

Abstract
A working hypothesis for the pathogenesis of myotonic dystrophy type 1 (DM1) involves the aberrant sequestration of an alternative splicing regulator, MBNL1, by expanded CUG repeats, r(CUG)exp. It has been suggested that a reversal of the myotonia and potentially other symptoms of the DM1 disease can be achieved by inhibiting the toxic MBNL1-r(CUG)exp interaction. Using rational design, we discovered an RNA-groove binding inhibitor (ligand 3) that contains two triaminotriazine units connected by a bisamidinium linker. Ligand 3 binds r(CUG)12 with a low micromolar affinity (Kd = 8 ± 2 μM) and disrupts the MBNL1-r(CUG)12 interaction in vitro (Ki = 8 ± 2 μM). In addition, ligand 3 is cell and nucleus permeable, exhibits negligible toxicity to mammalian cells, dissolves MBNL1-r(CUG)exp ribonuclear foci, and restores misregulated splicing of IR and cTNT in a DM1 cell culture model. Importantly, suppression of r(CUG)exp RNA-induced toxicity in a DM1 Drosophila model was observed after treatment with ligand 3. These results suggest ligand 3 as a lead for the treatment of DM1.
Introduction
The genetic origin and overall pathogenesis of myotonic dystrophy type 1 (DM1) was established more than two decades ago.1−4 Yet this multisystemic neuromuscular disease remains incurable despite the detailed understanding of its mechanism accumulated over the intervening years.5,6 What is known definitively is that DM1 originates in a progressive expansion of an unstable CTG triplet repeat in the 3′-untranslated region of the dystrophia myotonica protein kinase (DMPK) gene.1 Healthy individuals have <37 CTG repeats, whereas DM1 patients carry between 50 and many thousands of repeating units.1 The transcribed expanded CUG repeats, r(CUG)exp, have a toxic gain-of-function that affects the level of two alternative splicing regulators, muscleblind-like 1 (MBNL1) and CUG-binding protein 1 (CUGBP1).7 Thus, a decrease in free MBNL1 levels results from the protein sequestration by the r(CUG)exp transcript leading to formation of ribonuclear foci.8,9 The mechanism for the increased CUGBP1 level remains unclear.10 Other possible pathogenic mechanisms include the repeat-associated non-ATG-initiated (RAN) translation of the expanded RNA transcript11 and the dysregulation of various miRNAs.12
The studies described above suggest that DM1 may arise through a multimodal pathogenic mechanism. However, it is known that over 80% of the misregulated splicing events are directly related to the r(CUG)exp sequestration of MBNL1 in a DM1 mouse model.13 Thus, a promising approach to restore MBNL1 activity is to target the r(CUG)exp transcript thereby inhibiting its binding of MBNL1. This approach has shown success using antisense oligonucleotides (ASOs),14−16 peptoid-based oligomers,17−19 and small molecules.20−23 We reported a very different strategy that used Berglund’s reported X-ray structure of r(CUG)6 to rationally design a highly selective class of CUG ligands based on the triaminotriazine moiety, that may form base triplets with U–U mismatches.24,25 This heterocyclic, Janus-wedge recognition unit was linked to an acridine intercalator to provide a hydrophobic driving force for RNA binding (see 1, Figure 1). Ligand 1 showed highly selective, nanomolar affinity to oligonucleotides containing rCUG sequences24 and studies with analogues provided support for the Janus-wedge binding model.26 However, ligand 1 was poorly water-soluble, did not enter cells, and exhibited an unacceptably high level of cytotoxicity. Improvements in each of these areas were possible,27 and the inhibitory potency could be increased through dimerization.28 Nonetheless, the inherent cytotoxicity of the intercalator unit was a concern in comparison to other reported small molecule inhibitors (e.g., those in Figure 1).
Figure 1.
Structures of ligand 1, pentamidine, and a Hoechst-like compound (H1).
Herein, we describe a new structure-based approach that has led to a novel class of groove-binding ligands carrying two triaminotriazine units (e.g., 2–4, Figure 2). Whereas ligand 1 bound a single U–U mismatch, the new ligands are capable of targeting as many as three consecutive CUG units. The most potent inhibitor (ligand 3) exhibits negligible cytotoxicity yet dissolves MBNL1-r(CUG)exp ribonuclear foci and partially restores the cardiac troponin T (cTNT) and insulin receptor (IR) splicing defects in a DM1 cell culture model. More significantly, it suppresses the CUG-induced toxicity in a DM1 Drosophila model.
Figure 2.
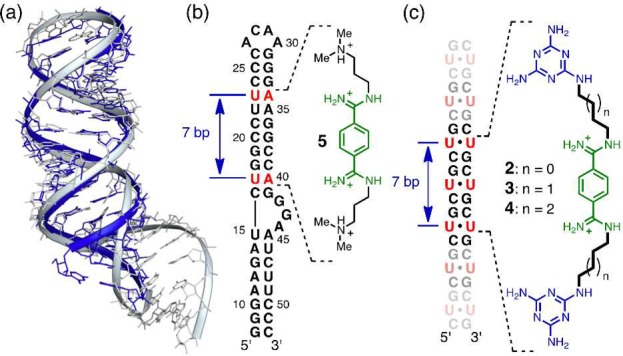
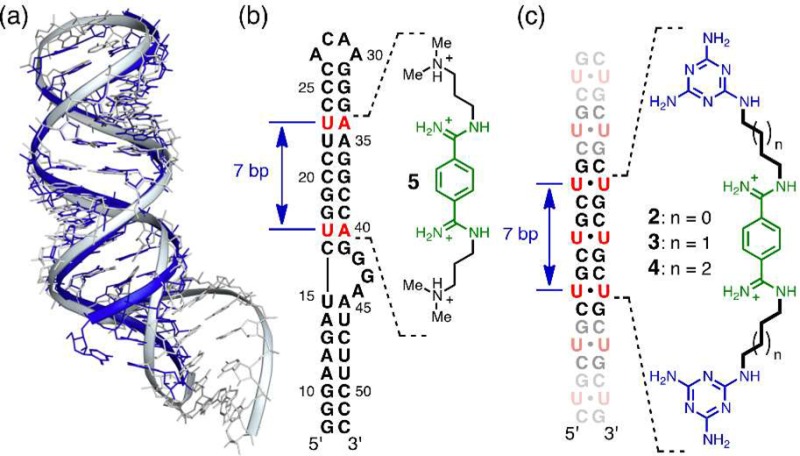
Design principles of the groove-binding ligands. (a) Superposition of the structures of the r(CUG)6 duplex (blue; PDB: 3GM7) and the HIV-1 FS RNA (gray; PDB: 2L94). (b) Schematic showing the ligand 5 binding site on the HIV-1 FS RNA sequence. (c) The proposed binding for the groove-binding ligands (2–4) on the CUG sequence.
Methods
Compounds, Materials, and General Methods
All compounds described herein gave NMR and mass spectral data in accord with their structures. The preparation of ligand 3 is representative and described below. The preparation of other compounds is described in the Supporting Information along with general methods used for their preparation and characterization. Details of the molecular dynamics simulations, isothermal titration calorimetry, and in vitro MBNL1-CUG inhibition experiments are also contained in the Supporting Information.
Synthesis of Ligand 3
A white suspension of 1.11 g (3.79 mmol) of diethyl terephthalimidate hydrochloride29 in 50 mL of anhydrous EtOH was cooled in an ice-water bath under a nitrogen atmosphere. To the suspension was added 1.10 mL (7.89 mmol) of Et3N to produce a colorless clear solution. A solution of 1.54 g (7.81 mmol) of N2-(4-aminobutyl)-1,3,5-triazine-2,4,6-triamine24 in 5 mL of ethylene glycol was added dropwise using a pipet. The reaction mixture was allowed to warm to 25 °C slowly and stirred for 18 h. The white suspension was filtered and washed with EtOH (20 mL). The white solid was purified by column chromatography on silica gel eluting with a starting gradient of CH2Cl2:n-BuOH:MeOH mixture (gradient from 1:5:4 to 1:3:6). Once the impurities were removed, the eluent was acidified with a gradient of 0.10–0.15 mL of 4 M HCl in dioxane (per liter of eluent) to afford the desired product. The product-containing fractions were combined, filtered, and concentrated in vacuo to afford 1.95 g (77%) of product as a white tetra·HCl salt (mp >230 °C (decomp.)). Rf(AcOH:H2O:MeOH = 3:6:1) = 0.30. 1H NMR (DMSO-d6): 10.25 (s, 2 H, NH), 9.81 (s, 2 H, NH), 9.43 (s, 2 H, NH), 7.98 (s, 4 H, ArH), 6.61 (s, 2 H, Het-NH), 6.19 (s, 4 H, Het-NH), 6.03 (s, 4 H, Het-NH), 3.47 (m, 4 H, N=CNHCH2), 3.22 (q, 4 H, J = 10 Hz, Het-NHCH2), 1.66 (q, 4 H, J = 10 Hz, CH2), 1.56 (q, 4 H, J = 10 Hz, CH2). 13C NMR (DMSO-d6): 167.2, 166.9, 166.4, 161.5, 133.0, 128.6, 42.7, 26.6, 24.8 (one overlapped). HR-ESI-MS for C22H35N16: 523.3231 found: 523.3233. ESI-MS [M + H]+: 30% [M + 2H]2+: 100%. Elemental analysis calculated for C22H34N16·4HCl C: 39.53, H: 5.73, N: 33.53, Cl: 21.21 found: C: 39.32, H: 5.89, N: 31.96, Cl: 21.77.
MBNL1 Expression and Purification
Expression and purification were performed as previously described.24 For full description, see Supporting Information.
Confocal Microscopy
Imaging of ribonuclear foci in DT960 transfected HeLa cells were performed as previously described.27 For full description, see Supporting Information.
Quantification of Foci Area
The areas of the foci occupied by the cells were quantified using the AxioVision software using an Automeasure module (Carl Zeiss, Obercohen, Germany), where the points having both red and green components were thresholded and quantified. The absolute green and red components were excluded.
Splicing Assays
Splicing assays on the IR pre-mRNA were performed as previously described.26 For full description, see Supporting Information.
Drosophila Genetics
Flies were raised at 25 °C on standard corn meal medium supplemented with dry yeast. Fly lines bearing UAS-(CTG)60 and UAS-(CTG)480(30) were a kind gift of Prof. Rubén Artero Allepuz (Universitat de València, Estudi General, Spain). UAS-DsRed-CAG100(31) and UAS-EGFP-CGG90(32) fly lines were obtained from Profs. Nancy Bonini (University of Pennsylvania, USA) and Stephen Warren (Emory University, USA), respectively.
Drug Treatment in Drosophila
Ligands 3 and 5 were dissolved in water and mixed with fly food. Final concentrations of the compounds were either at 200, 400, or 800 μM. Genetic crosses were set up in drug-containing fly food, and progeny flies of the correct genotypes were analyzed by light and scanning electron microscope analyses.
Examination of Adult Fly Eyes by Light and Electron Microscopies
Light microscopic examination was performed on an Olympus SZX-12 stereomicroscope. Eye images were captured using a SPOT Insight CCD camera (Diagnostic Instruments). Scanning electron microscopy (SEM) was performed according to a previously reported procedure.33 In brief, 2–3-day-old adult fly heads were fixed in 2.5% glutaraldehyde (EM grade, Electron Microscopy Sciences) in phosphate buffer (pH 7.4) for 4 h, then postfixed with 1% osmium tetroxide (Electron Microscopy Sciences), dehydrated to 100% ethanol, and critical-point dried with liquid CO2. Gold–palladium-coated specimens were examined with a JEOL JSM-6301FE microscope operated at 5 kV.
Determination of Maximum Tolerated Dose (MTD)
Three wild type C57/BL6 mice were dosed at 5, 10, 20, 50, and 100 mg/kg of ligand 3 (formulated in sterile water). A single dose was administered at the start of the study via intraperitoneal (i.p.) injection, and all the mice were monitored over the course of 24 h for signs of toxicity and weight loss.
Results
Structure-Based, Rational Design of Small Molecule Inhibitors
Key to the new strategy was the realization that all reported CUG repeat structures25,34−38 formed essentially A-form double-stranded (ds) helices whose structures closely resemble the HIV-1 frameshift site (FS) RNA stem-loop. For example, superposing the [r(CUG)6]2 structure (PDB: 3GM7)34 with the stem-loop region of the HIV-1 FS RNA (PDB: 2L94)29 shows that the two RNA constructs are strikingly similar (Figure 2a). Furthermore, the frameshifting stimulator, ligand 5 (known as DB213, Figure 2b) was shown by Butcher and co-workers to bind in the major groove of the HIV-1 FS RNA (Kd ≈ 360 μM).29 The NMR-determined complex structure showed ligand 5 to span a distance of about 7 base pairs from U17-A40 to U23-A34 (Figure 2b), nearly matching the distance from the first to the third U–U mismatch in the r(CUG)6 structure (Figure 2c).25 These observations suggested the bisamidinium unit of ligand 5 as a groove-binding scaffold for CUG recognition with two triaminotriazine-based recognition units in place of the dimethylammonium groups. It was anticipated that these Janus-wedge units would significantly increase the affinity and selectivity of the ligand because the X-ray analyses25,34−38 and molecular dynamics (MD) simulations26,38 of CUG hairpins indicate that the U–U mismatches are poorly paired with, at most, one direct interbase hydrogen bond.
Molecular Modeling and MD Simulations
Initial attempts to model a complex formed between [r(CUG)6]2 and ligand 2 did not result in a stable hydrogen-bonded triplet. Additionally, a 10 ns MD simulation suggested a progressive weakening of interactions between the two triaminotriazine units and the U–U mismatches. At 9 ns, both triaminotriazine rings became almost orthogonal to the mismatch, similar to the results observed in modeling (Figure S2). This result suggested that the trimethylene linker in 2 was too short to allow the designed interaction of the two triaminotriazine units and the U–U pairs. In contrast, modeling of ligand 3 indicated stable major groove binding with formation of two base triplets (Figure 3a); the greater flexibility allowed the two triaminotriazine units to interact with the U–U pairs forming a total of 5–6 hydrogen bonds (Figure 3b,c). Similar results were obtained for ligand 4. For ligands 3 and 4, the base triplets were stable during the course of the MD simulations. Nevertheless, all three ligands (2–4) were synthesized (see Supporting Information), and their biophysical and biological properties were evaluated.
Figure 3.
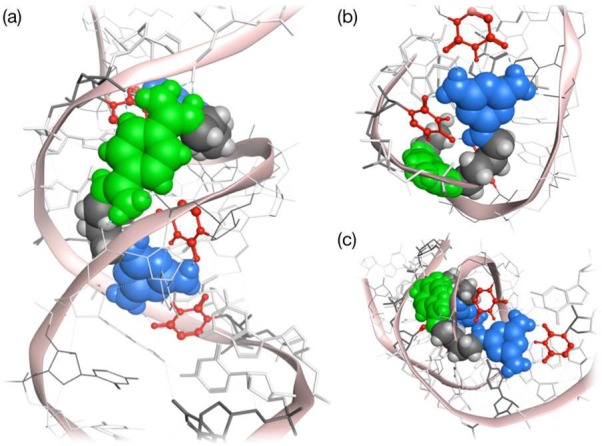
Molecular modeling of ligand 3 with [r(CUG)6]2 repeats. Energy minimized structures showing (a) the bisamidinium unit (in green) of ligand 3 binding to a CUG duplex in the major groove and (b and c) the recognition of two of the U–U pairs (in red) of ligand 3 through hydrogen bonding with triaminotriazine moieties (in blue). Internal U–U pairs not hydrogen bonded to triaminotriazine in gray.
Ligand 3 is a Potent Inhibitor of MBNL1-CUG Interaction in Vitro
The inhibitory activity of ligands 2–4 was studied by electrophoretic mobility shift assay (EMSA) using r(CUG)12 and MBNL1. Among the three ligands, 3 exhibited the best inhibitory ability with an IC50 value of 115 ± 14 μM (Figure 4a,b). Ligands 2 and 4 had measured IC50 values of 1.6 mM and 621 μM respectively, which were 14- and 5-fold less potent than ligand 3 (Figure S3a–d). The assays were performed under very stringent conditions; the MBNL1-r(CUG)12 complex had a low nanomolar dissociation constant (Kd = 7 ± 1 nM) in the presence of 0.05% (v/v) Triton X-100 as detergent. Taking this strong protein–RNA interaction into account, an apparent inhibition constant in the low micromolar range (Ki = 8 ± 2 μM) was obtained for ligand 3. A similar apparent inhibition constant was obtained (Ki = 14 ± 2 μM) at a higher concentration of detergent (0.1% Triton X-100), supporting the notion that the ligand is operating as a monomer rather than an aggregate.39
Figure 4.

In vitro inhibition of MBNL1-r(CUG)12 interaction by ligand 3. (a) Gel electrophoretic mobility shift assay of ligand 3 with r(CUG)12 RNA. First lane: RNA only; second lane: MBNL1-RNA complex with 10% DMSO. Conditions: [MBNL1] = 0.1 μM; [r(CUG)12] = 0.22 nM; [Tris·HCl] = 20 mM; pH = 8; 0.05% Triton X-100. (b) Inhibition plot of MBNL1-r(CUG)12 complex with ligand 3. Error bars indicate SEM of at least three independent measurements. (c) ITC study of the binding of ligand 3 to r(CUG)12. [r(CUG)12] = 10 μM; [NaCl] = 300 mM; [MOPS] = 20 mM; pH = 7.
How does ligand 3 compare to other small molecules, such as ligand 1,24 H1,21 and pentamidine22 (structures in Figure 1), reported to inhibit the MBNL1-r(CUG)12 interaction? Future drug discovery efforts might be facilitated by such comparisons. Comparing the Ki values of ligand 1 (Ki = 7 ± 1 μM)24 and 3 (Ki = 8 ± 2 μM), both compounds seem to be equally potent in inhibiting the MBNL1–r(CUG)12 interaction. Unfortunately, because IC50 values depend on the specific experimental conditions used, values from other reports21,22 cannot be compared directly. To compare the inhibitory ability of H1 and pentamidine, EMSA was performed on these compounds under the same stringent conditions. Under these stringent conditions, no inhibition of MBNL1–r(CUG)12 interaction by H1 or pentamidine was observed (Figure S3e–g). In repeating the EMSA study of pentamidine in the absence of Triton-X in the binding buffer (Figure S3h), conditions that better matched the original literature report,22 some inhibition was observed. After the current work was completed, a report appeared showing that pentamidine did not exhibit any inhibition in the absence of bromophenol blue in the loading dye40 consistent with our observation.
Ligand 3 Binds Tightly and Selectively to CUG Repeats
The binding affinity and selectivity of ligand 3 toward various targets was studied using isothermal titration calorimetry (ITC). It was found that ligand 3 bound r(CUG)12 with a low micromolar affinity (Kd = 8 ± 2 μM) (Figure 4c). In contrast, the binding of ligand 3 to bulk tRNA, GST-tagged MBNL1, HIV frameshift site RNA, and r(CCUG)8 was weak (Figure S4a), and only a lower limit of Kd > 200 μM could be assigned to these systems. These results indicate that ligand 3 inhibits the MBNL1–CUG complex by targeting the RNA, not by binding the protein. In addition, ligand 5 showed only very weak binding affinity (Kd > 200 μM) toward r(CUG)12 and GST-tagged MBNL1 (Figure S4b), the CUG affinity consistent with the Kd value (≈ 360 μM) of DB213 to an A-form RNA duplex reported by Butcher.29
Ligand 3 Reduces Ribonuclear Foci in a DM1 Cell Culture Model
One of the cellular hallmarks of DM1 is the presence of ribonuclear foci formed by the sequestration of MBNL1 by the toxic r(CUG)exp. As a cell culture model for DM1, HeLa cells were transfected with a plasmid containing DMPK exons 11–15, with 960 CTG repeats in exon 15 (DT960).41 In DT960 transfected cells, ribonuclear foci can be readily detected via immunofluorescent (IF) staining for MBNL1 and fluorescent in situ hybridization (FISH) using Cy3-labeled (CAG)10 probe (Figure 5). Treatment with 100 μM of ligand 3 led to a significant reduction in the amount of ribonuclear foci in DT960 transfected cells as early as 48 h post treatment (Figures 5 and S5A), whereas no effect was observed with ligand 5 at all time points treated (Figure S6). On the other hand, ligands 2 and 4 were considerably less effective than ligand 3 in similar experiments for 48 h (Figure S7). To more accurately quantify the reduction in the amount of ribonuclear foci in ligand 3 treated DT960-transfected cells, the area occupied by these foci was measured and compared to untreated DT960-transfected cells. Increasing the concentration of ligand 3 led to a steady reduction in the area of ribonuclear foci occupied by the cells (Figure S5b).
Figure 5.

Ligand 3 relieves MBNL1 sequestration and reduces ribonuclear foci in DT960-transfected HeLa cells. MBNL1 was visualized using mouse anti-MBNL1 and goat anti-mouse Alexa Fluor 488 antibodies; r(CUG)960 was imaged using FISH with 1 ng/μL Cy3-(CAG)10. Nuclei were stained with 10 μg/mL Hoechst 33342. Scale bar =10 μm.
Ligand 3 Partially Corrects Missplicing of cTNT and IR pre-mRNAs in a DM1 Cell Culture Model
Knowing that ligand 3 can relieve the sequestration of MBNL1 by r(CUG)exp, its ability to reverse the missplicing of two pre-mRNAs, cardiac troponin T (cTNT), and insulin receptor (IR) was examined in DT960-transfected HeLa cells.41,42 As a positive control, HeLa cells transfected with a plasmid containing DMPK exons 11–15 but no CTG repeats (DT0) was used. In the case of the cTNT pre-mRNA, inclusion of exon 5 was predominantly observed in DT960 cells, where 88% of exon 5 inclusion was observed compared to a 58% in DT0 cells (Figure 6). After a 72 h treatment with 100 μM of ligand 3, partial correction of cTNT missplicing was achieved (74% exon 5 inclusion; Figure 6a,b). Statistically significant reduction was also observed when DT960 cells were treated with 75 μM of ligand 3 (Figure 6c,d). Ligand 5 did not have a significant effect in these experiments.
Figure 6.
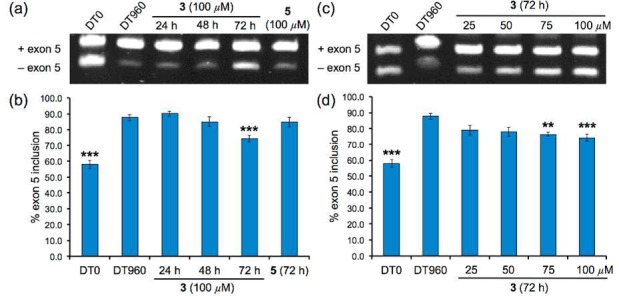
Ligand 3 partially corrects cTNT pre-mRNA missplicing in a DM1 cell culture model. Time-dependent correction of cTNT missplicing (a) determined via standard RT-PCR and (b) bar graph summarizing % exon 5 inclusion. Dose-dependent correction of cTNT missplicing (c) determined via standard RT-PCR and (d) bar graph summarizing % exon 5 inclusion. Error bars represent standard error of mean of at least 3 independent experiments. (* p < 0.05; ** p < 0.01; *** p < 0.001).
For the IR pre-mRNA, exclusion of exon 11 was predominantly observed in DT960 cells, where only 30% of exon 11 inclusion was observed compared to a 53% in DT0 cells (Figure 7). Treating DT960 cells with 100 μM of ligand 3 for 72 h partially corrected the missplicing of the IR pre-mRNA (43% exon 11 inclusion; Figure 7a,b). Statistically significant correction of missplicing was observed at 50 μM and 75 μM of ligand 3 (Figure 7c,d). These results demonstrate that ligand 3 can enter the cell nucleus and specifically bind r(CUG)exp and inhibit the sequestration of MBNL1, thereby improving the missplicing of two pre-mRNAs that are dependent on MBNL1 activity. Ligand 5 did not have a significant effect in these experiments.
Figure 7.

Ligand 3 partially corrects IR pre-mRNA missplicing in a DM1 cell culture model. Time-dependent correction of IR missplicing (a) determined via standard RT-PCR and (b) bar graph summarizing % exon 11 inclusion. Dose-dependent correction of IR missplicing (c) determined via standard RT-PCR and (d) bar graph summarizing % exon 11 inclusion. Error bars represent standard error of mean with at least 3 independent experiments. (* p < 0.05; ** p < 0.01; *** p < 0.001).
Ligand 3 Suppresses CUG-induced Toxicity in a DM1 Drosophila Model
The suppressive effect of ligand 3 on r(CUG)exp-induced toxicity in vivo was examined using a DM1 transgenic Drosophila model.30 Tissue-specific expression of the interrupted 480 CTG repeats, i(CTG)480, in flies using the gmr-GAL4 driver induces glossy and rough eye phenotypes with reduced eye size, which reflects severe morphological disturbance of the adult eye architecture (Figure 8a,e).30 Nuclear colocalization of the i(CUG)480 repeats and muscleblind (MBNL) proteins, Drosophila homologues of the human MBNL1 proteins,43 was also observed in this transgenic line.30 When flies were raised in food containing 200 μM of ligand 3, the glossy eye phenotype was slightly suppressed (Figure 8b). More significant improvement in the glossy phenotype was observed by increasing the concentration of ligand 3 to 400 μM and above (Figures 8c,d and S8a). However, mild phenotype mitigation was only observed when DM1-flies were treated with 800 μM of ligand 5 (Figure S8b), consistent with the in vitro and cell culture data reported above that ligand 5 is significantly less potent than ligand 3. SEM images also showed that the rough phenotype was partially reversed as evidenced by the improved regularity of the external eye structure (Figure 8f–h) compared to untreated flies.
Figure 8.
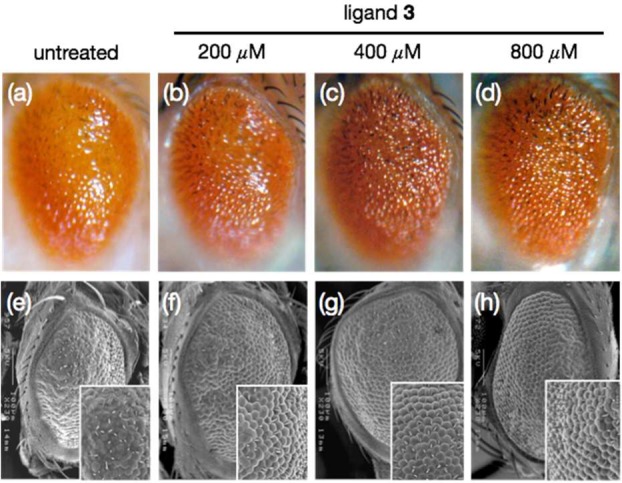
Effects of ligand 3 on the eye phenotypes in Drosophila expressing i(CUG)480 RNA observed under light microscopy (a–d) and SEM (e–h). Significant improvement in glossy and ommatidium defects was observed after treatment with ligand 3.
Flies expressing interrupted r(CUG)60 transcripts did not exhibit the severe morphological disturbance of the adult eye architecture observed in flies expressing longer repeats (Figure S9a). Treating these control flies with up to 800 μM of ligand 3 or 5, however, led to no observable differences in the eye morphology (Figure S9a). This shows that ligands 3 and 5 did not exert dominant deleterious effect on the (CTG)60 control flies. The compound’s ability to modify degenerative phenotypes induced by the expression of other trinucleotide expansion transcripts was also assessed. Similar to CTG480, expression of DsRed-CAG100(31) and EGFP-CGG90(32) transcripts induced rough eye phenotype. However, neither ligands 3 nor 5 suppressed the DsRed-CAG100 (Figure S9b) and EGFP-CGG90 rough eye phenotype (Figure S9c), demonstrating that ligand 3 specifically suppresses r(CUG)exp -induced toxicity.
Low-Toxicity Profile of Ligand 3
The toxicity of ligand 3 was studied in three cell lines: HeLa cells, human DM1 fibroblast cells, and 3T3 mouse fibroblast cells. No observable cell death was found for ligand 3 in these cell lines at concentrations as high as 100 μM for 72 h, whereas significant cytotoxicity was observed for ligand 1 after 24 h (Figure S10). In addition, the maximum tolerated dose (MTD) of ligand 3 in C57/BL6 mice (single i.p. injection formulated in sterile water) was found to be between 50 and 100 mg/kg.
Discussion
Finding small molecules that target expanded CUG RNA triplet-repeats is arguably the most promising current strategy toward myotonic dystrophy drug discovery.8,44 For example, one successful strategy involved screening libraries of small molecules or peptides leading to hits that showed efficacy in DM1 mouse model.45,46 Our efforts have focused on rational design, which led to a series of intercalator-based inhibitors.24,27,28 Despite being potent in vitro inhibitors of the MBNL1-r(CUG)exp interaction, these ligands may be limited by an inherently higher cytotoxicity. With the goal of developing an alternative class of high-potency inhibitors of MBNL1-r(CUG)exp interaction, a new structure-based design strategy was used that took advantage of the small molecule stimulator (DB213, ligand 5) of HIV-1 frameshifting. Although discovered by high-throughput screening and developed to recognize an entirely different target, the bisamidinium unit of ligand 5 appeared capable of serving as a general A-form RNA groove binder. Thus, the realization that the gross structure of CUG repeats is similar to that of the HIV-1 FS RNA stem-loop led to the design of ligands 2–4, wherein the triaminotriazine units were intended to provide high selectivity for r(CUG)exp. The success of this approach suggests the use of the bisamidinium unit as a general binding moiety to target A-form RNA and a suitable alternative to the aminoglycoside- and the Hoechst-based RNA-groove binders. Indeed, ligands 2–4 were highly water-soluble, an issue that has been a challenge in the Hoechst-based approach.
Molecular modeling suggests that groove-binding ligands 2–4 are able to span three consecutive CUG sites in the RNA duplex. Furthermore, MD simulations showed that the two triaminotriazine units in ligands 3 and 4 form stable base triplets with two of the U–U mismatches (Figure 3). Support for the importance of the triaminotriazine moieties comes from a comparison of ligand 3 and control compound ligand 5 (DB213). Thus, ligand 3, which was the most potent inhibitor studied, inhibited the MBNL1-r(CUG)12 complex with an apparent inhibition constant in the low micromolar range (Ki = 8 ± 2 μM), whereas ligand 2 exhibited negligible MBNL1-r(CUG)12 inhibition (Figure S3a, b).
Extensive studies on ligand 3 show that it is nontoxic to mammalian cells even after 72 h of incubation at 100 μM. Moreover, the inhibitory effect of ligand 3 was also observed in cell culture. Using an established cell culture model of DM1,41 disruption of ribonuclear foci due to the relief of MBNL1 sequestration by r(CUG)exp was observed as early as 48 h post treatment (Figure 5). The partial relief of MBNL1 sequestration by treatment with ligand 3 at 100 μM (Figure S5b) leads to a 46% and 56% correction for the missplicing of in the cTNT and IR pre-mRNAs, respectively. It had been shown in genetic studies that approximately 50% of unsequestered MBNL1 was sufficient to partially restore normal splicing patterns in DM1-mice.47,48 This finding suggests that ligand 3 is an excellent lead compound and that analogues with even modest improvements in r(CUG)exp binding may lead to full reversal of missplicing defects.
Using a DM1 Drosophila model, we were also able to demonstrate the efficacy of ligand 3 in reversing the glossy and rough eye phenotype in DM1-infected flies (Figures 8 and S8a). Ligand 3 is not effective in improving the neurodegenerative phenotypes observed in the eyes of Huntington-31 and Fragile X-infected32 fly models (Figure S9b,c), suggesting that direct targeting of the U–U mismatches is likely responsible for the phenotypic reversal observed in DM1-inflicted flies. To the best of our knowledge, this is the first report of a rationally designed small molecule inhibitor that achieves reversal of phenotypic defects observed in DM1-infected Drosophila.
Comparing ligand 3 head-to-head with previously reported inhibitors having similar molecular weights revealed ligand 3 to be similar to ligand 1,24 but significantly more effective than H121 and pentamidine22 in inhibiting the toxic MBNL1-r(CUG)12 interaction (Figures 4a and S3e–h). Combined with its promising activity profile in DM1 cell and animal models described above, the advantages of high water-solubility and low cytotoxicity make ligand 3 a promising candidate for the future development. Going forward, a powerful strategy to prepare more potent DM1 inhibitors was demonstrated by Disney and co-workers in linking multiple Hoechst 33258 monomers to a PNA backbone. Thus, a pentamer exhibited a potency (IC50 = 0.140 ± 0.040 μM) that was approximately 1000-fold higher than that of the analogous Hoechst monomer (IC50 = 110 ± 20 μM).17 There are obvious strategies for preparing oligomers of ligand 3. Likewise, applying the approach described herein to the other RNA-repeat-mediated diseases is of considerable interest. For example, structurally similar groove binders containing triaminopyrimdine units for recognizing CCUG-hairpin sequences49 may offer a strategy for developing therapeutic agents for myotonic dystrophy type 2 (DM2). Efforts along these lines are under active investigation and results will be reported in due course.
Acknowledgments
We thank Thomas Cooper (Baylor College of Medicine) for the cTNT, DT0, and DT960 minigene plasmids and Nicholas Webster (University of California, San Diego) for the IR minigene plasmid. This work was supported by National Institutes of Health (RO1AR058361). C.-H.W. is a recipient of a Croucher Foundation Scholarship (HKSAR).
Supporting Information Available
Synthesis of other ligands, the protocols of MD simulations, ITC, protein expression and purification, EMSA experiments, confocal microscopy experiments, and splicing and cytotoxicity assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# Department of Chemistry, University of California, Berkeley, Berkeley, CA 94720.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Brook J. D.; McCurrach M. E.; Harley H. G.; Buckler A. J.; Church D.; Aburatani H.; Hunter K.; Stanton V. P.; Thirion J.-P.; Hudson T.; Sohn R.; Zemelman B.; Snell R. G.; Rundle S. A.; Crow S.; Davies J.; Shelbourne P.; Buxton J.; Jones C.; Juvonen V.; Johnson K.; Harper P. S.; Shaw D. J.; Housman D. E. Cell 1992, 68, 799–808. [DOI] [PubMed] [Google Scholar]
- Harley H. G.; Brook J. D.; Rundle S. A.; Crow S.; Reardon W.; Buckler A. J.; Harper P. S.; Housman D. E.; Shaw D. J. Nature 1992, 355, 545–546. [DOI] [PubMed] [Google Scholar]
- Buxton J.; Shelbourne P.; Davies J.; Jones C.; Tongeren T. V.; Aslanidis C.; de Jong P.; Jansen G.; Anvret M.; Riley B.; Williamson R.; Johnson K. Nature 1992, 355, 547–548. [DOI] [PubMed] [Google Scholar]
- Aslandis C.; Jansen G.; Amemiya C.; Shutler G.; Mahadevan M.; Tsilfidis C.; Chen C.; Alleman J.; Wormskamp N. G. M.; Vooijs M.; Buxton J.; Johnson K.; Smeets H. J. M.; Lennon G. G.; Carrano A. V.; Korneluk R. G.; Wieringa B.; de Jong P. J. Nature 1992, 355, 548–551. [DOI] [PubMed] [Google Scholar]
- La Spada A. R.; Taylor J. P. Nat. Rev. Genet. 2010, 11, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyzosiak W. J.; Sobczak K.; Wojciechowska M.; Fiszer A.; Mykowska A. P. Nucleic Acids Res. 2012, 40, 11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatchel J. R.; Zoghbi H. Y. Nat. Rev. Genet. 2005, 6, 743–755. [DOI] [PubMed] [Google Scholar]
- Klein A. F.; Gasnier E.; Furling D. Biochimie 2011, 93, 2006–2012. [DOI] [PubMed] [Google Scholar]
- Fernandez-Costa J. M.; Llamusi M. B.; Garcia-Lopez A.; Artero R. Biol. Rev. 2011, 86, 947–958. [DOI] [PubMed] [Google Scholar]
- Kuyumcu-Martinez N. M.; Wang G.-S.; Cooper T. A. Mol. Cell 2007, 28, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T.; Gibbens B.; Doty N. S.; Gomes-Pereira M.; Huguet A.; Stone M. D.; Margolis J.; Peterson M.; Markowski T. W.; Ingram M. A.; Nan Z.; Forster C.; Low W. C.; Schoser B.; Somia N. V.; Clark H. B.; Schmechel S.; Bitterman P. B.; Gourdon G.; Swanson M. S.; Moseley M.; Ranum L. P. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Costa J. M.; Garcia-Lopez A.; Zuñiga S.; Fernandez-Pedrosa V.; Felipo-Benavent A.; Mata M.; Jaka O.; Aiastui A.; Hernandez-Torres F.; Aguado B.; Perez-Alonso M.; Vilchez J. J.; de Munain A. L.; Artero R. D. Hum. Mol. Genet. 2013, 22, 704–716. [DOI] [PubMed] [Google Scholar]
- Du H.; Cline M. S.; Osborne R. J.; Tuttle D. L.; Clark T. A.; Donohue J. P.; Hall M. P.; Shiue L.; Swanson M. S.; Thornton C. A.; Ares M. Jr. Nat. Struct. Mol. Biol. 2010, 17, 187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulders S. A. M.; van den Broek W. J. A. A.; Wheeler T. M.; Croes H. J. E.; van Kuik-Romeijn P.; de Kimpe S. J.; Furling D.; Platenburg G. J.; Gourdon G.; Thornton C. A.; Wieringa B.; Wansink D. G. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13915–13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler T. M.; Sobczak K.; Lueck J. D.; Osborne R. J.; Lin X.; Dirksen R. T.; Thornton C. A. Science 2009, 325, 336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler T. M.; Leger A. J.; Pandey S. K.; MacLeod A. R.; Nakamori M.; Cheng S. H.; Wentworth B. M.; Bennett C. F.; Thornton C. A. Nature 2012, 488, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushechnikov A.; Lee M. M.; Childs-Disney J. L.; Sobczak K.; French J. M.; Thornton C. A.; Disney M. D. J. Am. Chem. Soc. 2009, 131, 9767–9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan L.; Disney M. D. Angew. Chem., Int. Ed. 2013, 52, 10010–10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzuczek S. G.; Gao Y.; Tang Z.-Z.; Thornton C. A.; Kodadek T.; Disney M. D. ACS Chem. Biol. 2013, 8, 2312–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareiss P. C.; Sobczak K.; McNaughton B. R.; Palde P. B.; Thornton C. A.; Miller B. L. J. Am. Chem. Soc. 2008, 130, 16254–16261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkesh R.; Childs-Disney J. L.; Nakamori M.; Kumar A.; Wang E.; Wang T.; Hoskins J.; Tran T.; Housman D.; Thornton C. A.; Disney M. D. J. Am. Chem. Soc. 2012, 134, 4731–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf M. B.; Nakamori M.; Matthys C. M.; Thornton C. A.; Berglund J. A. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 18551–18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs-Disney J. L.; Stepniak-Konieczna E.; Tran T.; Yildirim I.; Park H.; Chen C. Z.; Hoskins J.; Southall N.; Marugan J. J.; Patnaik S.; Zheng W.; Austin C. P.; Schatz G. C.; Sobczak K.; Thornton C. A.; Disney M. D. Nat. Commun. 2013, 4, 2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arambula J. F.; Ramisetty S. R.; Baranger A. M.; Zimmerman S. C. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 16068–16073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooers B. H. M.; Logue J. S.; Berglund J. A. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 16626–16631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C.-H.; Richardson S. L.; Ho Y.-J.; Lucas A. M. H.; Tuccinardi T.; Baranger A. M.; Zimmerman S. C. ChemBioChem 2012, 13, 2505–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahromi A. H.; Nguyen L.; Fu Y.; Miller K. A.; Baranger A. M.; Zimmerman S. C. ACS Chem. Biol. 2013, 8, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahromi A. H.; Fu Y.; Miller K. A.; Nguyen L.; Luu L. M.; Baranger A. M.; Zimmerman S. C. J. Med. Chem. 2013, 56, 9471–9481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheschi R. J.; Tonelli M.; Kumar A.; Butcher S. E. ACS Chem. Biol. 2011, 6, 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Lopez A.; Monferrer L.; Garcia-Alcover I.; Vicente-Crespo M.; Alvarez-Abril M. C.; Artero R. D. PLoS One 2008, 3, e1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi H.; Lau T. C.-K.; Tsang S.-Y.; Lau K.-F.; Chan H. Y. E. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 13428–13433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P.; Zarnescu D. C.; Zhang F.; Pearson C. E.; Lucchesi J. C.; Moses K.; Warren S. T. Neuron 2003, 39, 739–747. [DOI] [PubMed] [Google Scholar]
- de Haro M.; Al-Ramahi I.; De Gouyon B.; Ukani L.; Rosa A.; Faustino N. A.; Ashizawa T.; Cooper T. A.; Botas J. Hum. Mol. Genet. 2006, 15, 2138–2145. [DOI] [PubMed] [Google Scholar]
- Kiliszek A.; Kierzek R.; Krzyzosiak W. J.; Rypniewski W. Nucleic Acids Res. 2009, 37, 4149–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Park H.; Fang P.; Parkesh R.; Guo M.; Nettles K. W.; Disney M. D. Biochemistry 2011, 50, 9928–9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamjar J.; Katorcha E.; Popov A.; Malinina L. J. Biomol. Struct. Dyn. 2012, 30, 505–523. [DOI] [PubMed] [Google Scholar]
- Coonrod L. A.; Lohman J. R.; Berglund J. A. Biochemistry 2012, 51, 8330–8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkesh R.; Fountain M.; Disney M. D. Biochemistry 2011, 50, 599–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B. Y.; Shoichet B. K. Nat. Protoc. 2006, 1, 550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coonrod L. A.; Nakamori M.; Wang W.; Carrell S.; Hilton C. L.; Bodner M. J.; Siboni R. B.; Docter A. G.; Haley M. M.; Thornton C. A.; Berglund J. A. ACS Chem. Biol. 2013, 8, 2528–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips A.; Timchenko L.; Cooper T. Science 1998, 280, 737–741. [DOI] [PubMed] [Google Scholar]
- Savkur R. S.; Philips A. V.; Cooper T. A. Nat. Genet. 2001, 29, 40–47. [DOI] [PubMed] [Google Scholar]
- Monferrer L.; Artero R. J. Hered. 2006, 97, 67–73. [DOI] [PubMed] [Google Scholar]
- Muntoni F.; Wood M. J. A. Nat. Rev. Drug Discovery 2011, 10, 621–637. [DOI] [PubMed] [Google Scholar]
- García-López A.; Llamusí B.; Orzáez M.; Pérez-Payá E.; Artero R. D. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 11866–11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofori L. O.; Hoskins J.; Nakamori M.; Thornton C. A.; Miller B. L. Nucleic Acids Res. 2012, 40, 6380–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X.; Miller J. W.; Mankodi A.; Kanadia R. N.; Yuan Y.; Moxley R. T.; Swanson M. S.; Thornton C. A. Hum. Mol. Genet. 2006, 15, 2087–2097. [DOI] [PubMed] [Google Scholar]
- Kanadia R. N.; Johnstone K. A.; Mankodi A.; Lungu C.; Thornton C. A.; Esson D.; Timmers A. M.; Hauswirth W. W.; Swanson M. S. Science 2003, 302, 1978–1980. [DOI] [PubMed] [Google Scholar]
- Wong C.-H.; Fu Y.; Ramisetty S. R.; Baranger A. M.; Zimmerman S. C. Nucleic Acids Res. 2011, 39, 8881–8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.