protein Cell. 2011, 2, 202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries M.; Ihrig J.; Brocklehurst K.; Shevchik V. E.; Pickersgill R. W. EMBO J. 2007, 26, 3879–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.