

Skeletal muscle is the major site of glucose disposal, accounting for 80% of glucose catabolism under insulin‐stimulated conditions in healthy subjects. In type 2 diabetes mellitus patients, muscular glucose uptake is decreased; therefore, skeletal muscle can be a major therapeutic target for the daily control of hyperglycemia and improvement in insulin sensitivity. To develop therapeutic approaches to insulin resistance, numerous researchers have shown its underlying mechanisms, and there is a great deal of evidence that abnormalities in muscular mitochondrial function can lead to the accumulation of toxic lipid metabolites and induce insulin resistance (the lipotoxicity paradigm). Furthermore, increasing evidence indicates that metabolic inflexibility leads to muscular insulin resistance. Indeed, Muoio et al.1 recently described a molecular mechanism by which metabolic inflexibility causes insulin resistance in skeletal muscle, and showed the efficacy of carnitine supplementation therapy in patients with insulin resistance and metabolic inflexibility.
Mitochondrial dysfunction in skeletal muscle is considered central to the development of insulin resistance and type 2 diabetes mellitus. Many studies show that decreased muscular lipid oxidation capacity results in the accumulation of toxic lipid intermediates, such as diacylglycerol, acyl‐coenzyme A (acyl‐CoA) and ceramides, and activates atypical protein kinase C isoforms. This is followed by serine phosphorylation of insulin receptor substrates and, eventually, impaired insulin signaling. According to these findings, researchers postulated the lipotoxicity hypothesis, which suggests that decreased mitochondrial function in the prediabetic state leads to the accumulation of toxic fatty metabolites, thereby contributing to the development of insulin resistance.
Kelley and Mandarino2 originally defined metabolic flexibility as the capacity to switch from predominant lipid oxidation and high rates of fatty acid uptake during fasting conditions to the suppression of lipid oxidation, and increased glucose uptake, oxidation and storage under insulin‐stimulation. In contrast, metabolic inflexibility observed in type 2 diabetes mellitus patients is characterized by an inability of the organism to shift mitochondrial energy sources promptly in response to changes in nutrient availability. However, the causal relationship between metabolic inflexibility and muscular insulin resistance remains unclear.
Carnitine is considered a key molecule associated with the mechanism of metabolic flexibility. Although carnitine is best known for its role in the import of long‐chain fatty acids (acyl groups) into the mitochondrial matrix for subsequent β‐oxidation, it is also necessary for the mitochondrial efflux of acyl groups in cases where substrate oxidation exceeds energy demands. Importantly, free carnitine is decreased in skeletal muscles of obese rats, whereas dietary L‐carnitine supplementation rescues metabolic flexibility. Therefore, deficient carnitine‐dependent efflux systems might be one of the mechanisms underlying metabolic inflexibility.
To test this hypothesis, Muoio et al.1 generated muscle‐specific carnitine acyltransferase (CrAT)‐knockout (CrATM−/−) mice. CrAT is a carnitine‐dependent acyltransferase enzyme that catalyzes the reversible conversion of short‐chain acyl‐CoA (mainly acetyl‐CoA) and carnitine to free CoA and short‐chain acylcarnitine (mainly acetylcarnitine). Unlike acyl‐CoA, these metabolites can cross mitochondrial and cellular membranes with ease. CrAT resides principally in the mitochondrial matrix, where it regulates the flux of acetyl‐CoA. In normal skeletal muscle, CrAT converts excessive acetyl‐CoA to acetylcarnitine and promotes its mitochondrial and cellular efflux (Figure 1a). However, in CrAT‐deficient muscle, acetyl‐CoA was not converted to its permeable form and was not excreted from mitochondria or cells. This led to the overaccumulation of acetyl‐CoA, which exerted an allosteric inhibiting effect on pyruvate dehydrogenase (PDH), a rate‐limiting enzyme for pyruvate entry into the tricarboxylic acid cycle, and subsequently impaired glucose utilization (Figure 1b). In addition, CrATM−/− mice showed glucose intolerance, insulin resistance and metabolic inflexibility. It is noteworthy that insulin‐stimulated phosphorylation of Akt in skeletal muscle did not differ between wild‐type and CrATM−/− mice, suggesting that glucose intolerance in CrATM−/− mice is not a result of lack of insulin sensitivity in skeletal muscle and might be a result of attenuation of glucose oxidation capacity through PDH inhibition. Taken together, CrAT inhibits mitochondrial acetyl‐CoA overload by regulating the efflux of acetyl‐CoA, thus maintaining PDH activity in skeletal muscle. Through these molecular mechanisms, muscular CrAT plays an important role in whole‐body glucose tolerance.
Figure 1.
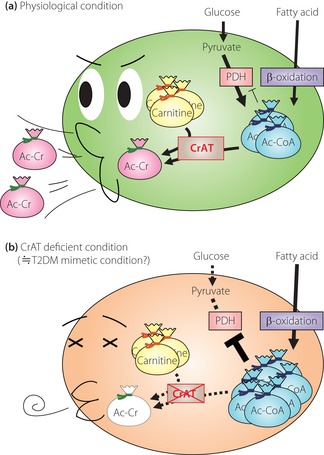
The roles of carnitine acyltransferase (CrAT) and carnitine in skeletal muscle mitochondria. (a) In physiological conditions, CrAT converts excessive acetyl‐coenzyme A (Ac‐CoA) to acetylcarnitine, and promotes its efflux out of mitochondria and cells. (b) In CrAT‐deficient conditions, Ac‐CoA is not converted to its permanent form, and is not excreted from mitochondria or cells. Excessive Ac‐CoA acts as an allosteric inhibitor of pyruvate dehydrogenase (PDH), a rate‐limiting enzyme for pyruvate entry into the tricarboxylic acid cycle, followed by impaired glucose utilization. Ac‐Cr, acetylcarnitine; T2DM, type 2 diabetes mellitus.
Muoio et al.1 also showed the accumulation of medium‐ and long‐chain acylcarnitines in muscles from CrATM−/− mice, which resembled the metabolite profile assayed in muscles from obese and diabetic rodents. In addition, pronounced suppression of CrAT mRNA expression was also observed in insulin‐resistant patients. As aforementioned, free carnitine levels are decreased in skeletal muscles of obese rats. Therefore, the pathological condition of type 2 diabetes mellitus patients might be similar to those of CrATM−/− mice, indicating that carnitine metabolism might be a therapeutic target for the treatment of insulin resistance.
The antidiabetic potential of long‐term L‐carnitine supplementation has not been tested in rigidly controlled human studies. In a clinical study by Muoio et al.1, L‐carnitine supplementation for 6 months increased serum free carnitine and acetylcarnitine concentrations, decreased plasma glucose and insulin levels, and decreased the homeostasis model assessment of insulin resistance (HOMA‐IR) index. Muscle PDH activity was also increased, and metabolic inflexibility was partially ameliorated by carnitine supplementation. These data provided the first firm evidence of a therapeutic effect of carnitine on insulin resistance, prompting the experts to carry out larger and more rigidly controlled studies.
Notably, Muoio et al.1 also showed that mitochondrial dysfunction can cause whole‐body and muscular insulin resistance. As aforementioned, several studies have addressed the role of mitochondria in insulin resistance. However, conflicting data suggest that mitochondrial dysfunction does not always lead to type 2 diabetes mellitus. For example, mitochondrial flavoprotein apoptosis‐inducing factor (Aifm1)‐knockout mice and muscle‐specific mitochondrial transcription factor A (Tfam)‐knockout mice showed increased glucose tolerance despite decreased mitochondrial oxidative phosphorylation capacity. Differences in terminology might at least partially explain the contradictory conclusions, as the term mitochondrial dysfunction is used interchangeably to describe alterations in mitochondrial content, capacity of oxidative phosphorylation and other mitochondrial inadequacies. Recently, systematic terminology to define the parameters of mitochondrial function has been proposed in a review article in Nature Reviews Endocrinology3 (Table 1). In light of these criteria, although maximal oxidative phosphorylation capacity was not impaired in muscle mitochondria from CrATM−/− mice, the loss of submaximal oxidative phosphorylation was suggested by insulin‐stimulated changes in the respiratory exchange ratio in vivo and oxidative phosphorylation in isolated mitochondria under limiting conditions. However, mitochondrial activity, plasticity or coupling were not measured in that study. Therefore, the parameters of mitochondrial function that are relevant to the development of insulin resistance remain unclear.
Table 1. Definition and classification of mitochondrial function.
| Parameter | Definition |
|---|---|
| Activity | Resting oxidative phosphorylation flux (ADP:ATP ratio) in distinct metabolic conditions |
| Oxidative phosphorylation capacity | Maximal ADP‐stimulated oxidative phosphorylation |
| Submaximal oxidative phosphorylation | In vivo non‐resting mitochondrial activity stimulated by energy depletion during standardized in vivo experiments |
| Plasticity | Changes of mitochondrial activity, mitochondrial content or oxidative phosphorylation capacity due to altered metabolic conditions |
| Coupling | The molar ratio of the yield of ATP per oxygen consumed or electrochemical coupling |
ADP, adenosine diphosphate; ATP, adenosine triphosphate.
In a recent article published in Nature Medicine, Kim et al.4 reported an intriguing mechanism that relates muscular mitochondrial dysfunction to insulin sensitivity. In skeletal muscle‐specific autophagy‐knockout (Atg7 knockout) mice with decreased oxidative phosphorylation capacity in skeletal muscle (one of the definitions of mitochondrial dysfunction), they observed resistance to diet‐induced obesity and amelioration of insulin resistance through activating transcription factor 4‐dependent induction of fibroblast growth factor (FGF) 21. These observations suggest that deficient autophagy and subsequent mitochondrial dysfunction promotes FGF21 expression, and subsequently affects whole‐body metabolism. Like FGF21 in that study, a cell‐non‐autonomous signal released in response to mitochondrial stress is termed as a ‘mitokine’5. Given the mitokine paradigm, increased glucose tolerance of Aifm knockout mice and Tfam knockout mice might reflect high circulating levels of FGF21. However, although muscle mitochondria were also impaired in CrATM−/− mice, the antidiabetic action of mitokine was not observed. Therefore, the relationship between mitochondrial dysfunction and FGF21 and other mitokine production will be the subject of future investigations.
References
- 1.Muoio DM, Noland RC, Kovalik JP, et al Muscle‐specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab 2012; 15: 764–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelley DE, Mandarino LJ. Fuel Selection in Human Skeletal Muscle in Insulin. Diabetes 2000; 49: 677–683 [DOI] [PubMed] [Google Scholar]
- 3.Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 2011; 8: 92–103 [DOI] [PubMed] [Google Scholar]
- 4.Kim KH, Jeong YT, Oh H, et al Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 2013; 19: 83–92 [DOI] [PubMed] [Google Scholar]
- 5.Durieux J, Wolff S, Dillin A. The cell‐non‐autonomous nature of electron transport chain‐mediated longevity. Cell 2011; 144: 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]