

Abstract
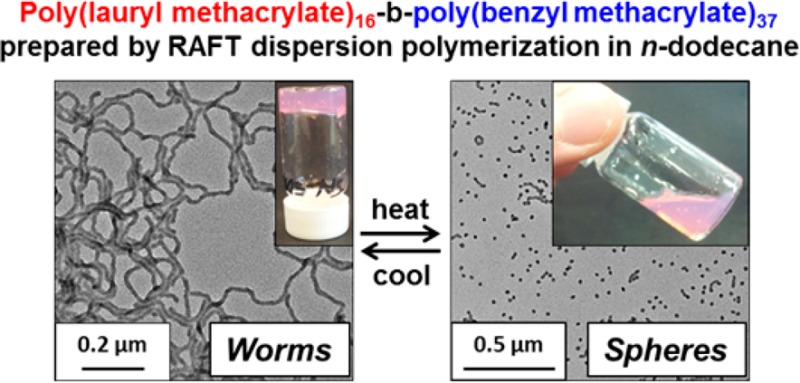
Benzyl methacrylate (BzMA) is polymerized using a poly(lauryl methacrylate) macromolecular chain transfer agent (PLMA macro-CTA) using reversible addition–fragmentation chain transfer (RAFT) polymerization at 70 °C in n-dodecane. This choice of solvent leads to an efficient dispersion polymerization, with polymerization-induced self-assembly (PISA) occurring via the growing PBzMA block to produce a range of PLMA–PBzMA diblock copolymer nano-objects, including spheres, worms, and vesicles. In the present study, particular attention is paid to the worm phase, which forms soft free-standing gels at 20 °C due to multiple inter-worm contacts. Such worm gels exhibit thermo-responsive behavior: heating above 50 °C causes degelation due to the onset of a worm-to-sphere transition. Degelation occurs because isotropic spheres interact with each other much less efficiently than the highly anisotropic worms. This worm-to-sphere thermal transition is essentially irreversible on heating a dilute solution (0.10% w/w) but is more or less reversible on heating a more concentrated dispersion (20% w/w). The relatively low volatility of n-dodecane facilitates variable-temperature rheological studies, which are consistent with eventual reconstitution of the worm phase on cooling to 20 °C. Variable-temperature 1H NMR studies conducted in d26-dodecane confirm partial solvation of the PBzMA block at elevated temperature: surface plasticization of the worm cores is invoked to account for the observed change in morphology, because this is sufficient to increase the copolymer curvature and hence induce a worm-to-sphere transition. Small-angle X-ray scattering and TEM are used to investigate the structural changes that occur during the worm-to-sphere-to-worm thermal cycle; experiments conducted at 1.0 and 5.0% w/w demonstrate the concentration-dependent (ir)reversibility of these morphological transitions.
Introduction
It is well-known that self-assembly of AB diblock copolymers occurs in appropriate selective solvents.1−13 This is typically conducted in dilute solution (<1%) and usually involves post-polymerization processing methods such as a solvent switch,4 a pH switch,14 or thin film rehydration.15 The precise diblock copolymer morphology that is generated mainly depends on the relative volume fractions of the two blocks, because this dictates the so-called packing parameter.16 The most commonly reported copolymer morphologies are spheres, worms/cylinders, or vesicles.6 However, there are many studies describing spheres and vesicles, while relatively few papers are focused on worms/cylinders. Presumably, this is simply because the worm/cylinder morphology occupies the narrowest region of the phase diagram.2,10,17 Nevertheless, there has been considerable recent activity focused on the production of block copolymer worms/cylinders, often based on semicrystalline core-forming blocks.18−21
We22−29 and others30−40 have recently shown that polymerization-induced self-assembly (PISA) offers a potentially decisive option for the formation of diblock copolymer nano-objects.41,42 For example, spherical, worm-like, or vesicular block copolymer morphologies can be efficiently generated directly in either water,22−25,28,31−34 alcohol,26,27,30,35−40 or n-alkanes29,43 at relatively high solids (up to 25%) during the synthesis of the copolymer chains. The worm phase is particularly interesting, because such particles can either act as thickeners or form free-standing gels, depending on the copolymer concentration. Recently, Blanazs et al. have shown that a semi-concentrated aqueous dispersion of methacrylic diblock copolymer worms undergoes a worm-to-sphere morphological transition on cooling from 20 to 5 °C.44 This results in thermo-reversible degelation, which allows facile sterilization via cold ultrafiltration because this protocol removes any micrometer-sized bacteria that may be present. Such worm gels are also highly biocompatible, and both the gel strength and the critical gelation temperature can be tuned;45 thus they are expected to have biomedical applications in the context of cell storage media.
In the present work, we revisit a new RAFT non-aqueous dispersion polymerization formulation that enables a range of poly(lauryl methacrylate)–poly(benzyl methacrylate) [PLMA–PBzMA] diblock copolymer nano-objects to be prepared in n-heptane.29 Working at a fixed solids concentration of 20% w/w in n-dodecane, a phase diagram has been constructed that reveals the effect of varying the mean degrees of polymerization (DP) of the stabilizer and core-forming blocks on the final copolymer morphology. Using n-dodecane in place of n-heptane facilitates studies of the worm gel phase, because the problem of in situ solvent evaporation is avoided. Furthermore, we demonstrate that PLMA-PBzMA diblock copolymer worm gels undergo reversible degelation on heating (see Figure 1). Transmission electron microscopy (TEM), dynamic light scattering (DLS), gel rheology, variable temperature 1H NMR spectroscopy, and small-angle X-ray scattering (SAXS) are used to characterize this phase transition, and a physical mechanism for worm disintegration is suggested.
Figure 1.
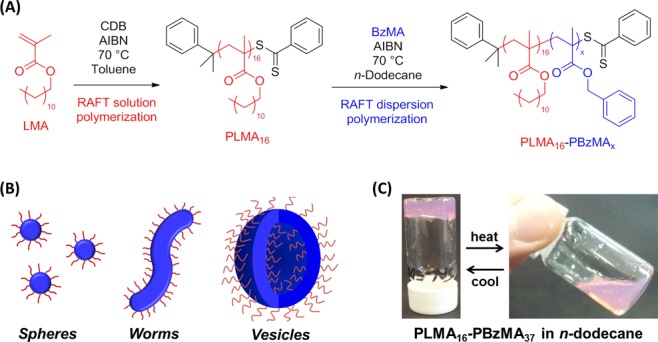
(A) RAFT synthesis of poly(lauryl methacrylate) (PLMA) macro-CTA via RAFT solution polymerization in toluene at 70 °C, followed by RAFT dispersion polymerization of benzyl methacrylate (BzMA) in n-dodecane at 70 °C. (B) Schematic representation of the change in morphology that occurs on increasing the PBzMA target degree of polymerization when using a relatively short PLMA macro-CTA. (C) Thermo-responsive solution behavior exhibited by 20% w/w PLMA16–PBzMA37 diblock copolymer nanoparticles in n-dodecane. A free-standing gel is formed at 20 °C, which becomes a free-flowing solution when heated to 70 °C (see main text for an explanation of this phase transition).
Experimental Section
Materials
Monomers were purchased from Sigma-Aldrich (UK) and passed through basic alumina prior to use. n-Dodecane (≥90% technical grade), CDCl3, and all other reagents were purchased from Sigma-Aldrich (UK) and were used as received, unless otherwise noted. THF and toluene were purchased from Fisher Scientific (UK), while CD2Cl2 and d26-dodecane were purchased from Goss Scientific (UK).
Synthesis of Poly(lauryl methacrylate) Macro-Chain Transfer Agent
The synthesis of PLMA macro-CTAs has been described in detail elsewhere,29 and therefore only one representative formulation is briefly discussed here. A typical synthesis of PLMA16 macro-CTA was conducted as follows. A 100 mL round-bottomed flask was charged with lauryl methacrylate (LMA; 30 g; 118 mmol), cumyl dithiobenzoate (CDB; 3.22 g; 11.8 mmol), 2,2′-azobis(isobutyronitrile) (AIBN; 387 mg, 2.37 mmol; CDB/AIBN molar ratio = 5.0), and toluene (50.4 g). The sealed reaction vessel was purged with nitrogen and placed in a preheated oil bath at 70 °C for 11 h. The resulting PLMA (LMA conversion = 79%; Mn = 4900 g mol–1, Mw = 5400 g mol–1, Mw/Mn = 1.19) was purified by precipitation into excess methanol. The mean degree of polymerization (DP) of this macro-CTA was calculated to be 16 using 1H NMR spectroscopy by comparing the integrated signals corresponding to the CDB aromatic protons at 7.1–8.1 ppm to that assigned to the two oxymethylene protons of PLMA at 3.7–4.2 ppm.
Synthesis of Poly(lauryl methacrylate)–Poly(benzyl methacrylate) (PLMA–PBzMA) Diblock Copolymer Particles
A typical RAFT non-aqueous dispersion polymerization synthesis of PLMA16–PBzMA37 diblock copolymer worms at 20% w/w solids was carried out as follows. Benzyl methacrylate (BzMA; 0.450 g; 2.55 mmol), AIBN initiator (4.50 mg; 0.027 mmol; dissolved at 1.0% w/w in BzMA), and PLMA16 macro-CTA (0.30 g; 1.10 mmol; macro-CTA/initiator molar ratio = 5.0) were dissolved in n-dodecane (4.0 mL; 3.00 g). The reaction mixture was sealed in a 10 mL round-bottomed flask and purged with nitrogen gas for 25 min while being immersed in an ice bath to reduce solvent evaporation. The deoxygenated solution was then placed in a preheated oil bath at 70 °C for 16 h (final BzMA conversion = 98%; Mn = 10 800 g mol–1, Mw/Mn = 1.25). The same diblock copolymer formulation was scaled up for rheological studies. In further syntheses, the mean DP of the PBzMA block was systematically varied by adjusting the amount of added BzMA monomer under otherwise identical reaction conditions.
Gel Permeation Chromatography
Molecular weight distributions were assessed by gel permeation chromatography (GPC) using THF eluent. The GPC setup comprised two 5 μm (30 cm) Mixed C columns, a WellChrom K-2301 refractive index detector operating at 950 ± 30 nm, a Precision detector PD 2020 light scattering detector (with scattering angles of 90° and 15°), and a BV400RT solution viscosity detector. The mobile phase contained 2.0% v/v triethylamine and 0.05% w/v butylhydroxytoluene, and the flow rate was fixed at 1.0 mL min–1. A series of 10 near-monodisperse poly(methyl methacrylate) standards (Mp values ranging from 1280 to 330 000 g mol–1) were used for calibration.
1H NMR Spectroscopy
1H NMR spectra were recorded in either CD2Cl2 or CDCl3 using a Bruker AV1-400 or AV1-250 MHz spectrometer. Typically 64 scans were averaged per spectrum. For variable-temperature 1H NMR studies, 0.50 mL of a 20% w/w dispersion of PLMA16–PBzMA37 worms was diluted to 10% w/w using n-dodecane (0.50 mL) prior to centrifugation at 8000 rpm for 8 h. The sedimented worms were then redispersed with the aid of an ultrasonic bath using d26-dodecane (2.0 mL) to produce a 5.0% w/w dispersion. 1H NMR spectra were recorded at various temperatures ranging from 25 to 149 °C (32 scans per spectrum, delay time = 0.10 s) using a Bruker AV1-500 MHz spectrometer.
Dynamic Light Scattering
Dynamic light scattering (DLS) studies were performed using a Zetasizer Nano-ZS instrument (Malvern Instruments, UK) at a fixed scattering angle of 173°. Copolymer dispersions were diluted in n-dodecane prior to light scattering studies at 25 °C. Temperature-dependent DLS studies were performed using the same Zetasizer Nano-ZS instrument, which was equipped with a Peltier cell. Copolymer dispersions were diluted in n-dodecane and equilibrated for 5 min at 10 °C intervals in a 20 °C–90 °C–20 °C thermal cycle. In both sets of experiments, the intensity-average diameter and polydispersity of the diblock copolymer particles were calculated at a given temperature by cumulants analysis of the experimental correlation function using Dispersion Technology Software version 6.20. Data were averaged over 13 runs each of 30 s duration.
Transmission Electron Microscopy
Transmission electron microscopy (TEM) studies were conducted using a Philips CM 100 instrument operating at 100 kV and equipped with a Gatan 1 k CCD camera. Diluted block copolymer solutions (< 0.50% w/w) were placed on carbon-coated copper grids and exposed to ruthenium(IV) oxide vapor for 7 min at 20 °C prior to analysis.46 This heavy metal compound acted as a positive stain for the core-forming PBzMA block to improve contrast. The ruthenium(IV) oxide was prepared as follows: ruthenium(II) oxide (0.30 g) was added to water (50 g) to form a black slurry; addition of sodium periodate (2.0 g) with stirring produced a yellow solution of ruthenium(IV) oxide within 1 min.
Rheology Measurements
An AR-G2 rheometer equipped with a variable-temperature Peltier plate and a 40 mL 2° aluminum cone was used for all experiments. The loss and storage moduli (G″ and G′, respectively) were measured as a function of temperature at a heating rate of 1.0 °C per minute, a fixed strain of 1.0%, and an angular frequency of 10 rad s–1 so as to assess the gel strength and critical gelation temperature (CGT). During temperature sweeps, the temperature was varied at 5 °C intervals, with an equilibration time of 5 min being allowed prior to each measurement. A frequency sweep from 0.1 to 100 rad s–1 was conducted at 20 °C using a fixed strain of 1.0% and an equilibration time of 1 min between each measurement. In all cases, the sample gap was 58 μm.
Small-Angle X-ray Scattering Measurements
SAXS patterns were collected at a synchrotron source (ESRF, station BM26, Grenoble, France) using monochromatic X-ray radiation (wavelength λ = 0.1 nm, with q ranging from 0.023 to 1.3 nm–1, where q = 4π sin θ/λ is the length of the scattering vector and θ is one-half of the scattering angle) and a 2D Pilatus 1M CCD detector. Glass capillaries of 2 mm diameter were used as a sample holder. Time-resolved SAXS patterns were recorded at a rate of 2 frames per minute during thermal cycles performed on the samples (heating from 20 °C to 160 °C at a rate of 5 °C min–1, equilibrating for 5 min, and then cooling back to 20 °C at 5 °C min–1). The sample temperature was controlled by a heating/cooling capillary holding stage (Linkam Scientific Instruments Ltd., Tadworth, England). Scattering data were reduced by Nika SAS macros for Igor Pro (integration, normalization, and background subtraction) and were further analyzed using Irena SAS macros for Igor Pro.47 Glassy carbon was used for the absolute intensity calibration.48 Measurements were conducted on 1.0% w/w and 5.0% w/w dispersions of PLMA16–PBzMA37 particles in n-dodecane.
Results and Discussion
Synthesis of PLMA Macro-CTAs
RAFT solution polymerization of LMA was conducted in toluene at 70 °C. This afforded low polydispersity PLMA macro-CTAs in high yield with a mean degree of polymerization (DP) of 16, 18, or 21 (see Supporting Information Table S1). Each macro-CTA was formed using cumyl dithiobenzoate as a CTA. In all LMA polymerizations, the reaction was quenched at 73%–84% conversion, so as to avoid monomer-starved conditions and hence ensure retention of the RAFT end-groups.49 This is a prerequisite for high blocking efficiencies and hence well-defined PLMA–PBzMA diblock copolymers. Each PLMA macro-CTA had a polydispersity (Mw/Mn) of less than 1.25, which is consistent with previous studies reporting well-controlled RAFT syntheses.29
Block Copolymer Syntheses and Phase Diagram
BzMA monomer was polymerized using each of the relatively short, low polydispersity PLMA macro-CTAs (DP = 16, 18, or 21) in turn via RAFT dispersion polymerization in n-dodecane to generate a series of well-defined PLMA–PBzMA diblock copolymers at 20% w/w solids (see Supporting Information Figure S1 and Table S2). In all cases, more than 94% BzMA conversion was achieved within 16 h at 70 °C, as judged by 1H NMR spectroscopy. TEM studies indicated that spherical, worm-like, or vesicular morphologies can be accessed when chain-extending PLMA16 and PLMA18 macro-CTAs (see Figure 2). However, only spherical morphologies were obtained when using a longer PLMA21 macro-CTA. This indicates that the upper limit degree of polymerization for the stabilizer block to form higher order morphologies (i.e., either worms or vesicles) is relatively low for this PLMA–PBzMA/n-dodecane formulation. We reported similar results for the synthesis of the same diblock copolymer via RAFT dispersion polymerization in n-heptane at 90 °C.29 In this earlier study, a detailed phase diagram was constructed for a fixed PLMA stabilizer DP of 17, with variables being the mean DP of the PBzMA core-forming block and the total solids at which the syntheses were conducted. It was shown that lower concentrations (≤15% w/w solids) typically led to mixed phases, with the formation of pure phases requiring somewhat higher concentrations (>17.5% w/w). Thus, in the present work, we chose to construct a different phase diagram, whereby all syntheses were conducted at 20% w/w solids and the two variables were the mean DPs of the PLMA and PBzMA blocks. Three PLMA macro-CTAs with DPs of 16, 18, and 21 were utilized, while the DP of the PBzMA block was systematically varied from 20 to 80 (see Figure 2 and Supporting Information Table S2). As anticipated, the worm phase region is relatively narrow (e.g., less than 10 BzMA units for PLMA18–PBzMAx). The worms have relatively well-defined mean widths (10–20 nm), but are rather polydisperse in length (100–1000 nm). This suggests that the mechanism of worm formation during the RAFT polymerization of BzMA most likely involves the one-dimensional aggregation and fusion of monomer-swollen spheres. Similar observations have been reported for various other diblock copolymer worm systems in the literature.23,27,29,30,34
Figure 2.
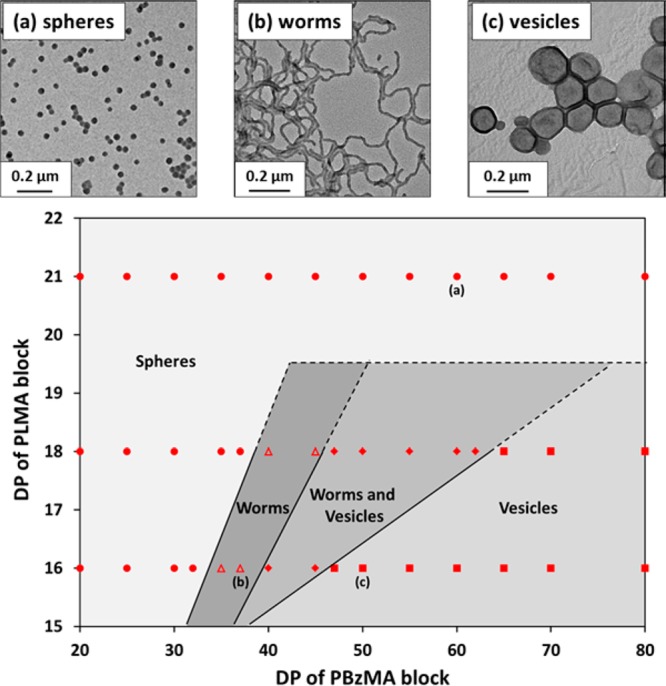
Phase diagram constructed for PLMAx–PBzMAy diblock copolymer nanoparticles prepared by RAFT dispersion polymerization of BzMA in n-dodecane at 20% w/w solids using AIBN at 70 °C (PLMA/AIBN molar ratio = 5.0). The post mortem diblock copolymer morphologies were determined by TEM. Note that higher order morphologies (worms and vesicles) can be obtained when using PLMA16 and PLMA18 macro-CTAs, but not when using the PLMA21 macro-CTA. TEM images (a), (b), and (c) correspond to examples of the three pure morphologies (spheres, worms, or vesicles), respectively, and the individual block copolymer compositions are indicated within the phase diagram.
Focusing on the worm phase boundary, we previously reported that PLMA17–PBzMAx worms were obtained in n-heptane at 20% w/w solids when x = 50–60.29 Inspecting Figure 2, the worm phase appears to be located within x = 35–40 for this hypothetical diblock composition at 20% w/w in n-dodecane. This implies a significant shift in the worm phase boundaries on switching from n-heptane to n-dodecane, which in turn suggests that each n-alkane solvent requires the construction of a detailed phase diagram to ensure reproducible targeting of pure diblock copolymer morphologies.
Tube inversion tests confirm that the worms form transparent free-standing physical gels at 20 °C, whereas the spherical and vesicular dispersions remain free-flowing, low-viscosity fluids (see Supporting Information Figure S2). Presumably, gelation is the result of multiple inter-worm interactions. These worm gels also proved to be thermo-responsive, with degelation occurring on heating the dispersion. The rest of this Article is focused on the detailed study of this unexpected thermal transition.
Characterization of Dilute Dispersions of Diblock Copolymer Worms
To examine the physical mechanism of degelation, DLS studies were conducted on a highly dilute (0.10% w/w) PLMA16–PBzMA37 worm dispersion in n-dodecane (see Figure 3). An apparent sphere-equivalent hydrodynamic diameter of 170 nm (DLS polydispersity = 0.37) was observed at 20 °C, which is consistent with the presence of worm-like particles. However, heating this dilute dispersion resulted in a dramatic reduction in the apparent hydrodynamic diameter, from 167 nm at 60 °C to around 60 nm at 90 °C. The nanoparticle polydispersity at 90 °C is also reduced to around 0.14, which is characteristic of spheres rather than worms. The dimensions of these particles remained relatively constant on cooling to 20 °C (hydrodynamic diameter = 46 nm; DLS polydispersity = 0.13), suggesting that this thermal transition is essentially irreversible when conducted in sufficiently dilute solution. TEM studies of the same dilute dispersion conducted before and after the 20–90–20 °C thermal cycle confirmed an irreversible change in morphology for the PLMA16–PBzMA37 particles (at least on a time scale of several hours). A well-defined worm morphology is observed at 20 °C before the heating cycle (see image (a) in Figure 3), whereas a predominantly spherical morphology exists at 20 °C after the heating cycle (see image (b) in Figure 3). Presumably, degelation occurs on heating once a critical fraction of worms has been converted into spheres, because this reduction in particle anisotropy inevitably leads to a catastrophic reduction in the number of inter-particle contacts.
Figure 3.
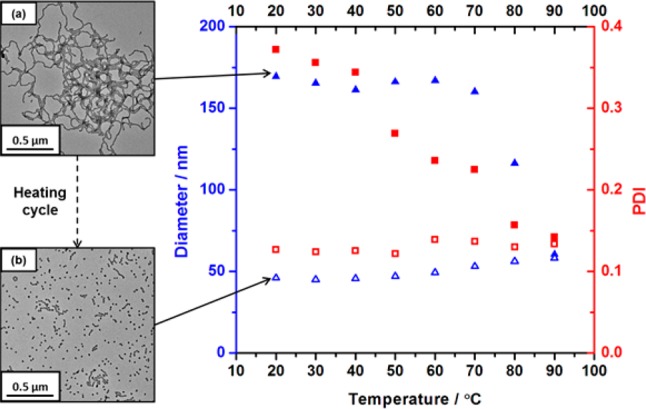
Variable-temperature dynamic light scattering (DLS) studies showing the variation of hydrodynamic diameter (blue ▲) and polydispersity (red ■) for a 0.10% w/w PLMA16–PBzMA37 dispersion in n-dodecane on heating from 20 to 90 °C (filled symbols) and on cooling from 90 to 20 °C (open symbols). Representative transmission electron microscopy (TEM) images obtained for the same dilute dispersion are shown as insets. These data confirm the irreversible nature of the worm-to-sphere transition that occurs on heating PLMA16–PBzMA37 under these conditions.
Characterization of Concentrated Dispersions of Diblock Copolymer Worms
Rheological studies were performed on a representative worm gel (20% w/w PLMA16–PBzMA37 in n-dodecane). At 20 °C, G′ was found to be relatively independent of frequency (see Supporting Information Figure S3), indicating that the worm gel can be considered a “true” gel. Additionally, variable-temperature studies were conducted to characterize the thermo-responsive behavior of this gel. The critical gelation temperature (CGT) is defined as the point at which the G′ and G″ curves cross over. On heating to 47 °C, a CGT was observed at approximately 47 °C (see Figure 4). The gel had reformed at 20 °C, with the G′ and G″ curves intersecting at approximately the same CGT during the cooling cycle. This suggests a reversible thermal transition. However, the final gel strength (G′ ≈ 87 Pa) was substantially reduced when compared to the original gel strength (G′ ≈ 2300 Pa), which indicates significant hysteresis under these conditions.
Figure 4.
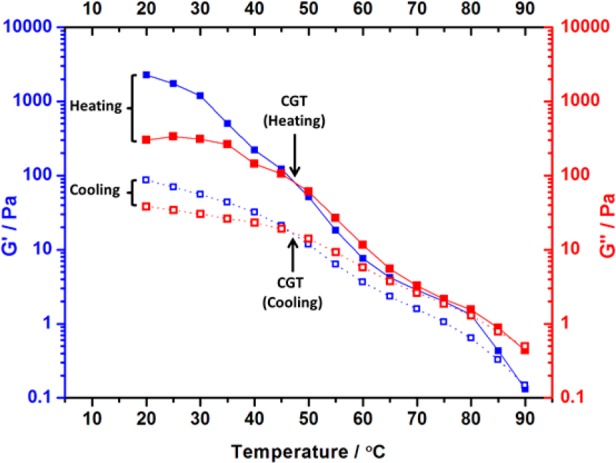
Variation of storage moduli (G′, blue squares) and loss moduli (G″, red squares) for a PLMA16–PBzMA37 worm gel at 20% w/w during a thermal cycle of heating from 20 to 90 °C (filled symbols) and cooling from 90 to 20 °C (open symbols) at a rate of 1.0 °C per minute. Data were recorded at 1.0% strain using an angular frequency of 10 rad s–1 and an equilibration time of 5 min between each measurement.
It is perhaps noteworthy that the critical temperature (>70 °C) required for the worm-to-sphere transition suggested by the DLS studies shown in Figure 3 is somewhat higher than that indicated by the rheological studies (CGT ≈ 47 °C) shown in Figure 4. It is possible that this discrepancy may be simply the result of the differing copolymer concentrations (0.10% w/w for DLS vs 20% w/w for rheology). However, the mean particle size reported by DLS will only decrease significantly once almost all of the worms have been converted into spheres, because the scattered light intensity is proportional to the sixth power of the particle dimensions. In contrast, it is likely that only a minor fraction of worms need to be converted into spheres to cause degelation in the rheology studies. Thus, these two techniques are understandably sensitive to different stages of the worm-to-sphere transition.
Systematic dilution of a 20% w/w PLMA16–PBzMA37 worm gel in n-dodecane was undertaken to estimate the critical gelation concentration (CGC). This dispersion failed a tube inversion test at approximately 11% w/w, indicating degelation at this concentration. This is significantly higher than the CGC of approximately 3–4% w/w observed for aqueous diblock copolymer worm gels reported by Verber et al.45 One possible explanation for this unexpected difference is that the aqueous worms may have a significantly greater mean contour length than the worms described herein.50,51
TEM studies of the original 20% w/w PLMA16–PBzMA37 worm gel (after dilution of this dispersion to 0.01% w/w using n-dodecane) reveal a pure worm phase at 20 °C, as expected (see Figure 5A). Heating this 20% w/w worm gel to 90 °C causes degelation, indicating a worm-to-sphere transition.52 Cooling this concentrated dispersion to 20 °C led to regelation. After dilution to 0.01% w/w solids, TEM studies confirmed the reformation of worms, albeit with a minor population of spheres (see Figure 5B). Thus, the worm-to-sphere thermal transition appears to be reasonably reversible for concentrated copolymer dispersions, in marked contrast to the irreversible behavior observed for highly dilute dispersions (see Figure 3). In unpublished work, we have recently observed a similarly strong concentration dependence for aqueous diblock copolymer worm gels/dispersions. Presumably, this reflects the highly cooperative nature of the sphere-to-worm transition, which requires the self-assembly of many spheres to form a single worm. Such multiple fusion events are much less likely to occur for highly dilute dispersions, at least within normal experimental time scales (hours/days).
Figure 5.

TEM images recorded for a 20% w/w PLMA16–PBzMA37 worm gel. Initially, worms are observed at 20 °C on dilution to 0.01% w/w solids [see image (A)]. On heating this 20% w/w gel to 90 °C, degelation occurs. Allowing the hot 20% w/w worm gel to cool from 90 to 20 °C (followed by dilution to 0.01% w/w solids prior to preparing a TEM grid) reveals mainly a worm phase [see image (B)], with a minor population of isolated spheres (see red arrows).
Variable-temperature 1H NMR spectroscopy studies were undertaken using d26-dodecane to examine the molecular basis for the worm-to-sphere transition. On heating above 70 °C, the aromatic signals assigned to the BzMA residues at 6.7–7.4 ppm become increasingly intense relative to the two oxyethylene protons assigned to the PLMA block at 4.0 ppm (see Figure 6). This spectral change proved to be reversible on cooling and suggests solvent plasticization of the core-forming block at elevated temperature. Similar effects have been reported by Price and co-workers for polystyrene-core diblock copolymer micelles dispersed in n-octane.53 The same workers also reported an irreversible worm-to-sphere transition on heating a dilute dispersion of polystyrene–polyisoprene diblock copolymer worms in N,N′-dimethylacetamide.1 However, no attempt was made to study this thermal transition for relatively concentrated copolymer gels/dispersions, where the change in morphology becomes more or less reversible (as in the present work).
Figure 6.
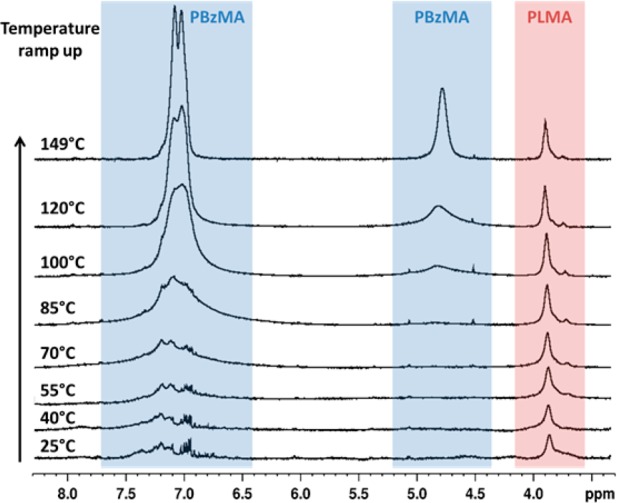
Variable-temperature 1H NMR spectra recorded for 5.0% w/w PLMA16–PBzMA37 diblock copolymer worms in d26-dodecane.
Thus, these variable-temperature 1H NMR studies confirm that the PBzMA block becomes partially plasticized in hot d26-dodecane, which must cause a subtle shift in the relative volume fractions of the stabilizer and core-forming blocks. Because the worm phase occupies only a narrow phase region, this in turn induces a morphological transition. However, these observations immediately suggest an apparent paradox. Assuming a constant degree of solvation for the PLMA stabilizer chains, greater solvation of the core-forming PBzMA block due to its plasticization would be expected to increase its relative volume fraction, and so reduce the molecular curvature of the copolymer chains. This should produce a worm-to-vesicle transition, rather than the worm-to-sphere transition that is actually observed (see Figure 3). We believe that this apparent discrepancy can be explained as follows. The partial solvation of the core-forming PBzMA block indicated by the variable-temperature 1H NMR studies most likely involves surface plasticization of the sterically stabilized worms. Thus, the ingress of solvent into the worms leads to those segments of the core-forming PBzMA block that are closest to the PLMA stabilizer chains becoming preferentially solvated. This causes an increase in the effective stabilizer block DP (and a concomitant reduction in the effective core-forming block DP) and hence higher molecular curvature for the diblock copolymer chains. This hypothesis is physically realistic and accounts for the observed worm-to-sphere transition; it is supported by 1H NMR studies of a polystyrene-based diblock copolymer in d18-octane reported by Heatley and co-workers.54 [N.B. Surface plasticization can also be invoked to account for the worm-to-sphere transition observed for the aqueous worm gel formulation previously reported by Blanazs et al.44]
More recently, LaRue and co-workers used a combined theoretical and experimental approach to explore the thermally induced worm-to-sphere transition exhibited by a polystyrene–polyisoprene diblock copolymer in n-heptane, which is a selective solvent for the polyisoprene block.55 Static light scattering (SLS) and atomic force microscopy (AFM) were used to characterize this transition, which was described as “reversible”. However, although the worm-to-sphere transition induced by heating from 25 to 35 °C was relatively fast, the reported SLS data actually showed that the sphere-to-worm transition was still substantially incomplete even after 36 days at 25 °C. Bearing in mind the DLS studies reported in the present work, this is almost certainly because of the dilute solution conditions (<0.10%) required for the SLS experiments. In contrast, our rheology studies summarized in Figure 4 were performed at 20% w/w and indicate relatively good reversibility for the worm-to-sphere-to-worm transition exhibited by PLMA16–PBzMA37 on much shorter time scales (minutes). More specifically, the CGT values observed for the heating and cooling cycles are very similar, but a somewhat weaker worm gel is obtained (final G′ ≈ 102 Pa, as compared to an initial G′ of ∼103 Pa).
SAXS is a powerful technique for characterizing the morphologies of various types of colloidal dispersions.44,56−58 It is statistically much more robust than TEM, because the X-ray scattering data are averaged over millions of particles in solution. Moreover, unlike SLS and DLS, SAXS can be performed at relatively high copolymer concentrations.44 Accordingly, a series of absolute intensity SAXS measurements were recorded for PLMA16–PBzMA37 copolymer particles at both 1.0 and 5.0% w/w.
In a typical I(q) versus q plot, the low q (Guinier) region is particularly useful for assessing the particle morphology. A gradient of zero indicates a spherical morphology, whereas a negative gradient of unity is obtained for rods.59 In the present study, the PLMA16–PBzMA37 worms are highly anisotropic but relatively flexible, so a negative gradient close to (but less than) unity is expected. Previously, Blanazs et al.44 utilized SAXS to study the thermally induced worm-to-sphere transition for PGMA–PHPMA diblock copolymer nanoparticles in aqueous solution and observed excellent reversibility at a copolymer concentration of 10% w/w.
SAXS patterns obtained for PLMA16–PBzMA37 diblock copolymer nano-objects in n-dodecane as a function of temperature are shown in Figure 7A. At 20 °C, the scattering pattern obtained at 5.0% w/w copolymer has a negative gradient slightly lower than unity in the low q region and also a local minimum in intensity at high q (∼0.50 nm–1), indicating a mean worm cross-section of approximately 13 nm. Heating to 160 °C leads to a substantial change in this SAXS pattern: the gradient at low q tends to zero, and the feature at q ≈ 0.50 nm–1 disappears. On returning to 20 °C, a negative gradient at low q is again observed (albeit marginally lower than the original gradient), and the minimum at q ≈ 0.50 nm–1 is almost completely recovered.
Figure 7.
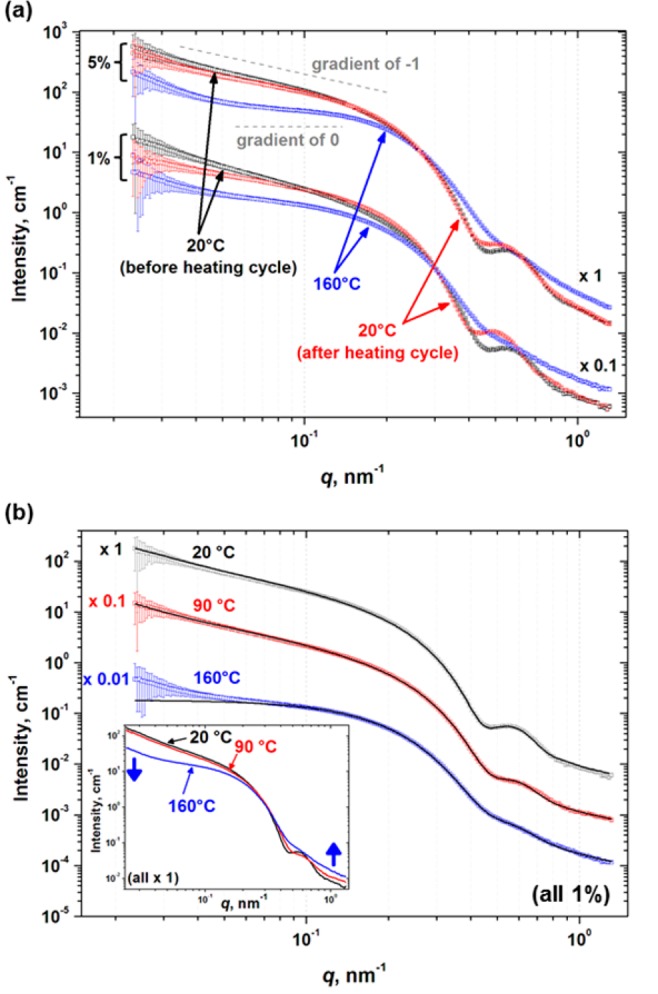
(a) Representative SAXS patterns recorded for 5.0 and 1.0% w/w PLMA16–PBzMA37 copolymer dispersions in n-dodecane recorded during a 20–160–20 °C thermal cycle. The 1.0% w/w data are offset by a factor of 0.1 for clarity. Gradients of zero and negative unity (dashed gray lines) are also shown as a guide to the eye. (b) Representative SAXS patterns (symbols) for the same 1.0% w/w PLMA16–PBzMA37 copolymer dispersion in n-dodecane recorded during heating from 20 °C to 160 °C. In this case, SAXS patterns are offset a factor of 0.1 (90 °C) and 0.01 (160 °C) for clarity, fits to the data (solid lines) are shown, and the results of this analysis are summarized in Table 1. The inset shows all three scattering patterns plotted on the same scale.
These observations suggest fairly good reversibility for the worm-to-sphere-to-worm transition over the experimental time scale (hours). In contrast, the same thermal cycle conducted at 1.0% w/w provides good evidence for the same worm-to-sphere transition, albeit accompanied by a significant reduction in reversibility (as judged by the less steep gradient at low q and a small but discernible shift in the local minimum at high q; see lower set of three SAXS patterns shown in Figure 7A).
Notwithstanding the above qualitative observations, detailed SAXS analysis requires relatively low copolymer concentrations to avoid inter-particle interactions, which suppress the scattering intensity at low q.60,61 In practice, the scattering patterns can be satisfactorily fitted at a copolymer concentration of 1.0% w/w using an established model62,63 for worm-like micelles (see the Supporting Information for further details).
Figure 7B shows SAXS data obtained for 1.0% w/w PLMA16–PBzMA37 particles on heating from 20 °C to 160 °C. At 20 °C, the scattering pattern can be analyzed using the aforementioned worm model. Table 1 shows the key parameters obtained from the data fit using this model. Notably, the total worm cross-section is 15.4 nm and the worm contour length exceeds 600 nm, indicating highly anisotropic particles with a mean aspect ratio of more than 39; see Table 1. These values correlate well with TEM observations (see Figure 5A). Another interesting observation is that these PLMA16–PBzMA37 worms are relatively stiff, because their Kuhn length (160 nm) is an order of magnitude greater than the total worm cross-section. This is consistent with the bottle brush-like structure of the PLMA stabilizer block. Furthermore, both 1H NMR and SAXS studies indicate little or no solvation of the worm core-forming PBzMA block at 20 °C (see Figure 6 and Table 1, final column). The radius of gyration of 1.3 nm for the coronal PLMA block indicated by SAXS is close to that estimated on the basis of the mean DP of the PLMA block (∼1.0 nm; see the Supporting Information). On heating to 90 °C, the mean worm cross-section does not vary (although the minimum at q ≈ 0.50 nm–1 virtually disappears). However, the worm contour length is reduced from more than 600 to ∼350 nm, indicating partial worm disintegration. At this elevated temperature, the degree of core solvation (xsol) increases to 0.29, suggesting ingress of n-dodecane into the PBzMA cores (also indicated by the 1H NMR studies shown in Figure 6). On further heating to 160 °C, the SAXS pattern can now only be satisfactorily fitted using parameters that approximate to isotropic particles (i.e., a total worm cross-section = 15.1 nm, worm contour length = 17.3 nm, and worm Kuhn length = 16.8 nm). This indicates that the vast majority of particles in the dispersion are now actually spherical micelles, with only a relatively minor population of worms remaining at 160 °C (as indicated by the upturn in the scattering curve at very low q). At this temperature, the degree of solvation in the cores of these copolymer particles is relatively high (xsol = 0.48), which supports our hypothesis that surface plasticization of the PBzMA core-forming block is a key factor for the conversion of worms into spheres.
Table 1. Structural Parameters Obtained from Data Fitting to SAXS Patterns Recorded for 1.0% w/w PLMA16–PBzMA37 Diblock Copolymer Particles in n-Dodecane at Various Temperatures: Mean Worm Core Cross-Section (2Rsw), Total Worm Cross-Section [2(Rsw + Rg)], Worm Contour Length (Lw), Aspect Ratio [Lw/2(Rsw + Rg)], Worm Kuhn Length (bw), and Solvent Fraction in the Core of the Particle (xsol).
| temp/°C | mean worm core cross-section (2Rsw)/nm | total worm cross-sectiona [2(Rsw + Rg)]/nm | worm contour length (Lw)/nm | aspect ratio [Lw/2(Rsw + Rg)] | worm Kuhn length (bw)/nm | solvent fraction in particle corec (xsol) |
|---|---|---|---|---|---|---|
| 20 | 12.8 ± 1.7 | 15.4 ± 1.8 | >600b | >39.0 | 160 ± 20 | ∼0 |
| 90 | 12.0 ± 1.9 | 14.7 ± 2.0 | 350 ± 48 | 23.8 ± 6.5 | 143 ± 7 | 0.29 |
| 160 | 12.5 ± 2.8 | 15.1 ± 2.9 | 17.3 ± 0.5 | 1.15 ± 0.25 | 16.8 ± 1.7 | 0.48 |
The radius of gyration of the PLMA corona block (Rg) was found to be 1.3 nm in all cases.
The q range available for worm contour length analysis means that a reliable upper limit value cannot be calculated.
Uncertainties are smaller than the number of significant figures quoted.
In principle, the PLMA16–PBzMA37 worms could either undergo “sequential budding” of spheres or a series of “random worm cleavage” events (see routes A and B in Figure 8). However, the SAXS observations described above combined with the appearance of a minor population of spheres in Figure 5B suggest that the former mechanism is more likely to be prevalent.
Figure 8.
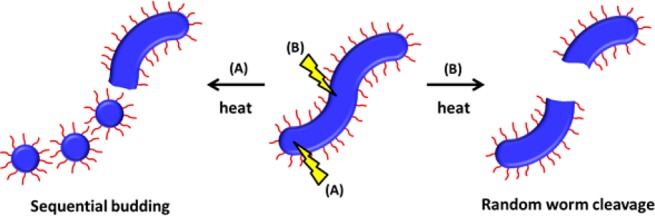
Schematic representation of two possible mechanisms for the worm-to-sphere transition that occurs on heating a dispersion of PLMA16–PBzMA37 diblock worms in n-dodecane: (A) sequential budding and (B) random worm cleavage.
It is emphasized that the worm-to-sphere transition described herein, which occurs on heating a worm dispersion in an n-alkane solvent, is wholly complementary to that previously reported for aqueous worms, which form spheres on cooling. This is presumably related to the well-known upper critical solution temperature (UCST) effect for hydrophobic polymers in organic solvents (e.g., polystyrene in cyclohexane64), as opposed to the lower critical solution temperature (LCST) effect that is widely reported for many non-ionic water-soluble polymers in aqueous solution.8,65−68 Finally, in view of the scalable nature of RAFT polymerization chemistry,69,70 such thermo-sensitive block copolymer formulations may offer potential applications as viscosity modifiers (thickeners) or lubricants for next-generation engine oils.71
Conclusions
In summary, PLMA–PBzMA diblock copolymer spheres, worms, or vesicles can be readily prepared via polymerization-induced self-assembly at 20% w/w solids in n-dodecane at 70 °C, provided that the mean degree of polymerization of the PLMA stabilizer block is relatively low. The worms form free-standing gels at 20 °C, but undergo degelation on heating via a worm-to-sphere order–order transition. Variable-temperature 1H NMR and SAXS studies indicate that this is the result of a subtle change in the relative volume fractions occupied by the stabilizer and core-forming blocks caused by surface plasticization of the core-forming block. This thermally induced change in copolymer morphology is essentially irreversible on an experimental time scale of hours when conducted in highly dilute solution (∼0.1% w/w), as judged by DLS and TEM studies. This is because the self-assembly of each worm from the fusion of multiple spheres is highly inefficient under these conditions. In contrast, much more reversible behavior is observed at higher copolymer concentrations (5.0–20% w/w), as judged by 1H NMR spectroscopy, TEM, gel rheology, and SAXS studies. However, the latter technique indicates a significant reduction in mean contour length for the reformed worms, which is consistent with the reduced gel strength observed for the reconstituted worm gel as judged by gel rheology.
Acknowledgments
Susan Bradshaw is thanked for her assistance with the variable-temperature 1H NMR studies. EPSRC (EP/J007846/1) is acknowledged for providing postdoctoral support for L.A.F., and The University of Sheffield is thanked for funding M.J.D. S.P.A. also acknowledges an ERC Advanced Investigator grant (PISA 320372). We are grateful to ESRF for providing synchrotron beam-time and thank the personnel of BM26 for their assistance.
Supporting Information Available
Gel permeation chromatograms, digital photographs of a selection of block copolymer dispersions, a rheology frequency sweep, summary tables for various (co)polymers, and a description of the SAXS worm model. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Price C.; Chan E. K. M.; Hudd A. L.; Stubbersfield R. B. Polym. Commun. 1986, 27, 196. [Google Scholar]
- Discher D. E.; Eisenberg A. Science 2002, 297, 967. [DOI] [PubMed] [Google Scholar]
- Xu R. L.; Winnik M. A.; Hallett F. R.; Riess G.; Croucher M. D. Macromolecules 1991, 24, 87. [Google Scholar]
- Zhang L. F.; Eisenberg A. Science 1995, 268, 1728. [DOI] [PubMed] [Google Scholar]
- Zhang L. F.; Yu K.; Eisenberg A. Science 1996, 272, 1777. [DOI] [PubMed] [Google Scholar]
- Mai Y. Y.; Eisenberg A. Chem. Soc. Rev. 2012, 41, 5969. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Pickard S.; Deng N. J.; Barlow R. J.; Attwood D.; Booth C. Macromolecules 1994, 27, 2371. [Google Scholar]
- Booth C.; Attwood D. Macromol. Rapid Commun. 2000, 21, 501. [Google Scholar]
- Discher B. M.; Won Y. Y.; Ege D. S.; Lee J. C. M.; Bates F. S.; Discher D. E.; Hammer D. A. Science 1999, 284, 1143. [DOI] [PubMed] [Google Scholar]
- Jain S.; Bates F. S. Science 2003, 300, 460. [DOI] [PubMed] [Google Scholar]
- Won Y. Y.; Davis H. T.; Bates F. S. Science 1999, 283, 960. [DOI] [PubMed] [Google Scholar]
- Lee J. C. M.; Bermudez H.; Discher B. M.; Sheehan M. A.; Won Y. Y.; Bates F. S.; Discher D. E. Biotechnol. Bioeng. 2001, 73, 135. [DOI] [PubMed] [Google Scholar]
- Qin S. H.; Geng Y.; Discher D. E.; Yang S. Adv. Mater. 2006, 18, 2905. [Google Scholar]
- Bütün V.; Armes S. P.; Billingham N. C. Polymer 2001, 42, 5993. [Google Scholar]
- Howse J. R.; Jones R. A. L.; Battaglia G.; Ducker R. E.; Leggett G. J.; Ryan A. J. Nat. Mater. 2009, 8, 507. [DOI] [PubMed] [Google Scholar]
- Antonietti M.; Förster S. Adv. Mater. 2003, 15, 1323. [Google Scholar]
- Blanazs A.; Armes S. P.; Ryan A. J. Macromol. Rapid Commun. 2009, 30, 267. [DOI] [PubMed] [Google Scholar]
- Wang X. S.; Guerin G.; Wang H.; Wang Y. S.; Manners I.; Winnik M. A. Science 2007, 317, 644. [DOI] [PubMed] [Google Scholar]
- Gadt T.; Ieong N. S.; Cambridge G.; Winnik M. A.; Manners I. Nat. Mater. 2009, 8, 144. [DOI] [PubMed] [Google Scholar]
- Petzetakis N.; Dove A. P.; O’Reilly R. K. Chem. Sci. 2011, 2, 955. [Google Scholar]
- Kouwer P. H. J.; Koepf M.; Le Sage V. A. A.; Jaspers M.; van Buul A. M.; Eksteen-Akeroyd Z. H.; Woltinge T.; Schwartz E.; Kitto H. J.; Hoogenboom R.; Picken S. J.; Nolte R. J. M.; Mendes E.; Rowan A. E. Nature 2013, 493, 651. [DOI] [PubMed] [Google Scholar]
- Li Y.; Armes S. P. Angew. Chem., Int. Ed. 2010, 49, 4042. [DOI] [PubMed] [Google Scholar]
- Blanazs A.; Madsen J.; Battaglia G.; Ryan A. J.; Armes S. P. J. Am. Chem. Soc. 2011, 133, 16581. [DOI] [PubMed] [Google Scholar]
- Sugihara S.; Blanazs A.; Armes S. P.; Ryan A. J.; Lewis A. L. J. Am. Chem. Soc. 2011, 133, 15707. [DOI] [PubMed] [Google Scholar]
- Blanazs A.; Ryan A. J.; Armes S. P. Macromolecules 2012, 45, 5099. [Google Scholar]
- Jones E. R.; Semsarilar M.; Blanazs A.; Armes S. P. Macromolecules 2012, 45, 5091. [Google Scholar]
- Semsarilar M.; Jones E. R.; Blanazs A.; Armes S. P. Adv. Mater. 2012, 24, 3378. [DOI] [PubMed] [Google Scholar]
- Semsarilar M.; Ladmiral V.; Blanazs A.; Armes S. P. Langmuir 2012, 28, 914. [DOI] [PubMed] [Google Scholar]
- Fielding L. A.; Derry M. J.; Ladmiral V.; Rosselgong J.; Rodrigues A. M.; Ratcliffe L. P. D.; Sugihara S.; Armes S. P. Chem. Sci. 2013, 4, 2081. [Google Scholar]
- Zhang X.; Rieger J.; Charleux B. Polym. Chem. 2012, 3, 1502. [Google Scholar]
- An Z.; Shi Q.; Tang W.; Tsung C.-K.; Hawker C. J.; Stucky G. D. J. Am. Chem. Soc. 2007, 129, 14493. [DOI] [PubMed] [Google Scholar]
- Boisse S.; Rieger J.; Belal K.; Di-Cicco A.; Beaunier P.; Li M.-H.; Charleux B. Chem. Commun. 2010, 46, 1950. [DOI] [PubMed] [Google Scholar]
- Rieger J.; Grazon C.; Charleux B.; Alaimo D.; Jerome C. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 2373. [Google Scholar]
- Zhang X.; Boisse S.; Zhang W.; Beaunier P.; D’Agosto F.; Rieger J.; Charleux B. Macromolecules 2011, 44, 4149. [Google Scholar]
- Zhang X. W.; Boisse S.; Bui C.; Albouy P. A.; Brulet A.; Li M. H.; Rieger J.; Charleux B. Soft Matter 2012, 8, 1130. [Google Scholar]
- Wan W.-M.; Pan C.-Y. Polym. Chem. 2010, 1, 1475. [Google Scholar]
- Wan W.-M.; Sun X.-L.; Pan C.-Y. Macromol. Rapid Commun. 2010, 31, 399. [DOI] [PubMed] [Google Scholar]
- Huang C.-Q.; Pan C.-Y. Polymer 2010, 51, 5115. [Google Scholar]
- Huang C.-Q.; Wang Y.; Hong C.-Y.; Pan C.-Y. Macromol. Rapid Commun. 2011, 32, 1174. [DOI] [PubMed] [Google Scholar]
- Cai W.; Wan W.; Hong C.; Huang C.; Pan C. Soft Matter 2010, 6, 5554. [Google Scholar]
- Monteiro M. J.; Cunningham M. F. Macromolecules 2012, 45, 4939. [Google Scholar]
- Charleux B.; Delaittre G.; Rieger J.; D’Agosto F. Macromolecules 2012, 45, 6753. [Google Scholar]
- Houillot L.; Bui C.; Save M.; Charleux B.; Farcet C.; Moire C.; Raust J. A.; Rodriguez I. Macromolecules 2007, 40, 6500. [Google Scholar]
- Blanazs A.; Verber R.; Mykhaylyk O. O.; Ryan A. J.; Heath J. Z.; Douglas C. W. I.; Armes S. P. J. Am. Chem. Soc. 2012, 134, 9741. [DOI] [PubMed] [Google Scholar]
- Verber R.; Blanazs A.; Armes S. P. Soft Matter 2012, 8, 9915. [Google Scholar]
- Trent J. S. Macromolecules 1984, 17, 2930. [Google Scholar]
- Ilavsky J.; Jemian P. R. J. Appl. Crystallogr. 2009, 42, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Ilavsky J.; Long G.; Quintana J. G.; Allen A.; Jemian P. Metall. Mater. Trans. A 2010, 41, 1151. [Google Scholar]
- Cacioli P.; Hawthorne D. G.; Laslett R. L.; Rizzardo E.; Solomon D. H. J. Macromol. Sci. Chem. 1986, A23, 839. [Google Scholar]
- Otten R. H. J.; van der Schoot P. J. Chem. Phys. 2011, 134, 094902. [DOI] [PubMed] [Google Scholar]
- Chatterjee A. P. J. Chem. Phys. 2010, 132, 224905. [DOI] [PubMed] [Google Scholar]
- We attempted to prepare TEM grids directly from the hot 0.10% w/w dispersion at 90 °C. However, this temperature is well above the glass transition (Tg) of the core-forming PBzMA block (Tg ≈ 55 °C), so only extensive film formation was observed and no copolymer morphology was discernible.
- Candau F.; Heatley F.; Price C.; Stubbersfield R. B. Eur. Polym. J. 1984, 20, 685. [Google Scholar]
- Godward J.; Heatley F.; Price C. J. Chem. Soc., Faraday Trans. 1993, 89, 3471. [Google Scholar]
- LaRue I.; Adam M.; Pitsikalis M.; Hadjichristidis N.; Rubinstein M.; Sheiko S. S. Macromolecules 2005, 39, 309. [Google Scholar]
- Balmer J. A.; Mykhaylyk O. O.; Armes S. P.; Fairclough J. P. A.; Ryan A. J.; Gummel J.; Murray M. W.; Murray K. A.; Wiliams N. S. J. J. Am. Chem. Soc. 2011, 133, 826. [DOI] [PubMed] [Google Scholar]
- Balmer J. A.; Mykhaylyk O. O.; Fairclough J. P. A.; Ryan A. J.; Armes S. P.; Murray M. W.; Murray K. A.; Williams N. S. J. J. Am. Chem. Soc. 2010, 132, 2166. [DOI] [PubMed] [Google Scholar]
- Warren N. J.; Mykhaylyk O. O.; Mahmood D.; Ryan A. J.; Armes S. P. J. Am. Chem. Soc. 2013, 136, 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatter O.; Kratky O.. Small-angle X-ray Scattering; Academic Press: London, 1982. [Google Scholar]
- Garamus V. M.; Pedersen J. S.; Kawasaki H.; Maeda H. Langmuir 2000, 16, 6431. [Google Scholar]
- Arleth L.; Bergström M.; Pedersen J. S. Langmuir 2002, 18, 5343. [Google Scholar]
- Pedersen J. J. Appl. Crystallogr. 2000, 33, 637. [Google Scholar]
- Pedersen J. S.; Schurtenberger P. Macromolecules 1996, 29, 7602. [Google Scholar]
- Brandrup J.; Immergut E. H.. Polymer Handbook, 2nd ed.; John Wiley & Sons: New York, 1975. [Google Scholar]
- Wang X. H.; Qiu X. P.; Wu C. Macromolecules 1998, 31, 2972. [Google Scholar]
- Forder C.; Patrickios C. S.; Armes S. P.; Billingham N. C. Macromolecules 1996, 29, 8160. [Google Scholar]
- Li H.; Yu G. E.; Price C.; Booth C.; Hecht E.; Hoffmann H. Macromolecules 1997, 30, 1347. [Google Scholar]
- Bütün V.; Lowe A. B.; Billingham N. C.; Armes S. P. J. Am. Chem. Soc. 1999, 121, 4288. [Google Scholar]
- Price D.; Mosier P. E.; Vilardo J. S.; Baum M.. Antiwear polymer and lubricating composition thereof. Patent US8507422 B2, 2008.
- Beebe R. L.; Brzytwa A. J.; Hilker C. D.; Johnson J. R.; Yodice R.. Process for making substituted trithiocarbonate derivatives. Patent WO2009035793 A1, 2008.
- Zheng R.; Liu G.; Devlin M.; Hux K.; Jao T.-C. Tribol. Trans. 2010, 53, 97. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.