

Abstract
Hibernating myocardium is an adaptive response to repetitive myocardial ischemia that is clinically common, but the mechanism of adaptation is poorly understood. Here we compared the proteomes of hibernating versus normal myocardium in a porcine model with 24 biological replicates. Using the ion-current-based proteomic strategy optimized in this study to expand upon previous proteomic work, we identified differentially expressed proteins in new molecular pathways of cardiovascular interest. The methodological strategy includes efficient extraction with detergent cocktail; precipitation/digestion procedure with high, quantitative peptide recovery; reproducible nano-LC/MS analysis on a long, heated column packed with small particles; and quantification based on ion-current peak areas. Under the optimized conditions, high efficiency and reproducibility were achieved for each step, which enabled a reliable comparison of 24 the myocardial samples. To achieve confident discovery of differentially regulated proteins in hibernating myocardium, we used highly stringent criteria to define “quantifiable proteins”. These included the filtering criteria of low peptide FDR and S/N > 10 for peptide ion currents, and each protein was quantified independently from ≥2 distinct peptides. For a broad methodological validation, the quantitative results were compared with a parallel, well-validated 2D-DIGE analysis of the same model. Excellent agreement between the two orthogonal methods was observed (R = 0.74), and the ion-current-based method quantified almost one order of magnitude more proteins. In hibernating myocardium, 225 significantly altered proteins were discovered with a low false-discovery rate (∼3%). These proteins are involved in biological processes including metabolism, apoptosis, stress response, contraction, cytoskeleton, transcription, and translation. This provides compelling evidence that hibernating myocardium adapts to chronic ischemia. The major metabolic mechanisms include a down-regulation of mitochondrial respiration and an increase in glycolysis. Meanwhile, cardioprotective and cytoskeletal proteins are increased, while cardiomyocyte contractile proteins are reduced. These intrinsic adaptations to regional ischemia maintain long-term cardiomyocyte viability at the expense of contractile function.
Keywords: myocardial ischemia, proteomics, biomarker discovery, ion current-based quantification, label-free proteomics
Introduction
Hibernating myocardium is a clinical condition in which patients with chronic coronary artery narrowings develop viable, chronically dysfunctional myocardium in response to repetitive reversible myocardial ischemia.1,2 In its purest state, fibrosis and infarction are absent, and thus cardiac function often improves following bypass surgery or placement of a coronary stent.2 While identification of hibernating myocardium is important for directing therapeutic efforts, the protein pathways and physiological mechanisms responsible for the many intrinsic adaptations arising from reversible ischemia remain unclear. We have previously demonstrated that coronary flow and myocardial function were dissociated from the usual determinants of myocardial oxygen demand in response to stress in hibernating myocardium.3,4 This suggests that hibernation may prevent the development of irreversible ischemic injury after submaximal stress by limiting regional energy utilization. On the basis of this observation as well as others,5,6 we hypothesized that hibernating myocytes intrinsically down-regulate their metabolic needs to achieve a balance between supply and demand by reducing regional workload.
Recently, state-of-the-art proteomic strategies have been applied to the study of cardiovascular systems.7−13 For proteomic expression profiling, 2D gel electrophoresis14 or LC/MS-based methods are generally employed. Quantitative LC/MS-based methods include isotope labeling by metabolic incorporation (e.g., SILAC),15 chemical/enzymatic labeling (e.g., ICAT, iTRAQ, and 18O-incorporation),16,17 and, more recently, label-free protein expression profiling approaches.18−21 Adapting these techniques to achieve extensive and reliable discovery of protein alterations responsible for hibernating myocardium poses challenges. First, for a preclinical study involving a chronic large-animal model, a relatively large number of biological replicates (e.g., n = 12 animals per group were used in this study) is desirable to alleviate false-positive discovery arising from interanimal variability. The use of many biological replicates enhances the reliability of biomarker discovery while taking into account biological variability in a fashion that a single pooled sample analysis from multiple subjects cannot.18−20 Unfortunately, it is technically challenging (and costly) to use isotope-labeling methods for this purpose. Label-free methods afford a promising alternative to quantify multiple biological replicates, but these approaches do not typically employ an internal standard. Thus, they require highly quantitative and reproducible sample preparation and LC/MS analysis. These requirements are often difficult to achieve in large-scale experiments.19,21,22 Additionally, on the basis of our previous findings using 2D-DIGE, many chronic enzyme changes in the hibernating myocardium are modest (generally within ±50% of normal values).20,23 This underscores the need to achieve precise quantification to evaluate multiple dysregulated candidate proteins.
It remains challenging to achieve extensive coverage of a tissue proteome using current strategies due to the high complexity and wide dynamic range found in whole tissue samples.24 While utilization of prefractionation techniques such as multidimensional chromatography significantly enhances coverage,25,26 it is not practical to adapt this approach for the analysis of multiple biological replicates. In addition, myocardial tissue contains many hydrophobic membrane proteins,10,11,27 which can be challenging to extract and analyze efficiently using conventional proteomic methods.28 Many of these proteins are critically involved in ion channels, cardiac excitation, and myocardial contraction, yet they remain underrepresented when the proteome is characterized with existing approaches. Finally, false-positive discoveries of significantly changed proteins can result in false biological leads that need to be evaluated and controlled.29−31 Increasing biological sample size and using a reproducible well-defined experimental model can help to minimize this. While statistical approaches have also been developed to address this,32−34 there is no practical method to evaluate or control the false-positive discovery rate experimentally.
With these considerations in mind, we developed and optimized a proteomics approach to perform an extensive, reproducible and relatively large-scale proteomic profiling (24 animals) to identify multiple pathways and proteins that are altered in hibernating myocardium. Myocardial proteins were extracted uniformly with a strong buffer containing high concentrations of detergent cocktails, followed by a precipitation/on-pellet-digestion procedure,20,35 which provides a high yield and quantitative peptide recovery. We employed a nano-LC/nanospray flow setup with low void-volume, large loading capacity, high separation efficiency, and high chromatographic reproducibility for LC/MS analysis. Samples were efficiently resolved on a long (75 cm in length), heated nano-LC column with a 7 h shallow gradient, which markedly enhanced our ability to analyze low-abundance peptides. Orbitrap MS enhanced by an ion-overfilling strategy was employed for the sensitive acquisition of precursor signals for the ion-current-based quantification. Each step was rigorously optimized and evaluated to achieve high run-to-run reproducibility. The false discovery of altered proteins was experimentally measured by preparing and analyzing “decoy” sample groups interspaced with the 24 biological samples. Finally, the quantitative results were compared with parallel results using established 2D-DIGE to provide a broad validation of relative protein changes identified in swine with hibernating myocardium.
Experimental Section
Animal Experiments
Procedures and protocols conformed to institutional guidelines for the care and use of animals in research and have been previously detailed.1,36,37 In brief, juvenile Yorkshire pigs (n = 12) underwent surgical implantation of a fixed diameter Delrin stenosis (1.5 to 2.0 mm inner diameter) on the proximal left anterior descending (LAD) coronary artery. Hibernating myocardium developed as the chronic stenosis progressed to occlusion over 3 to 4 months. This is associated with chronic contractile dysfunction, a critical limitation of coronary blood flow, which occurs in the absence of fibrosis and infarction. Sham swine underwent anesthesia and thoracotomy but did not receive a coronary stenosis. Regional myocardial function was assessed by M-mode echocardiography in the closed-chest anesthetized state and blood flow with fluorescent microspheres. Myocardial tissue harvesting was performed at least 3 days after the physiological studies to circumvent effects of pharmacological agents on acute protein expression. On the day of tissue harvesting, animals were rapidly anesthetized, and subendocardial samples from the inner third of the left ventricular wall were flash-frozen in liquid nitrogen and stored at −80 °C until analyzed.
Protein Extraction and the Precipitation/On-Pellet Digestion Procedure
Frozen samples (0.1 to 0.2 g) were quickly minced and mechanically homogenized with a Polytron in 2 mL of lysis buffer (50 mM Tris pH 8, 150 mM NaCl, 2% NP-40, 0.5% sodium deoxycholate, 1% SDS, protease and phosphatase inhibitors (Roche cat. no. 04693132 and cat. no. 04906837)) on ice. Following centrifugation at 5000g for 10 min, samples were stored at −80 °C. Prior to quantitation with BSA standards in a Pierce BCA Protein Assay Kit, samples were centrifuged 40 min at 140 000g at 4 °C. Solutions were diluted with the lysis buffer to a concentration of 3.3 mg/mL prior to further preparation.
In an Eppendorf Thermomixer (oscillating at 200 rpm), protein disulfide bonds were reduced with 4 mM Tris(2-carboxyethyl) phosphine for 20 min, and free thiols were alkylated with 20 mM iodoacetamide at 37 °C for 30 min. Proteins were precipitated by an optimized two-step approach: one volume of chilled acetone (at −20 °C) was slowly added so that the mixture was cloudy, but no visible particulate was observed. The mixture was vortexed thoroughly to extract the detergents and nonprotein matrix components in acetone–water; subsequently, 5 volumes of chilled acetone were added with vigorous vortex. After incubation at −20 °C, overnight samples were centrifuged at 20 000g for 30 min. Supernatant was gently removed, and the pellet was washed with 800 μL of chilled acetone/water mixture (85/15, v/v %). Next, a two-phase, on-pellet-digestion procedure was performed; phase one was the digestion-aided pellet dissolution: 100 μL of Tris buffer (50 mM, pH 8.5) containing trypsin at an enzyme/substrate ratio of 1:30 (w/w) was added to the pellet and incubated at 37 °C for 6 h with vigorous vortexing at 700 rpm in the Eppendorf Thermomixer. In phase two, complete cleavage was attained by adding another batch of trypsin solution at an enzyme/substrate ratio of 1:25 (w/w) followed by incubation at 37 °C overnight (12 h) with vortexing. Digestion was stopped with 1 μL of formic acid. Supernatant from each sample containing tryptic peptides derived from 6 μg of proteins was used for each LC/MS analysis.
Quantitative 2D-DIGE Analysis
Quantitative 2D-DIGE was performed as previously described.23,37,38 In brief, protein extracted from hibernating myocardial tissue (n = 22) was labeled with Cy3 or Cy5 and compared with age-matched sham myocardial samples (n = 11), which were pooled, labeled with Cy2, and loaded on each gel. The first dimension included isoelectric focusing on pH 3–10 nonlinear 24 cm Immobiline dry strips followed by separation on 12% SDS-PAGE gels. Scanned images of the gels were matched and analyzed with DeCyder v6.5 software (GE Healthcare). We manually extracted the spot volume data and calculated average LAD/sham ratios and unpaired t tests for all matched spots. Protein identification was accomplished with MALDI-TOF and nano-LC/MS.
Nanoflow, Reversed-Phase LC/MS
A Spark Endurance autosampler (AS1, Emmen, Holland) and an ultrahigh pressure Eksigent (Dublin, CA) Nano-2D Ultra capillary/nano-LC instrument were used for nano-LC analysis. A nano-LC/nanospray setup devised in house was employed, which utilized a direct trap-column connection and homogeneous heating; the large-ID trap not only enabled a large loading volume but also dampened pump noise to promote reproducible gradient delivery. This feature, in conjunction with homogeneous heating and programmed tip washing, affords exceptional run-to-run reproducibility. Heart tissue digests were separated on a 75 cm column (50 μm I.D. and packed with 3 μm C18 particles in lab) to permit extensive analysis.
Mobile phase A and B were 0.1% formic acid in 2% acetonitrile and 0.1% formic acid in 82% acetonitrile, respectively. Digests containing 6 μg of peptides were loaded onto a large-ID trap (300 μm ID × 0.5 cm, packed with Zorbax 3-μm C18 material) with 1% B at a flow rate of 10 μL/min, and the trap was washed for 3 min before being switched in line with the nanoflow path. A series of optimized nanoflow gradients (with a flow rate at 250 nL/min, 3 to 8% B over 10 min; 8 to 25.5% B over 220 min; 24 to 36% B over 115 min; 38 to 63% B over 55 min; 63 to 97% B in 5 min, and isocratic at 97% B for 15 min) were used for separation.
An LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, San Jose, CA) was used for detection. The instrument was operating under data-dependent product ion mode. One scan cycle included an MS1 survey scan (m/z 310–2000) at a resolution of 60 000 to acquire the precursor peaks of peptides, followed by seven MS2 scans at CID activation mode, to fragment the top seven most abundant precursors in the survey scan. Dynamic exclusion was used with one repeat count, 35 s repeat duration, and 60 s exclusion duration. The target value for MS1 by Orbitrap was 8 × 106, under which the Orbitrap was calibrated for mass accuracy and FT transmission. The use of high target value on the Orbitrap enabled a highly sensitive detection without compromising the mass accuracy and resolution. The activation time was 30 ms, the isolation width was 1.5 Da for ITMS, the normalized activation energy was 35%, and the activation q was 0.25.
Protein Identification and Ion-Current-Based Quantification
The LC/MS raw data were searched against the reviewed Sus scrofa Uniprot-Swissprot protein database (released September 2012) with 1412 protein entries using SEQUEST-based Proteome Discoverer (version 1.2.0.208, Thermo-Scientific). Raw files were imported into the package, and DTA files were generated from MS2 spectra. Given the limited coverage of the pig database, the human database from the same source was combined for searching. Mass tolerances for precursor and fragment ion mass were 15 ppm and 0.5 amu. One missed cleavage was permitted for fully tryptic peptides. Carbamidomethylation of cysteines was set as a fixed modification, and a variable modification of methionine oxidation was allowed. The false discovery rate (FDR) of identification was estimated by a target-decoy search strategy, using a concatenated database containing both forward and reversed sequences. The search results were later imported into SIEVE (version 2.1, Thermo Fisher Scientific) for matching with quantitative information and data summary/visualization. To achieve a confident identification, we employed a set of strict cutoff thresholds to yield a FDR of 0.8% at peptide level. “Quantifiable Proteins” had to have at least two distinct peptides quantified, with high-quality ion current (S/N > 10) without missing data in any replicate on a protein level.
Ion-current-based label-free quantification was performed in two steps. First, the SIEVE package was employed to import the raw files and to perform chromatographic alignment and detection, extraction, and integration of the peak areas of peptide ion currents. The LC–MS runs were aligned with an adaptive tiled algorithm;39 then, quantitative frames, each containing a group of ion-current AUC (area-under-the-curve) data from the same peptide, were defined using a set of stringent criteria, including the requirement of S/N > 10 for frame detection and the threshold windows of m/z <±10 ppm and retention time <±1 min for frame definition. Normalization of peak areas for individual samples was carried out based on the sum of total ion-current (TIC) area in each run. A peptide ID was assigned to a frame by linking the identification information from the MS2 scans associated with the frame. After integration of AUC at frame level, a Perl script was developed for further quantitative analysis. Protein ratios in hibernating versus sham groups were computed by aggregating the AUC data on frame levels to peptide and then protein levels using a relative-variance-based weighting model. The formula for aggregation of ratios (R) is R = (∑iN(Ri/σi))/(∑iN(1/σi)), where σ is defined as (standard deviation (SD)/ratio), and Ri is the lower level of ratios (e.g., frame or peptide ratios). The statistical significance of difference between groups were assessed by the Student’s t test at frame levels; then, the corresponding p values on protein levels were determined by Fisher’s exact test.
Results and Discussion
1. Optimization of the Analytical Strategy
Although ion-current-based methods may provide higher sensitivity and quantitative accuracy over other label-free approaches such as spectral counting,19,40,41 they are more technically demanding when analyzing large experimental sample sizes because of their requirement for a sensitive and selective MS1 detection and highly reproducible sample preparation and chromatographic separation.18,42,43.19,40 Furthermore, extensive proteomic coverage is desirable to enhance the quantification of lower abundance proteins that may be regulators of high interest.19,40,44 To achieve these goals, we thoroughly optimized the analytical strategy using whole myocardial tissue samples.
1.1. Reproducible and Efficient Preparation and Digestion of the Myocardial Samples
For reliable profiling of cardiac tissue, it is necessary to conduct quantitative protein extraction with effective removal of nonprotein components from the tissue matrix and extraction buffer while maintaining reproducible peptide recovery. To accomplish this, we homogenized tissue in a buffer containing a relatively high concentration of detergent cocktail. (See the Experimental Section.) These components were selected by balancing the considerations of extraction efficiency, the desire to identify hydrophobic membrane proteins, and compatibility with removal procedure during subsequent sample preparation/precipitation steps. Using this buffer, a highly reproducible and efficient extraction was achieved, with protein yield of 92 ± 2.1 mg/g of the wet tissue mass (Supporting Information, SI Figure 1A).
The precipitation/on-pellet-digestion procedure is composed of two steps. First, an overnight cold acetone precipitation was employed to eliminate nonprotein matrix components (e.g., lipids and small-molecule nucleic acids) and detergents while maintaining high protein recovery. The precipitation procedure was carefully optimized to avoid enclosure of detergents within large chunks of sediment. Second, the protein pellet was subjected to a two -phase tryptic digestion procedure under constant agitation. Phase 1 brings the protein pellet into solution by adding trypsin to cleave the precipitated proteins into large, soluble tryptic fragments, while phase 2 involves reduction, alkylation, and completion of enzymatic digestion. The on-pellet digestion procedure circumvents the need of pellet reconstitution, which is required for a conventional in-solution digestion procedure but difficult to accomplish for pellets containing membrane proteins, unless high concentrations of detergents and urea are used.20,35,44 To achieve highly reproducible, complete digestion of all samples in the experimental set, key conditions for the two-phase digestion were extensively optimized. As measured by a modified BCA (bicinchoninic acid assay) method described previously,20 high and reproducible peptide recoveries were observed across the 24 samples, as shown in SI Figure 1B in the Supporting Information. Such a high level of recovery and reproducibility greatly contributes to accurate ion-current-based quantification.
1.2. Extensive, Sensitive, and Reproducible Nano–LC/MS Analysis
Because of the high complexity of the myocardial proteome,7,23 a large number of tryptic peptides are retrieved, which renders it challenging to achieve in-depth proteomic investigation. Here we optimized a method for extensive reversed-phase chromatographic separation to enhance the likelihood of quantifying low-abundance peptides that may include regulatory proteins of high interest. Because the quantitative strategy requires the accurate match of peptide ion currents among parallel runs, high run-to-run reproducibility of retention times and MS signal intensities are critical for reliable profiling.18,21 To address these requirements, we utilized a nano-LC/nanospray configuration with low void volume, high separation efficiency and reproducibility, and a long, heated nano-LC column with a shallow, 7 h gradient for analysis of the myocardial digests. As to the MS detection, an Orbitrap analyzer operating under “ion-overfilling” conditions was employed to achieve high quantitative sensitivity.
The chromatographic strategy was thoroughly optimized for the separation of myocardial digests and holds several salient advantages. First, the chromatographic setup eliminates the in-valve void volume between trap and column that is typical in a more conventional configuration,45 significantly improving peak shapes and reducing tailing.20,21 Second, a long reversed-phase nanocolumn (75 cm in length and packed with 3 μm materials) and a 7 h shallow gradient were used for separation to enhance the analysis of low abundance peptides, as suggested by our experiments and by others.20,35,41,46 Moreover, the sufficient separation between peptide peaks and interferences also enhanced the success rate of matching among many runs, resulting in minimal missing data, as found in our pilot study. Heating the long column homogenously during separation decreased bandwidth (fwhm) on average by >40% for peptides eluted within the first quarter of the chromatographic window when the temperature was elevated from room temperature to 50 °C. By balancing the considerations of resolution gain and thermostability of C18 materials, a temperature at 52 °C was selected for separation.
A third advantage of the chromatographic approach is that it provides good reproducibility in terms of both retention times and the area under the curve (AUC) of peptide ion currents. This was achieved with the constant separation temperature and a large I.D. (300 μm I.D.) trap along with a bidirectional flow path in the nano-LC analysis cycle (Experimental Section). A previous study indicated that poor chromatographic reproducibility during the long, shallow gradient could occur due to the incomplete mixing of mobile phases and inaccuracy/variation in mobile phase delivery (i.e., the pump noise).20,47 We found that a 300 μm I.D. trap substantially increased the reproducibility of separation by providing improved gradient mixing and dampening pump noise, which results in reproducible gradient delivery to the downstream nanocolumn. An additional benefit of our nano-LC/MS strategy is that the large-I.D. trap provides a large loading capacity for peptides without column overloading, thus enhancing sensitivity for peptide identification/quantification. The optimal loading amount was identified experimentally. SI Figure 2 in the Supporting Information shows that hydrophilic peptides were more susceptible to trap overcapacity than hydrophobic peptides, so these peptides determined the optimal loading mass. On the basis of the data in SI Figure 2 in the Supporting Information, a peptide loading mass of 6 μg was chosen for each run.
A high-resolution mass analyzer is preferred for ion-current-based profiling because it permits accurate and facile matching of the ion-current peaks of the same peptide among parallel runs as well as high quantitative specificity and confident peptide identification.19,48 In this study, an Orbitrap MS was utilized to produce the ion-current data. Because of the unique features of its electrostatic fields, the Orbitrap analyzer is much less prone to space charge effect than most other types of MS analyzers.49,50 In previous studies, we demonstrated markedly increased analytical sensitivity for proteomic analysis by overfilling the Orbitrap with 10 to 20 times more ions under the dynamic injection control by AGC (automatic gain control).20,51,52 This advance is useful when analyzing highly complex proteomic samples, where peptides with low abundance may escape detection/quantification due to insufficient sensitivity. It was observed that the overfilling approach (8E6) detected an additional15% of qualified peptide precursors in myocardial samples and improved signal-to-noise ratios of lower abundance peptides, with <6 ppm mass error.
A representative base peak chromatogram for the analysis of myocardial samples under the optimized nano-LC/MS conditions is shown in Figure 1. An extended peptide elution window of ∼340 min and peak capacity of ∼1120 were achieved. This high level of separation facilitated the peak alignment and quantification of low-abundant proteins. To investigate the performance of the analytical method for the expression profiling of the myocardial proteome at 24 replicates, we evaluated reproducibility of chromatographic separation and Orbitrap analysis with 24 repeated injections of the same pooled myocardial sample over a 7 day period. The reproducibility of chromatographic separation and precursor ion-current AUC was evaluated using 20 peptides randomly selected at different retention times in the peptide elution window (SI Figure 3 in the Supporting Information). The relative standard deviation (RSD%) for retention times and AUC were, respectively, in the range of 0.6–2.2% and 4.9–15.4% for the 20 replicate runs. This high level of reproducibility could be attributed to two factors: (i) the reproducible chromatographic configuration previously described and (ii) the efficient and reproducible precipitation/on-pellet-digestion procedure.
Figure 1.

Representative base peak chromatogram of swine heart digest. The column was 75cm -long with 50 μm I.D. and 3 μm C18 particles and heated to 52 °C. A wide peptide elution window of ∼340 min and a peak capacity >1100 (estimated based on average fwhm of peptide peaks) were achieved.
2. Comparison of the Proteomes of Hibernating versus Normal Myocardium
2.1. Ion-Current-Based Quantification
In this study, the optimized analytical strategy allowed reproducible sample preparation and nano-LC separation, with sensitive Orbitrap MS detection of peptides in complex myocardial samples. To minimize quantitative false-positives arising from time-dependent factors such as potential drifts in nano-LC/ionization performance and possible instability of some tryptic peptides,53 samples from the 24 animals were prepared and analyzed in a random order.
Several ion-current AUC quantification software packages, including SIEVE (Thermo Fisher Scientific), Decyder MS (GE Healthcare), and Progenesis LC–MS (nonlinear) were evaluated, and Sieve was finally chosen for its ability to smoothly analyze the very large data sets of 24 runs generated by the long gradient nano-LC separation and the overfilled Orbitrap analyzer. The optimal parameters for peak alignment, peptide ion matches, and normalization were identified by analyzing 24 repetitive runs of the pooled tissue digest. Because of the reproducible and efficient sample preparation steps, chromatographic separation, and MS detection, high analytical reproducibility was achieved, as expressed by the excellent Sieve alignment scores (0.86 to 0.92, with 1 being the maximum) across the 24 runs.
We compared the myocardial proteomes of hibernating animals (n = 12) versus nonhibernating sham controls (n = 12). To perform confident proteomic quantification, we employed stringent criteria to define “quantifiable proteins”, including (i) strict criteria for peak detection and frame generation, so that only peptides with high-quality peptide ion-current data (e.g., S/N > 10) are quantified; (ii) high cutoff thresholds for protein identification resulting in a low peptide FDR of 0.8%; and (iii) each “quantifiable protein” must be quantified independently by two or more unique “quantifiable peptides” that meet both of the previous two requirements. Under these criteria, 911 unique protein groups were quantified with high confidence. All proteins were quantified in the 24 biological samples without any missing data at the protein level. Given the very stringent criteria for quantification, the fact that every protein was quantified in each of the 24 biological replicates and that the swine protein database is very incomplete (only 1412 total pig protein entries in the reviewed, nonredundant Uniprot database), this study achieved relatively extensive proteomic quantification. All quantified proteins are listed in Supplemental Table 1 in the Supporting Information.
2.2. Quantitative Assessment of False Discovery of Altered-Proteins
The false-positive discovery of significantly altered proteins represents a common problem for proteomics experiments.29,31,54 It arises primarily from two sources: biological variability and technical variability.29 To reduce biological variability, the current study conducted well-controlled animal experiments (cf. Experimental Section) and employed a relatively large number of biological replicates (n = 12 animals for each group). To minimize technical variability, we employed a reproducible sample preparation and LC/MS strategy to enhance quantitative precision and set stringent criteria to define “quantifiable proteins.”
To experimentally estimate the false-positive discovery, we utilized a decoy sample set (i.e., the “experimental null”) consisting of 24 samples from the sham subjects. Among these, 12 were randomly assigned as the “experimental group” and the rest were assigned as the “control group”. The decoy samples were prepared and analyzed by LC/MS randomly interspaced with the hibernating/sham sample sets. Obviously, the “significantly-altered” proteins discovered in the decoy sample set are false-positives due to both biological and technical variations. The false-positive discovery under a given set of cutoff thresholds (p value and fold change) was calculated by uniformly applying the thresholds to both the hibernating/sham sample set and the decoy sample set. The false-positive discovery rate was defined as the ratio of the number of false-positives in the decoy set versus that in the hibernating/sham set. This experimental approach provides a straightforward and promising alternative to statistical approaches that mostly address the “multiple hypothesis testing” problem.32−34,55
Finally, the cutoff criteria of >1.25-fold change (up- or down-regulated) and p < 0.01 between hibernating myocardium and healthy controls were determined as optimal. The volcano plots (fold change vs p value) under the optimal thresholds are shown in Figure 2. In the sham data set, the vast majority of proteins showed a ratio close to 1 (i.e., 0 on Y axis), indicating the high quantitative accuracy of the overall proteomics strategy. There were only six false-positives in the sham data set using these thresholds; by comparison, under the same thresholds, 225 significantly altered proteins were discovered in the hibernating/sham set (shown in SI Table 1 in the Supporting Information), resulting in a low FDR of ∼3%. Therefore, the developed ion-current-based method is capable of reliably identifying small changes, largely due to the high precision of the well-controlled, ion-current-based approach.
Figure 2.
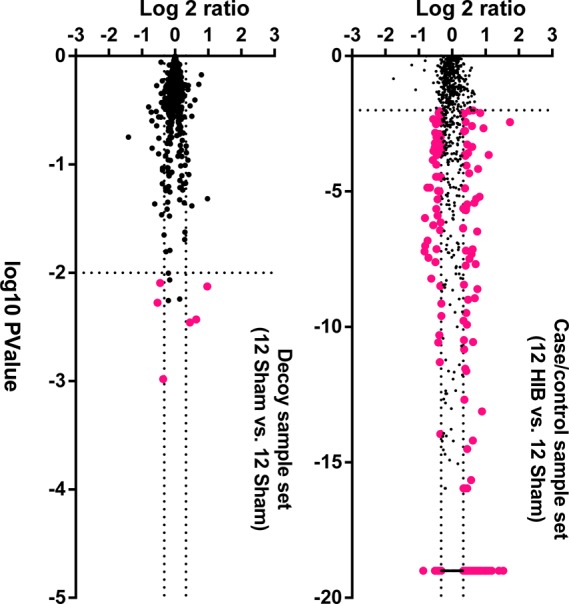
Volcano plots of the (A) decoy and (B) HIB versus sham sample sets. The decoy set consists of only sham subjects, with 12 samples randomly assigned as the experiment group and the other 12 as the control group. The Y and X axes, respectively, show the protein level ratios between two groups and the p values for the comparison. Each dot represents a unique protein group, and the dotted lines denote the optimized cutoff thresholds that define significantly altered proteins (shown as red dots).
3. Comparison of Protein Quantification by the Ion-Current-Based Approach versus 2D-DIGE
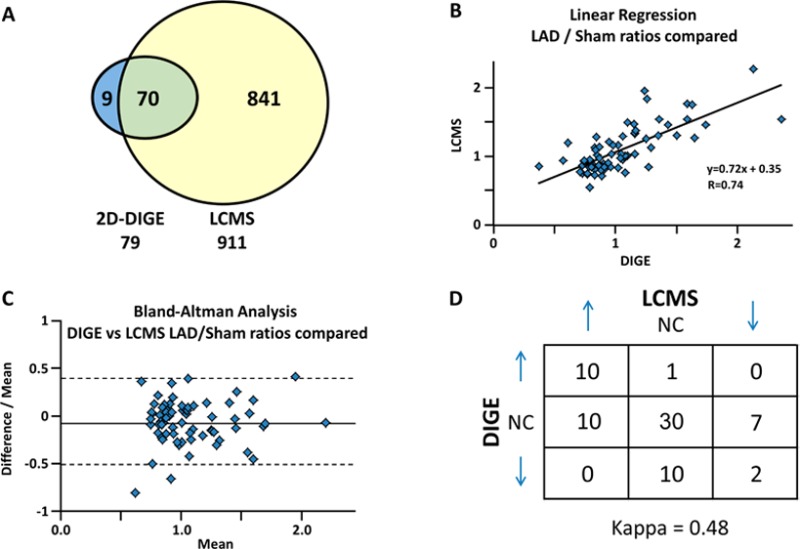
As a means of cross-validation, we compared the quantitative results obtained in this study with those by 2D-DIGE. We have evaluated proteomic quantitation using 2D-DIGE in swine with chronic hibernating myocardium in the same animal model as used here.23,37 Here we show that the ion-current-based quantitation not only confirms most of the previously reported relative protein changes between hibernating and normal myocardium but also greatly increases the number of quantified proteins in porcine myocardial tissue. While 2D-DIGE quantified 79 proteins, the ion-current-based method quantified 911 protein groups with high confidence. A total of 70 proteins were common to both proteomic approaches (Figure 3A). Figure 3B demonstrates good correlation between the altered proteins by the two independent approaches. Correlation between the two methods was further confirmed with a Bland–Altman analysis (shown in Figure 3C), showing no systematic variation between the two approaches. Importantly, we found no discordant results, that is, proteins that were altered in different directions by the two methods (Figure 3D). Representative comparison of selected proteins from 2D-DIGE and ion-current-based methods is shown in Figure 4. The favorable comparison of results between the two orthogonal methods using two independent experimental data sets supports the reliability of the developed ion-current-based approach to quantify regional protein level changes in this reproducible animal model.
Figure 3.

Comparison of the quantitative results by 2D-DIGE and the ion-current-based (ICB) method for the comparison of hibernating myocardium (HIB) versus sham (N = 12/group). (A) Overlap of the proteins quantified and identified by 2D DIGE and ICB. Despite the incomplete swine sequence database, ICB quantified nine times more proteins than 2D-DIGE; of these 911 quantified proteins, 70 were common to each proteomic approach. (B) Linear regression plot of the common proteins shows good agreement of the two methods. (C) Bland–Altman analysis (95% confidence intervals) plot of the difference between each data pair divided by the mean versus the mean of the pair. 95% of the time, there was <45% variability between the two measurements, confirming good agreement between the two methods. (D) Measurement of the concordance of the two methods, observed as a proportion of the maximum possible, provided a good kappa score of 0.48.
Figure 4.

Representative data for the relative quantification of key myocardial proteins in hibernating (HIB) versus sham hearts using DIGE and ion-current-based (ICB) methods.
4. Physiological Features of Chronic Hibernating Myocardium Corresponding to the LC–MS Proteomic Profiling
Hibernating myocardium is an adaptive state characterized by regional down-regulation in myocardial function and metabolism in response to repetitive myocardial ischemia,56 which contrasts with the contractile dysfunction secondary to a myocardial infarction in that the tissue is viable with no evidence of myocardial scar. An important aspect of this unique adaptation is that revascularization and alleviation of chronic ischemia results in the improvement of ventricular function over time. Figure 5 shows the salient pathophysiological features of chronic hibernating myocardium in this model.1 Three or more months after surgical instrumentation, the LAD coronary artery develops a critical stenosis or total occlusion with collateral dependent myocardium (Figure 5A). The stenosis limits the ability of coronary blood flow to increase in response to stress (Figure 5B). Measurements of regional myocardial perfusion using microspheres demonstrate an inability to increase blood flow above the resting level in response to pharmacological vasodilation with adenosine (coronary flow reserve) in hibernating LAD regions as compared with the usual five- to six-fold increase in flow to the normally perfused remote myocardium. As a result of repetitive reversible ischemia from the stenosis, myocardial contractile function as assessed using echocardiographic measurements of regional wall thickening is depressed in the LAD region in comparison with normally perfused myocardium (Figure 5C).
Figure 5.
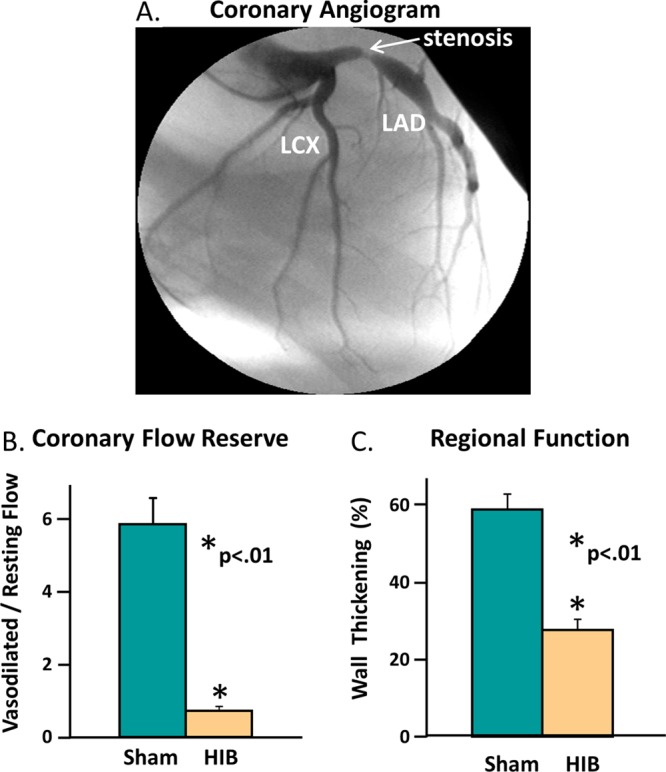
Important pathophysiological features of chronic hibernating myocardium (HIB) observed in the swine models. (A) Coronary angiogram shows that the left anterior descending coronary artery (LAD) develops a critical stenosis 3 months after surgical instrumentation. (B) Comparison of sham and HIB animals for LAD coronary flow reserve in response to vasodilator stress. Vasodilated flow in hibernating myocardium did not increase above resting levels. (C) Comparison of sham and HIB animals for contractile function in the LAD region. Regional LAD wall thickening was depressed at rest distal to the chronic coronary stenosis.
While there is no evidence of myocardial infarction by vital tissue staining in this model, microscopic evaluation demonstrates a regional increase in cardiomyocyte diameter indicating cellular hypertrophy (Figure 6). These changes are restricted to the LAD region and arise from a low rate of myocyte loss secondary to programmed cell death or apoptosis.57 There is also a slight increase in interstitial connective tissue in hibernating myocardium versus normal remote regions. Previous studies have summarized additional ultrastructural findings, which include myofibrillar loss and increased glycogen content in the absence of sarcolemmal disruption.58
Figure 6.

Microscopic images of chronic hibernating myocardium versus normal myocardium. Top panels show trichrome stained normal and hibernating myocardium at low power. Blue areas denote increasing interstitial connective tissue between red-stained myocytes in hibernating regions. High-power views of H&E stained myocytes (lower panels) show enlarged hypertrophied myocytes in hibernating myocardium as compared with normal sham controls.
5. Functional Characterization of Significantly Altered Proteins in Chronic Hibernating Myocardium
Significantly altered proteins discovered by the ion-current-based method were annotated and characterized. We categorized the 225 differentially altered proteins (Supplemental Table 2 in the Supporting Information) by their biological processes using Gene Ontology analysis; the major categories are: metabolism (35), apoptosis and cell death (39), organization of cytoskeleton (40), transcription and translation (25), protein processing (18), cell differentiation and proliferation (7), muscle contraction (20), ion transport (6), heat shock proteins (9), and signal transduction (8). A pictorial view of the findings is shown in Figure 7. The major GO categories and selected differentially expressed proteins are discussed in the subsequent sections and summarized in the Tables. The pattern of differential protein expression in chronic hibernating myocardium revealed many molecular pathways through which myocytes adapt to repetitive ischemia. A small subset of the major differentially altered proteins is discussed later.
Figure 7.
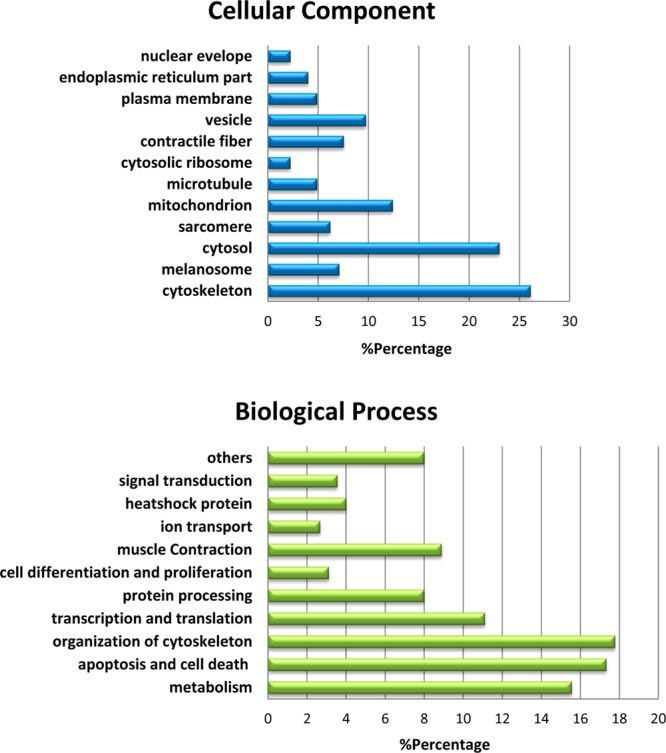
Distribution of the 225 significantly altered proteins in hibernating versus sham animals by cellular components and biological processes.
5.1. Differentially Altered Energetics and Enzymes Involved in Mitochondrial Electron Transport, Glycolysis, and Myocardial Metabolism in Hibernating Myocardium
Previous physiological studies demonstrated reduced regional myocardial oxygen consumption3 and mitochondrial function in hibernating myocardium.59 These alterations suggest down-regulation of myocardial oxidative metabolism, which reduces energy requirements and prevents an oxygen supply and demand imbalance.2,60 Table 1 summarizes significantly altered mitochondrial proteins discovered in this category.
Table 1. Significantly-Altered Mitochondrial Enzymes Involved in Aerobic Metabolism.
| name | ID | description | HIB/sham |
|---|---|---|---|
| Tricarboxylic Acid Cycle, Mitochondrial | |||
| MDHM | P00346 | malate dehydrogenase | 0.76 |
| FUMH | P07954 | fumarate hydratase | 0.74 |
| SUCB2 | P53590 | succinyl-CoA ligase, subunit beta | 0.70 |
| Electron Transport, Mitochondrial | |||
| NDUV2 | P19404 | NADH dehydrogenase flavoprotein 2 | 0.73 |
| NDUS2 | O75306 | NADH dehydrogenase iron-sulfur protein 2 | 0.78 |
| QCR2 | P22695 | cytochrome b-c1 complex subunit 2 | 0.72 |
| UCRIL | P0C7P4 | putative cytochrome b-c1 complex subunit Rieske-like protein 1 | 0.78 |
| Fatty Acid Metabolism, Mitochondrial | |||
| CPT1B | Q92523 | carnitine O-palmitoyltransferase 1, muscle isoform | 0.71 |
| PCCB | P79384 | propionyl-CoA carboxylase beta chain | 0.76 |
| ACADL | P79274 | long-chain specific acyl-CoA dehydrogenase | 0.75 |
| ACADS | P79273 | short-chain specific acyl-CoA dehydrogenase | 0.76 |
| ACADV | P49748 | very long-chain specific acyl-CoA dehydrogenase | 0.79 |
| Amino Acid Catabolism, Mitochondrial | |||
| IVD | P26440 | isovaleryl-CoA dehydrogenase | 0.55 |
| MMSA | Q02252 | methylmalonate-semialdehyde dehydrogenase [acylating] | 0.61 |
Out of 113 quantifiable proteins localizing to mitochondrion, 17 were down-regulated and all but one (PRDX3) were directly involved in oxidative metabolism (Table 1). While many other enzymes involved in oxidative metabolism are also decreased in hibernating myocardium, the extent of changes did not reach the magnitude we established for selection. Similar reductions of a more limited number of mitochondrial enzymes involved in aerobic respiration were previously reported using 2D-DIGE by our laboratory as well as others.13,23,61 Reductions in electron transport chain proteins, fatty acid oxidation proteins, and amino acid catabolism proteins shown here are compatible with previous in vitro observations of reduced mitochondrial oxidative metabolism.59
The down-regulation of mitochondrial aerobic metabolism proteins is not a reflection of a global reduction in mitochondrial content in myocytes because we have previously shown that total mitochondrial protein per gram of myocardial tissue is not changed in chronic hibernating versus normal myocardium.23 In this study, the proteomics results confirm this as the majority of mitochondrial proteins found here, which are not involved in oxidative respiration, were not altered (SI Table 1 in the Supporting Information).
Metabolic enzymes listed in Table 2 were altered in a fashion that promotes glycolysis as well as glycogen storage, which are new proteomic observations for chronic hibernating myocardium. For example, hibernating myocardium had increased expression levels of the glycolytic enzymes alpha-enolase, beta-enolase (ENO1/3), GAPDH, and 6-phosphofructokinase (PFKM). This would facilitate additional ATP production without additional oxygen, and glycolytic ATP is critical to maintain sarcolemmal ion channel function. Under aerobic conditions, the resultant pyruvate from glycolysis enters the Krebs cycle via the pyruvate dehydrogenase complex (PDC), which is reduced along with reduced PDC activity in hibernating myocardium.23 Our ion-current-based proteomics data shows an increase in LDHA and a decrease in LDHB, which would facilitate the conversion of pyruvate to lactate during anaerobic conditions produced during stress because increases in oxygen delivery are limited in hibernating myocardium. A decrease in LDHB was also previously demonstrated with 2D-DIGE.23 The ion-current-based proteomics method also confirmed reductions in both the alpha and beta subunits of pyruvate dehydrogenase E1 component.
Table 2. Significantly Changed Enzymes of Anaerobic Glycolysis and Glycogenesis.
| name | ID | description | HIB/sham |
|---|---|---|---|
| Glycolysis | |||
| ENO1 | P06733 | alpha-enolase | 1.46 |
| ENO3 | Q1KYT0 | beta-enolase | 1.30 |
| GAPDH | P00355 | glyceraldehyde-3-phosphate dehydrogenase | 1.35 |
| PFKM | Q2HYU2 | 6-phosphofructokinase, muscle type | 1.31 |
| Glycogen Biosynthesis | |||
| GYS1 | P13807 | glycogen synthase, muscle | 1.43 |
| UGP2 | P79303 | UTP-glucose-1-phosphate uridylyltransferase | 1.28 |
| Pyruvate Metabolism | |||
| PDHA | P29804 | pyruvate dehydrogenase E1 component subunit alpha | 0.74 |
| PDHB | P11177 | pyruvate dehydrogenase E1 component subunit beta | 0.72 |
| LDHA | P00339 | l-lactate dehydrogenase A chain | 2.13 |
Increased myocardial glycogen has been reported in hibernating myocardium and serves to increase substrate availability in the event of a sudden increase in myocardial oxygen consumption. Up-regulation of glycogen synthase and UTP-glucose-1-phosphate uridylyltransferase found here supports this model. Increases in glycogen have also been hypothesized to be a sign of a switch to a fetal myocyte phenotype profile in hibernating myocardium.62 Kim et al. noted that the development of hibernating myocardium is associated with reductions in glycogen synthase kinase.63 Phosphorylation of glycogen synthase causes reduced activity, which is consonant with the shift from fatty acid to glucose utilization.
Collectively, these findings are not only compatible with previously observed reductions in the rate of ATP depletion during simulated acute ischemia in tissue excised from hibernating myocardium versus normal hearts59 but also consistent with in vivo findings demonstrating an attenuated increase in myocardial oxygen consumption during beta-adrenergic stimulation3 and a reduction in the baseline and stimulated energetic state of hibernating myocardium using MR spectroscopy.64 These findings provide further support for the notion that the intrinsic adaptive response to ischemia, as profiled in this proteomic study, reduces myocardial oxygen utilization and preserves myocyte viability by reducing an imbalance between oxygen delivery and metabolic demand.
5.2. Up-Regulation of Anti-apoptotic and Cell Survival Programs in Hibernating Myocardium
Mitochondrial proteins play a critical role in respiration but can also produce deleterious reactive oxygen species (ROS) under hypoxic conditions, resulting in oxidative stress that may reduce myocyte survival. The current study suggests that hibernating myocardium counteracts the deleterious effects of ROS by increasing the production of a variety of cell-survival molecules listed in Table 3. These regulators play prominent roles in scavenging ROS and decreasing superoxide formation to protect cells from ROS-induced damage and apoptosis. This finding correlates with limited observations from 2D-DIGE, where peroxiredoxin-2 and superoxide dismutase were increased in hibernating myocardium.23
Table 3. Significantly Altered Stress and Cardioprotective Proteins.
| name | ID | description | HIB/sham |
|---|---|---|---|
| Cardioprotection | |||
| CARP | Q865U8 | cardiac ankyrin repeat protein | 1.86 |
| DSP | P15924 | desmoplakin | 1.26 |
| MIF | P80928 | macrophage migration inhibitory factor | 1.49 |
| Cellular Response to Oxidative Stress | |||
| TRAP1 | Q12931 | heat shock protein 75 kDa, mitochondrial | 1.33 |
| PRDX | Q06830 | peroxiredoxin-1 | 1.52 |
| TXN | P82460 | thioredoxin | 1.81 |
| GPX1 | Q8MJ14 | glutathione peroxidase 1 | 1.63 |
| Anti-Apoptosis | |||
| CRYAB | Q7M2W6 | alpha crystallin B chain | 2.28 |
| APOA1 | P18648 | apolipoprotein A-I | 1.29 |
| HSPB6 | O14558 | heat shock protein beta-6 | 1.54 |
| HSPB1 | Q5S1U1 | heat shock protein 27 kDa | 1.54 |
| 1433Z | P63104 | 14-3-3 protein zeta/delta | 1.27 |
Hibernating myocardium is characterized by a low rate of myocyte apoptosis that leads to myocyte loss with compensatory cellular hypertrophy.57,58 Once adapted, the myocyte apoptosis declines. We identified several upregulated proteins that are involved in inhibiting oxidative stress-induced apoptosis, thus promoting cell survival. Using 2D-DIGE, we previously demonstrated the up-regulation of several antiapoptotic and stress proteins, including HSP27, HSP20, alpha crystalline B chain (CRYAB), and apolipoprotein A-1 (APOA1), in hibernating myocardium.23 In this study, additional up-regulated proteins having a potential role in cardioprotection were discovered, including cardiac ankyrin repeat protein (CARP), macrophage migration inhibitory factor (MIF), and desmoplakin (DSP) as well as peroxiredoxin-1 (PRDX), thioredoxin (TXN), and glutathione peroxidase-1 (GPX1). CARP is a nuclear transcriptional cofactor that increases the resistance to hypoxia-induced apoptosis.65 MIF acts in an autocrine or paracrine fashion to reduce ischemia/reperfusion injury via association with AMP kinase and JNK pathways.66,67 There is also in vivo evidence supporting a cardioprotective role of MIF by reducing oxidative stress in the postischemic heart68 as well as in hypertension and cardiac hypertrophy.69 DSP is an important component of desmosomes and when deficient is associated with increased myocardial fibrosis, myocyte apoptosis, and cardiac dysfunction.70 Also newly identified in this proteomic study are several isoforms of 14-3-3 protein (each were uniquely identified by characteristic peptides), known to regulate cellular processes including apoptosis and cell-cycle reentry. Collectively, the upregulation of these proteins may play important cardioprotective roles in preventing progressive myocyte loss and apoptosis in chronic hibernating myocardium.
5.3. Increased Cytoskeleton and Reduced Contractility
As shown in Figure 6, increased interstitial connective tissue and myocyte hypertrophy or enlargement are among the prominent pathological findings of cellular adaptation in hibernating myocardium. Myocyte cellular hypertrophy is regional and arises from transient myocyte apoptosis in the absence of infarction to maintain normal myocardial wall thickness despite the loss of cardiomyocytes. At the same time, remodeling of the extracellular matrix results in increased interstitial structural proteins.71 We identified a significant increase in multiple cytoskeletal proteins (Table 4), consistent with an expanded internal cytoskeleton required by hypertrophied myocytes.
Table 4. Changes in Cytoskeleton and Contractile Proteins.
| name | ID | description | HIB/sham |
|---|---|---|---|
| Cytoskeleton | |||
| TBB5 | P07437 | tubulin beta | 1.64 |
| TBA1B | P68363 | tubulin alpha-1B | 2.07 |
| DESM | P02540 | desmin | 1.76 |
| VIME | P08670 | vimentin | 1.96 |
| VINC | P26234 | vinculin | 1.28 |
| ACTC | P68032 | actin, alpha cardiac muscle 1 | 1.26 |
| TLN1 | Q9Y490 | talin-1 | 1.28 |
| ACTN4 | O43707 | alpha-actinin 4 | 1.36 |
| FLNC | Q14315 | filamin-C | 1.69 |
| COF1 | P23528 | cofilin-1 | 1.68 |
| PLSL | P13796 | plastin-2 | 3.32 |
| Contraction | |||
| MYH8 | P13535 | myosin-8 | 0.80 |
| TNNT2 | P45379 | troponin T | 0.75 |
| MYPC3 | Q14896 | myosin-binding protein C | 0.78 |
| MYOM1 | P52179 | myomesin-1 | 0.80 |
| MYL3 | P08590 | myosin light chain 3 | 0.77 |
| MYH11 | P35749 | myosin-11 | 1.41 |
In this regard, alpha and beta tubulin, which form the microtubule framework of the cell, increased to presumably provide mechanical stability in the face of increased stress.72 Desmin and vimentin are intermediate filaments that also increased and link the contractile apparatus to the cytoskeleton and other intracellular organelles. Models of pressure overload hypertrophy and heart failure having globally increased myocyte size also report increased desmin.73,74 Vinculin and alpha actin are also known to affect cell morphology, as defects in these proteins can lead to cardiomyopathies and ventricular hypertrophy. Like desmin, an increase in these proteins in hibernating myocardium may be a protective mechanism to stabilize the myocytes. In agreement with early reports by Hein et al. in 2000, talin-1 and alpha-actinin 4 are up-regulated.75 Both proteins bind to vinculin, which in turn binds actin to the cell membrane. Filamin, cofilin, and plastin are cytoskeletal actin-binding proteins, which also have been elevated in hibernating myocytes, and plastin-2 was markedly increased (3.32 fold). Collectively, these cytoskeletal adaptations likely arise from dysynchronous regional contraction in hibernating myocardium and serve to prevent systolic overstretching of hibernating cardiomyocytes by normally functioning myocytes in the remote normally perfused myocardium. The increased cytoskeletal proteins would also stabilize myocytes in the dysfunctional region and prevent the ventricular wall from becoming dyskinetic in response to superimposed acute ischemia.76
Contractile proteins were also reduced in hibernating myocardium with decreases in myosin 8 (MYH8), troponin T (TNNT2), myosin binding protein C (MYPC3), myomesin (MYOM1), and myosin light chain 3 (MYL3) (Table 4). MYH8 is a myosin II isoform integral to myocyte contraction because it uses its ATPase motor to move along the actin filaments. Troponin T regulates the binding of myosin to actin in the presence of Ca2+; MYPC3 holds actin and myosin filaments together, and myomesin is thought to anchor myosin to other filaments such as titan. Myosin light chains are part of the macromolecular myosin enzyme complexes. Consistent with this, both the previous 2D-DIGE and ion-current-based data are indicative of decreasing contractile proteins replaced by cytoskeletal structures.37
Taken together, the increases in cytoskeletal proteins and reductions in contractile proteins produce a regional molecular phenotype that is reminiscent of global hypertrophy seen in diseases such as ventricular pressure overload. Hein et al. outlined a scenario of hypertrophy leading to heart failure, beginning with reversible elevation in cytoskeletal proteins in response to increased stress on the cells, followed by an irreversible stage, as contractile filaments are lost and microtubules and associated proteins produce the densified “stiff” cells observed in failing hearts.75 These changes along with reductions in myocyte metabolism and calcium handling proteins such as the sarcoplasmic reticulum calcium ATPase raise the possibility that the adaptive changes seen in hibernating myocytes primarily represent a consequence of myocyte cell loss and a regional phenotype of cellular hypertrophy. Further comparative proteomic profiling will be required to test this hypothesis.
Conclusions
Using an ion-current-based strategy, we have developed a much more extensive profile of the proteome of hibernating myocardium that has identified new intrinsic adaptations of the heart to repetitive ischemia. Our proteomic method has the following salient features: (i) a highly reproducible sample preparation and nano-LC/MS analysis that enables accurate relative quantification using many biological replicates; (ii) separation on a long column allowing accurate ion-current match and a more in-depth proteomic analysis; and (iii) an approach that can discriminate relatively modest as well as large changes in relative protein abundance. When quantitative results were compared among common proteins from a more limited data set using 2D-DIGE, good agreement between the two orthogonal methods was observed. Of equal importance, even with highly stringent criteria, the ion-current-based strategy quantified nearly an order of magnitude more proteins than 2D-DIGE.
The results from this quantitative proteomic approach not only expand upon known pathophysiological changes in hibernating myocardium including metabolism and stress response but also identify new insights into the mechanisms of adaptation to chronic ischemia in hibernating myocardium, such as the elevation in glycolytic enzymes, increases in intracellular cytoskeleton proteins, and alterations in cardiac contractile proteins. The extensive number of concordant protein changes across multiple pathways highlights the complexity of the intrinsic physiological response of the heart to ischemia as well as underscoring potential limitations related to focusing on single pathways in a complex physiological adaptation. Further studies will be required to examine whether these protein changes are reversible or contribute to persistent contractile dysfunction in the absence of myocardial scar following coronary revascularization.
Acknowledgments
This work was supported by NIH grants HL55324, HL61610, and HL75324 (J.M.C.) and U54HD071594 (J.Q.), the Center for Protein Therapeutics (J.Q.), the Department of Veterans Affairs (J.M.C.), and American Heart Association (AHA) award 12SDG9450036 (J.Q.).
Supporting Information Available
Efficient and reproducible extraction of the LAD heart tissues and protein digestion in the sham and hibernating pigs. The average peptide peak area responses versus injection amounts of a heart tissue digest. Reproducibility of retention times and areas-under-the-curves of the 20 representative peptides randomly selected across the entire retention time window for 24 runs of a pooled tissue sample. 911 unique protein groups confidently quantified by the ion-current-based strategy. Dysregulated proteins in hibernating animals versus sham. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
○ J.Q., R.Y., and B.J.P. contributed equally to this work
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fallavollita J. A.; Perry B. J.; Canty J. M. Jr. 18F-2-deoxyglucose deposition and regional flow in pigs with chronically dysfunctional myocardium: Evidence for transmural variations in chronic hibernating myocardium. Circulation 1997, 95, 1900–1909. [DOI] [PubMed] [Google Scholar]
- Kelly R. F.; Sluiter W.; McFalls E. O. Hibernating myocardium: is the program to survive a pathway to failure?. Circ. Res. 2008, 10213–5. [DOI] [PubMed] [Google Scholar]
- Fallavollita J. A.; Malm B. J.; Canty J. M. Jr. Hibernating myocardium retains metabolic and contractile reserve despite regional reductions in flow, function, and oxygen consumption at rest. Circ. Res. 2003, 92148–55. [DOI] [PubMed] [Google Scholar]
- Fallavollita J. A. Spatial heterogeneity in fasting and insulin-stimulated 18F-2-deoxyglucose uptake in pigs with hibernating myocardium. Circulation 2000, 1028908–914. [DOI] [PubMed] [Google Scholar]
- McFalls E. O.; Baldwin D.; Palmer B.; Marx D.; Jaimes D.; Ward H. B. Regional glucose uptake within hypoperfused swine myocardium as measured by positron emission tomography. Am. J. Physiol. 1997, 2721H343–H349. [DOI] [PubMed] [Google Scholar]
- Wiggers H.; Noreng M.; Paulsen P. K.; Bottcher M.; Egeblad H.; Nielsen T. T.; Botker H. E. Energy stores and metabolites in chronic reversibly and irreversibly dysfunctional myocardium in humans. J. Am. Coll. Cardiol. 2001, 371100–108. [DOI] [PubMed] [Google Scholar]
- Van Eyk J. E. Overview: the maturing of proteomics in cardiovascular research. Circ. Res. 2011, 1084490–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnetti G.; Husberg C.; Van Eyk J. E. Divide and conquer: the application of organelle proteomics to heart failure. Circ. Res. 2011, 1084512–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stastna M.; Abraham M. R.; Van Eyk J. E. Cardiac stem/progenitor cells, secreted proteins, and proteomics. FEBS Lett. 2009, 583111800–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Ge Y. Comprehensive analysis of protein modifications by top-down mass spectrometry. Circ.: Cardiovasc. Genet. 2011, 46711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Guy M. J.; Norman H. S.; Chen Y. C.; Xu Q.; Dong X.; Guner H.; Wang S.; Kohmoto T.; Young K. H.; Moss R. L.; Ge Y. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J. Proteome Res. 2011, 1094054–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Zhang H.; Ayaz-Guner S.; Chen Y. C.; Dong X.; Xu Q.; Ge Y. Phosphorylation, but not alternative splicing or proteolytic degradation, is conserved in human and mouse cardiac troponin T. Biochemistry 2011, 50276081–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly R. F.; Cabrera J. A.; Ziemba E. A.; Crampton M.; Anderson L. B.; McFalls E. O.; Ward H. B. Continued depression of maximal oxygen consumption and mitochondrial proteomic expression despite successful coronary artery bypass grafting in a swine model of hibernation. J. Thorac. Cardiovasc. Surg. 2011, 1411261–268. [DOI] [PubMed] [Google Scholar]
- Lilley K. S.; Razzaq A.; Dupree P. Two-dimensional gel electrophoresis: recent advances in sample preparation, detection and quantitation. Curr. Opin. Chem. Biol. 2002, 6146–50. [DOI] [PubMed] [Google Scholar]
- Ong S. E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 15376–386. [DOI] [PubMed] [Google Scholar]
- Gygi S. P.; Rist B.; Gerber S. A.; Turecek F.; Gelb M. H.; Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 1710994–999. [DOI] [PubMed] [Google Scholar]
- DeSouza L.; Diehl G.; Rodrigues M. J.; Guo J.; Romaschin A. D.; Colgan T. J.; Siu K. W. Search for cancer markers from endometrial tissues using differentially labeled tags iTRAQ and cICAT with multidimensional liquid chromatography and tandem mass spectrometry. J. Proteome Res. 2005, 42377–386. [DOI] [PubMed] [Google Scholar]
- Wang G.; Wu W. W.; Zeng W.; Chou C. L.; Shen R. F. Label-free protein quantification using LC-coupled ion trap or FT mass spectrometry: Reproducibility, linearity, and application with complex proteomes. J. Proteome Res. 2006, 551214–1223. [DOI] [PubMed] [Google Scholar]
- Domon B.; Aebersold R. Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 2010, 287710–721. [DOI] [PubMed] [Google Scholar]
- Duan X.; Young R. F.; Straubinger R. M.; Page B. J.; Cao J.; Wang H.; Yu Y.; Canty J. M. Jr.; Qu J. A straightforward and highly efficient precipitation/on-pellet digestion procedure coupled to a long gradient nano-LC separation and Oprbitrap mass spectrometry for label-free expression profiling of the swine heart mitochondrial proteome. J. Proteome Res. 2009, 862838–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C.; Li J.; Bu Y.; Hangauer D.; Qu J. An ion-current-based, comprehensive and reproducible proteomic strategy for comparative characterization of the cellular responses to novel anti-cancer agents in a prostate cell model. J. Proteomics 2012, 77, 187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M.; Shitashige M.; Honda K.; Isobe T.; Kuwabara H.; Matsuzuki H.; Hirohashi S.; Yamada T. Label-free quantitative proteomics using large peptide data sets generated by nanoflow liquid chromatography and mass spectrometry. Mol. Cell. Proteomics 2006, 571338–1347. [DOI] [PubMed] [Google Scholar]
- Page B.; Young R.; Iyer V.; Suzuki G.; Lis M.; Korotchkina K.; Patel M.; Blumenthal K.; Fallavollita J. A.; Canty J. M. Jr. Persistent regional downregulation in mitochondrial enzymes and upregulation of stress proteins in swine with chronic hibernating myocardium. Circ. Res. 2008, 102, 103–112. [DOI] [PubMed] [Google Scholar]
- Calvo S. E.; Mootha V. K. The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 2010, 11, 25–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bildl W.; Haupt A.; Muller C. S.; Biniossek M. L.; Thumfart J. O.; Huber B.; Fakler B.; Schulte U. Extending the dynamic range of label-free mass spectrometric quantification of affinity purifications. Mol. Cell. Proteomics 2012, 112M111 007955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler J.; Nothwang H. G. Aqueous polymer two-phase systems: effective tools for plasma membrane proteomics. Proteomics 2006, 6205409–5417. [DOI] [PubMed] [Google Scholar]
- Bindoff L. Mitochondria and the heart. Eur. Heart J. 2003, 243221–224. [DOI] [PubMed] [Google Scholar]
- Leth-Larsen R.; Lund R. R.; Ditzel H. J. Plasma membrane proteomics and its application in clinical cancer biomarker discovery. Mol. Cell. Proteomics 2010, 971369–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr S. A.; Anderson L. Protein quantitation through targeted mass spectrometry: the way out of biomarker purgatory?. Clin. Chem. 2008, 54111749–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifai N.; Gillette M. A.; Carr S. A. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotechnol. 2006, 248971–983. [DOI] [PubMed] [Google Scholar]
- Diamandis E. P. Cancer biomarkers: can we turn recent failures into success?. JNCI, J. Natl. Cancer Inst. 2010, 102191462–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diz A. P.; Carvajal-Rodriguez A.; Skibinski D. O. Multiple hypothesis testing in proteomics: a strategy for experimental work. Mol. Cell. Proteomics 2011, 103M110 004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp N. A.; McCormick P. S.; Russell M. R.; Lilley K. S. Experimental and statistical considerations to avoid false conclusions in proteomics studies using differential in-gel electrophoresis. Mol. Cell. Proteomics 2007, 681354–1364. [DOI] [PubMed] [Google Scholar]
- Margolin A. A.; Ong S. E.; Schenone M.; Gould R.; Schreiber S. L.; Carr S. A.; Golub T. R. Empirical Bayes analysis of quantitative proteomics experiments. PLoS One 2009, 410e7454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J.; Lesse A. J.; Brauer A. L.; Cao J.; Gill S. R.; Murphy T. F. Proteomic expression profiling of Haemophilus influenzae grown in pooled human sputum from adults with chronic obstructive pulmonary disease reveal antioxidant and stress responses. Anal. Chem. 2010, 10, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm B. J.; Suzuki G.; Canty J. M. Jr.; Fallavollita J. A. Variability of contractile reserve in hibernating myocardium: Dependence on the method of stimulation. Cardiovasc. Res. 2002, 56, 422–433. [DOI] [PubMed] [Google Scholar]
- Page B. J.; Young R. F.; Suzuki G.; Fallavollita J. A.; Canty J. M. Jr. The physiological significance of a coronary stenosis differentially affects contractility and mitochondrial function in viable chronically dysfunctional myocardium. Basic Res. Cardiol. 2013, 1084354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unlu M.; Morgan M. E.; Minden J. S. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis 1997, 18112071–2077. [DOI] [PubMed] [Google Scholar]
- Sadygov R. G.; Maroto F. M.; Huhmer A. F. ChromAlign: A two-step algorithmic procedure for time alignment of three-dimensional LC–MS chromatographic surfaces. Anal. Chem. 2006, 78248207–8217. [DOI] [PubMed] [Google Scholar]
- Nilsson T.; Mann M.; Aebersold R.; Yates J. R. 3rd; Bairoch A.; Bergeron J. J. Mass spectrometry in high-throughput proteomics: ready for the big time. Nat. Methods 2010, 79681–685. [DOI] [PubMed] [Google Scholar]
- Tu C.; Li J.; Jiang X.; Sheflin L. G.; Pfeffer B. A.; Behringer M.; Fliesler S. J.; Qu J. Ion-current-based Proteomic Profiling of the Retina in a Rat Model of Smith-Lemli-Opitz Syndrome. Mol. Cell. Proteomics 2013, 12123583–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicker L.; Lin X.; Ivanov A. R. Increased power for the analysis of label-free LC–MS/MS proteomics data by combining spectral counts and peptide peak attributes. Mol. Cell. Proteomics 2010, 9122704–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin N. M.; Yu J.; Long F.; Oh P.; Shore S.; Li Y.; Koziol J. A.; Schnitzer J. E. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat. Biotechnol. 2010, 28183–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J.; Jusko W. J.; Straubinger R. M. Utility of cleavable isotope-coded affinity-tagged reagents for quantification of low-copy proteins induced by methylprednisolone using liquid chromatography/tandem mass spectrometry. Anal. Chem. 2006, 78134543–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premstaller A.; Oberacher H.; Walcher W.; Timperio A. M.; Zolla L.; Chervet J. P.; Cavusoglu N.; van Dorsselaer A.; Huber C. G. High-performance liquid chromatography-electrospray ionization mass spectrometry using monolithic capillary columns for proteomic studies. Anal. Chem. 2001, 73112390–2396. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Zhang R.; Moore R. J.; Kim J.; Metz T. O.; Hixson K. K.; Zhao R.; Livesay E. A.; Udseth H. R.; Smith R. D. Automated 20 kpsi RPLC–MS and MS/MS with chromatographic peak capacities of 1000–1500 and capabilities in proteomics and metabolomics. Anal. Chem. 2005, 77103090–3100. [DOI] [PubMed] [Google Scholar]
- Qu J.; Straubinger R. M. Improved sensitivity for quantification of proteins using triply charged cleavable isotope-coded affinity tag peptides. Rapid Commun. Mass Spectrom. 2005, 19192857–2864. [DOI] [PubMed] [Google Scholar]
- America A. H.; Cordewener J. H. Comparative LC–MS: a landscape of peaks and valleys. Proteomics 2008, 84731–749. [DOI] [PubMed] [Google Scholar]
- Makarov A.; Denisov E.; Kholomeev A.; Balschun W.; Lange O.; Strupat K.; Horning S. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal. Chem. 2006, 7872113–2120. [DOI] [PubMed] [Google Scholar]
- Gorshkov M. V.; Good D. M.; Lyutvinskiy Y.; Yang H.; Zubarev R. A. Calibration function for the Orbitrap FTMS accounting for the space charge effect. J. Am. Soc. Mass Spectrom. 2010, 21111846–1851. [DOI] [PubMed] [Google Scholar]
- Duan X.; Engler F. A.; Qu J. Electron transfer dissociation coupled to an Orbitrap analyzer may promise a straightforward and accurate sequencing of disulfide-bridged cyclic peptides: a case study. J. Mass Spectrom. 2010, 45121477–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C.; Li J.; Young R.; Page B. J.; Engler F.; Halfon M. S.; Canty J. M. Jr.; Qu J. Combinatorial peptide ligand library treatment followed by a dual-enzyme, dual-activation approach on a nanoflow liquid chromatography/orbitrap/electron transfer dissociation system for comprehensive analysis of swine plasma proteome. Anal. Chem. 2011, 83124802–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X.; Dai L.; Chen S. C.; Balthasar J. P.; Qu J. Nano-scale liquid chromatography/mass spectrometry and on-the-fly orthogonal array optimization for quantification of therapeutic monoclonal antibodies and the application in preclinical analysis. J. Chromatogr., A 2012, 1251, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White F. M. The potential cost of high-throughput proteomics. Sci. Signal. 2011, 4160pe8. [DOI] [PubMed] [Google Scholar]
- Xia Q.; Wang T.; Taub F.; Park Y.; Capestany C. A.; Lamont R. J.; Hackett M. Quantitative proteomics of intracellular Porphyromonas gingivalis. Proteomics 2007, 7234323–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canty J. M. Jr.; Suzuki G. Myocardial perfusion and contraction in acute ischemia and chronic ischemic heart disease. J Mol Cell Cardiol 2012, 524822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H.; Fallavollita J. A.; Hard R.; Kerr C. W.; Canty J. M. Jr. Profound apoptosis-mediated regional myocyte loss and compensatory hypertrophy in pigs with hibernating myocardium. Circulation 1999, 100, 2380–2386. [DOI] [PubMed] [Google Scholar]
- Canty J. M. Jr.; Fallavollita J. A. Hibernating myocardium. J. Nucl Cardiol. 2005, 121104–119. [DOI] [PubMed] [Google Scholar]
- Hu Q.; Suzuki G.; Young R. F.; Page B. J.; Fallavollita J. A.; Canty J. M. Jr. Reductions in mitochondrial O(2) consumption and preservation of high-energy phosphate levels after simulated ischemia in chronic hibernating myocardium. Am. J. Physiol.: Heart Circ. Physiol. 2009, 2971H223–H232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassiotis C.; Rajabi M.; Taegtmeyer H. Metabolic reserve of the heart: the forgotten link between contraction and coronary flow. Prog. Cardiovasc. Dis. 2008, 51174–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFalls E. O.; Sluiter W.; Schoonderwoerd K.; Manintveld O. C.; Lamers J. M.; Bezstarosti K.; van Beusekom H. M.; Sikora J.; Ward H. B.; Merkus D.; Duncker D. J. Mitochondrial adaptations within chronically ischemic swine myocardium. J. Mol. Cell. Cardiol. 2006, 41, 980–988. [DOI] [PubMed] [Google Scholar]
- Rajabi M.; Kassiotis C.; Razeghi P.; Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev. 2007, 123–4331–343. [DOI] [PubMed] [Google Scholar]
- Kim S. J.; Peppas A.; Hong S. K.; Yang G.; Huang Y.; Diaz G.; Sadoshima J.; Vatner D. E.; Vatner S. F. Persistent stunning induces myocardial hibernation and protection: flow/function and metabolic mechanisms. Circ. Res. 2003, 92111233–1239. [DOI] [PubMed] [Google Scholar]
- McFalls E. O.; Kelly R. F.; Hu Q.; Mansoor A.; Lee J.; Kuskowski M.; Sikora J.; Ward H. B.; Zhang J. The energetic state within hibernating myocardium is normal during dobutamine despite inhibition of ATP-dependent potassium channel opening with glibenclamide. Am. J. Physiol.: Heart Circ. Physiol 2007, 2935H2945–H2951. [DOI] [PubMed] [Google Scholar]
- Han X. J.; Chae J. K.; Lee M. J.; You K. R.; Lee B. H.; Kim D. G. Involvement of GADD153 and cardiac ankyrin repeat protein in hypoxia-induced apoptosis of H9c2 cells. J. Biol. Chem. 2005, 2802423122–23129. [DOI] [PubMed] [Google Scholar]
- Miller E. J.; Li J.; Leng L.; McDonald C.; Atsumi T.; Bucala R.; Young L. H. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature 2008, 4517178578–582. [DOI] [PubMed] [Google Scholar]
- Qi D.; Hu X.; Wu X.; Merk M.; Leng L.; Bucala R.; Young L. H. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J. Clin. Invest. 2009, 119123807–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga K.; Kenessey A.; Powell S. R.; Sison C. P.; Miller E. J.; Ojamaa K. Macrophage migration inhibitory factor provides cardioprotection during ischemia/reperfusion by reducing oxidative stress. Antioxid. Redox Signaling 2011, 1471191–1202. [DOI] [PubMed] [Google Scholar]
- Li H. W.; Gao Y. X.; Qi Y. F.; Katovich M. J.; Jiang N.; Braseth L. N.; Scheuer D. A.; Shi P.; Sumners C. Macrophage migration inhibitory factor in hypothalamic paraventricular nucleus neurons decreases blood pressure in spontaneously hypertensive rats. FASEB J. 2008, 2293175–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gras E.; Lombardi R.; Giocondo M. J.; Willerson J. T.; Schneider M. D.; Khoury D. S.; Marian A. J. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Invest. 2006, 11672012–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalla N. S.; Saini-Chohan H. K.; Rodriguez-Leyva D.; Elimban V.; Dent M. R.; Tappia P. S. Subcellular remodelling may induce cardiac dysfunction in congestive heart failure. Cardiovasc. Res. 2009, 813429–438. [DOI] [PubMed] [Google Scholar]
- White E. Mechanical modulation of cardiac microtubules. Pfluegers Arch. 2011, 4621177–184. [DOI] [PubMed] [Google Scholar]
- Collins J. F.; Pawloski-Dahm C.; Davis M. G.; Ball N.; Dorn G. W. 2nd; Walsh R. A. The role of the cytoskeleton in left ventricular pressure overload hypertrophy and failure. J. Mol. Cell Cardiol. 1996, 2871435–1443. [DOI] [PubMed] [Google Scholar]
- Rappaport L.; Samuel J. L. Microtubules in cardiac myocytes. Int. Rev. Cytol. 1988, 113, 101–143. [DOI] [PubMed] [Google Scholar]
- Hein S.; Kostin S.; Heling A.; Maeno Y.; Schaper J. The role of the cytoskeleton in heart failure. Cardiovasc. Res. 2000, 452273–278. [DOI] [PubMed] [Google Scholar]
- Fassett J. T.; Hu X.; Xu X.; Lu Z.; Zhang P.; Chen Y.; Bache R. J. AMPK Attenuates Microtubule Proliferation in Cardiac Hypertrophy. Am. J. Physiol.: Heart Circ. Physiol 2013, 304, H749–H758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.