

Abstract
160 nm nanocapsules containing up to 60% of camptothecin in the core and 7–8 polyelectrolyte bilayers in the shell were produced by washless layer-by-layer assembly of heparin and block-copolymer of poly-L-lysine and polyethylene glycol. The outer surface of the nanocapsules was additionally modified with polyethylene glycol of 5 kDa or 20 kDa molecular weight to attain protein resistant properties, colloidal stability in serum and prolonged release of the drug from the capsules. An advantage of the LbL coated capsules is the preservation of camptothecin lactone form with the shell assembly starting at acidic pH and improved chemical stability of encapsulated drug at neutral and basic pH, especially in the presence of albumin that makes such formulation more active than free camptothecin. LbL nanocapsules preserve the camptothecin lactone form at pH 7.4 resulting in triple activity of the drug toward CRL2303 glioblastoma cell.
Keywords: Layer-by-layer, Nanocapsules, Camptothecin, Lactone, Glioblastoma, Cytotoxic
1. Introduction
Among delivery methods for poorly soluble substances with antitumor activity, encasing drugs into micro and nanoparticles is a promising strategy to design implantable devices or injected directly at the tumor site (Marriott et al., 2006; Tsuzuki, 2009; Sivasankar and Kumar, 2010). Several formulations based on nanoparticle technology (Chen et al., 2011; Keck et al., 2008; Lee et al., 2008; Kahlweit, 2008; Parikh and Selvaraj, 2008; Müller et al., 2008) have been developed for oral or intramuscular injections, but only few of them are intended for intravenous injection as albumin-based nanoparticles containing paclitaxel (Abraxane®) (Kim et al., 2008; Cho et al., 2004; Wei et al., 2010). Another strategy, namely, injectable nanocapsules based on crystalline core of poorly soluble anticancer drugs stabilized with a thin shell of biopolyelectrolytes assembled via layer-by-layer (LbL) assembly technique, was recently reported (Shutava et al., 2012).
LbL assembly provides an opportunity to form a nano-thick coating in a controllable manner on the surfaces of variable curvature and size from interacting oppositely charges polyelectrolyte components (Shutava et al., 2012; Lvov, 2000; Sukhorukov, 2002; De Geest et al., 2009; Schneider and Decher, 2004; Chodanowski and Stoll, 2001; Li et al., 1994; Kong et al., 1998; Vertegel et al., 2004; Mu et al., 2012; Lu and Liu, 2012). Anchored to the core surface, the thin shell of hydrophilic polyelectrolytes allow exchange of excipients used on the core preparation. It concentrates nanocolloids without aggregation or recrystallization and supports their high stability in physiologically relevant buffers. Major advantages of LbL based formulation are: a high, up to 70%, drug loading capacity and an ease of further modifying the outer shell to enhance the stealth properties of the nanoparticles (Shutava et al., 2012).
To intensify preparation of LbL capsules on small, less than 200 nm, cores, the washless LbL technique has been developed (Shutava et al., 2012; Lvov et al., 2012; Bantchev et al., 2009). Polyanions and polycations are sequentially added to drug nanocores in the amounts that completely adsorb on the surface of the particles recharging them. No intermediate washing of non-reacted polyelectrolytes is needed in such approach. This minimizes loss of all substances, like those in conventional centrifugation or filtration-based LbL technique using step-wise sample washing. High hydrophilicity of used polymers, block-copolymers of polyaminoacids with polyethylene glycol and polysaccharides, and also low molecular weight i.e. <60 kDa of the shell wall components prevented colloidal aggregation during the LbL formation (Shutava et al., 2012; Schneider and Decher, 2004; Chodanowski and Stoll, 2001; Li et al., 1994; Vertegel et al., 2004).
Camptothecin (CPT) combines low solubility in aqua-based media with low stability of its active form, lactone, which easily hydrolyses into carboxylate already at neutral and slightly alkaline pH (Hatefi and Amsden, 2002; McCarron et al., 2008; Sanna et al., 2009; Dora et al., 2006; Hausheer et al., 1998). Among other nanoparticulated formulations of camptothecin, such as polymer-based delivery systems, lipid, liposome, and solid lipid nanoparticles, polyelectrolyte modified LbL-based nanocapsules of CPT seems to be a promising route to obtain a concentrated dispersion, 0.5–1.0 mg/mL, stable in physiologically relevant buffers and that can be injected intravenously with minimal side effects. We expect that the LbL-coated CPT nanocapsules (Fig. 1) will improve the chemical stability of camptothecin and preserve the active lactone form of the drug by reducing its hydrolysis to an inactive carboxylic form at neutral and alkaline conditions. It makes such nano-formulation more active and less toxic for living body due to lower drug dose.
Fig. 1.
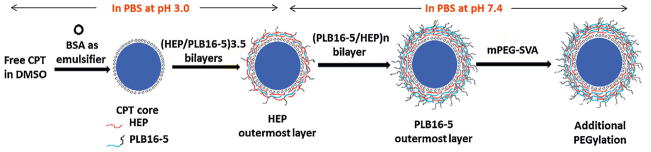
Scheme of CPT nanocapsules preparation.
2. Materials and methods
2.1. Materials
Camptothecin (CPT) was obtained from LC Laboratories, USA. Heparin sodium salt (Hep), bovine serum albumin (BSA), polystyrene sulfonate (PSS), polyethyleneimine (PEI), polyvinylpyrrolidone (PVP), Polysorbate 80, phosphate buffered saline (PBS), dimethyl sulfoxide (DMSO), and acetonitrile were obtained from Sigma–Aldrich and used as received. Fetal bovine serum (FBS) was obtained from Atlanta Biologicals, USA. Block-copolymers of poly-L-lysine with polyethylene glycol (PLB) of different molecular weights (PLL[16 kDa]-b-PEG[5 kDa] (PLB16-5) and PLL [16 kDa]-b-PEG[20 kDa] (PLB16-20)) were obtained from Alamanda Polymers, USA. Methoxypolyethylene glycol 5000 pentanoic acid N-succinimidyl ester (mPEG5kDa-SVA) and methoxypolyethylene glycol 20,000 pentanoic acid N-succinimidyl ester (mPEG20kDa-SVA) were obtained from Laysan Bio Inc., USA. For cell culture studies, rat brain glioblastoma cells CRL 2303 were obtained from American Type Culture Collection (Manassas, VA), DMEM from ATCC-30-2002, Thiazolyl Blue tetrazolium bromide, 98% (MTT) from Alfa Aesar, USA.
2.2. Drug nanocapsule preparation
2.2.1. Core preparation
Under continuous sonication, 200 μL of freshly prepared CPT solution in DMSO (7 mg/mL) was added to 2.58 mL of PBS buffer (pH 3) containing 0.64 mg/mL BSA and 1.44 mg/mL PVP and further sonicated for 15–20 min.
For optimization of nanoparticles preparation conditions, in one series of experiments the concentration of BSA in the mixture was varied from 0.35 to 2.50 mg/mL at C(PVP) = 1.44 mg/mL, while in another, the concentration of PVP was varied from 0 to 2.2 mg/mL and the C(BSA) was fixed at 0.64 mg/mL. Upon sonication, ζ potential (in DI water) and hydrodynamic diameter (in PBS buffer, pH 3) of the nanocores were measured using a ZetaPlus Brookhaven instrument.
2.2.2. Polyelectrolyte shell formation on nanocores
By alternating addition of 20 μL aliquots of Hep or PLB16-5 (both 60 mg/mL in acidic PBS, pH 3), 3.5 pairs of the polyelectrolyte layers were deposited on the cores, with heparin being the outermost layer. Each polyelectrolyte solution was added to the nanoparticles dispersion under constant sonication that continues for another 30 s. The obtained dispersion was kept for 5 min before addition of next polyelectrolyte. No intermediate separation of nanoparticles from supernatant or rinsing the nanoparticles with buffer was made. The assembly of polyelectrolytes was followed by the measurements of ζ potential (in DI water) and hydrodynamic diameter of the nanoparticles. The nanocapsules with Hep as the top layer (−20 mV) were separated by centrifugation at 10,000 rpm for 10 min (Eppendorf 5804R ultracentrifuge) and redispersed in the same volume of PBS buffer, pH 7.4. More pairs of layers were assembled at pH 7.4 using 60 mg/mL solutions of polyelectrolytes by sequentially adding 20 μL aliquots of Hep and a copolymer of PEG and PLL (PLB16-5 or PEG16-20).
2.2.3. Additional PEGylation of polyelectrolyte shell
The powder of mPEG5kDa-SVA or mPEG20kDa-SVA was directly added to the dispersion of nanoparticles with a positively charged outermost layer (PLB16-5) in PBS buffer at pH 7.4 to achieve the PEGylator concentration of 40 mg/mL and the mixture was vigorously shaken and sonicated for 30 s to dissolve the PEGylator. The dispersion was kept for 10 h at 4 °C. The nanoparticles were separated by centrifugation at 14,000 rpm for 10 min and the pellet was re-suspended in PBS, pH 7.4.
2.3. Influence of PVP on the amounts of polyelectrolytes needed for charge reversal
In this series of experiments, the dispersions of CPT cores were obtained as described above but the concentration of PVP varied from 0 to 2.2 mg/mL in different batches. Each polyelectrolyte was stepwise added to the dispersions containing a given amount of surfactants in small aliquots, 20 μL of a 6 mg/mL solution in PBS, pH 3.0. This was continued until the ζ potential of the nanoparticles reached a value of ±25 mV. The amount of polyelectrolyte needed to complete one layer was calculated as a sum of that added in all aliquots. Then, the polyelectrolyte with an opposite charge was added in a similar way. Two pairs of layers were assembled for each dispersion.
2.4. Analytical techniques
2.4.1. Amount of BSA adsorbed on nanocores
The amount of BSA remaining on CPT nanocores on different stages of shell preparation was evaluated using FITC-labeled BSA. The concentrations of BSA-FITC from 0.24 to 2.50 mg/mL were used for core preparation; a Hep/PLB16-5 bilayer was coated by adding 20 μL of 60 mg/mL solutions of each polyelectrolyte to the obtained dispersion at pH 3 as described above. In another series of experiments, 3.5 Hep/PLB16-5 bilayers were assembled on nanocores at pH 3. The nanocapsules were separated from supernatant by centrifugation, washed once with PBS buffer pH 7.4, redispersed in the buffer, and then coated with one more PLB16-5/Hep bilayer. The nanoparticles were separated from supernatant, and the concentration of BSA-FITC in the supernatant was estimated using FITC absorbance at 490 nm (Agilent 5893 UV-vis spectrophotometer).
2.4.2. Drug concentration in nanoparticles
The content of CPT in the obtained dispersions of nanocapsules was measured after extraction of the drug with a 1:1 DMSO: acetonitrile mixture (McCarron et al., 2008). The insoluble remains of polyelectrolyte shell were removed from the medium by centrifugation. The concentration of CPT in the solution was measured using the drug absorbance band at 381 nm (ε= 1.92 ×104 M−1 cm−1) (Hatefi and Amsden, 2002; McCarron et al., 2008; Sanna et al., 2009; Dora et al., 2006; Hausheer et al., 1998).
2.5. Characterization of CPT chemical stability
The chemical stability of CPT in the core of (Hep/P16-5)5.0 nanocapsules was studied both in PBS buffer and the buffer with BSA (25 mg/mL) at pH 3, 5, 7.4, and 9 and room temperature (22 °C). CPT dissolved in DMSO was used for comparison. The CPT nanocapsules were dispersed in a medium at the concentration of 0.04 mg/mL. At different time intervals, 20 μL aliquots of the mixture mentioned above were added to 1.98 mL of a 1:1 DMSO: acetonitrile solvent (McCarron et al., 2008) and absorbance of the extract at 354 nm was measured.
The lactone fraction at time t (LF) was calculated as percentage of total drug content:
| (1) |
where Alactone and Acarboxylate are absorbances of the solutions containing CPT only in the lactone and carboxylate form, At – current absorbance of the extract.
The percentage of lactone CPT that remains in the cores obtained at different pH was estimated using the ratio of absorbances at 366 nm and 381 nm at a given pH and pH 3.
| (2) |
2.6. Characterization of colloidal stability of CPT nanoparticles
A dispersion of CPT nanocapsules with a shell of given architecture was concentrated to 0.5 mg/mL in PBS buffer and the hydrodynamic diameter of the nanoparticles was measured over 7 days. During the test, all samples were kept at 22 °C.
2.7. Quartz crystal microbalance: analysis of PLB16-5/Hep film thickness, PEGylation, and binding of serum proteins
The films of PLB16-5 and Hep were formed on flat gold working surfaces of 5 MHz quartz crystal resonators from 0.5 mg/mL polyelectrolyte solutions in water in a liquid flow cell and the film assembly was followed by QCM-R technique with motional resistance monitoring (QCM200, Stanford Research System) (QCM 200, 2005; Marx, 2003; Voros, 2004). After adsorption of each polyelectrolyte, the cell was rinsed with DI water. After assembly of desired number of PLB16-5/Hep layers on a resonator, a 50 mg/mL solution of mPEG5kDa-SVA or mPEG20kDa-SVA in water was injected into the cell for 24 h. The unreacted PEGylator was removed from the cell by rinsing it with water. The attachment of proteins from fetal bovine serum was monitored for 20 min, and finally the cell was washed with excessive amount of water. The shifts of resonance frequency ΔF and motional resistance ΔR were calculated for a resonator with a film immersed in water relatively to the empty resonator. The mass of the deposit was calculated using the Sauerbray equation for rigid films (QCM 200, 2005; Marx, 2003; Voros, 2004).
2.8. Characterization of release study
The release of CPT from the LbL-coated capsules with and without additional PEG layer was studied in PBS buffer, pH 7.4 or 5.0, supplemented with 2% Polysorbate 80, or in FBS. The solubility of CPT in PBS-2% Polysorbate 80 is ca. 1.6 at pH 7.4 and 0.9 μg/mL at pH 5. The solubility of CPT in FBS is ca. 6.7 μg/mL. The dispersion of the nanoparticles under study was added to the release medium which was continuously stirred at 1050 rpm at 37.4 °C. With 0.5–12 h intervals, 1 mL/2 mL aliquots were withdrawn from the mixtures of 10 mL/30 mL, as total volumes for release in PBS/FBS, respectively. They were then replaced with the same volumes of fresh medium. The release was followed for 24 h. The nanocapsules with undissolved CPT were separated from the aliquots by centrifugation. The concentration of CPT dissolved in supernatant was estimated by UV–vis spectroscopy using the absorbance peaks of CPT at 370 nm (in PBS with 2% Tween80, ε= 4.0 104 M−1 cm−1). All experiments were run in triplicates and the data were averaged. In the control experiment, CPT dissolved in DMSO was used.
2.9. TEM sample preparation
10 μL of concentrated dispersion of CPT nanoparticles in DI water was dropped on a copper grid. After 30 s it was stained with 10 μL of 1% ammonium molybdate. The excess amount of stain was absorbed by a tip of filter paper. The sample was allowed to dry in air for half an hour. The TEM images were taken on a Zeiss EM912 at 120 kV.
2.10. In vitro cell culture study
2.10.1. Cell culture
Rat brain glioblastoma cells CRL2303 (American Type Culture Collection) were cultured in 24 well plates with DMEM medium supplemented with 10% Fetal Bovine Serum (ATCC) and penicillin/streptomycin. It was maintained at 37C in a 5% CO2 incubator. The incubation was done overnight to get a cell density of 40,000 cells/mL for MTT assay and 15,000 cells/mL for morphological studies (Alarifi et al., 2013; Bulcke et al., 2013).
2.10.2. Treatment of cells
After required incubation, the cells were treated with varying concentrations (0.01–10 μM) of CPT as nanoparticles coated with (Hep/PLB16-5)7 and (Hep/PLB16-5)7-mPEG shells, or free drug. To obtain a stock solution of free drug, its powder was sonicated for 15 min in PBS, pH 7.4 and then centrifuged out to obtain a particle-free supernatant. It had the CPT concentration of 26.9 μM and a LF value of 96.4%. For positive control, the treatment of cells was done with 50 μg/mL copper (II) oxide nanoparticles (CuO NPs) that have high cell toxicity (Alarifi et al., 2013; Bulcke et al., 2013). The morphological features were seen under an Olympus Ix51 inverted microscope. MTT cell viability assay (Joshi et al., 2012) was performed at two time points, 16 and 40 h. Four hundred μL of 1.25 mg/mL, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution was made in Locke’s solution at pH 7.2. It was initially pre-warmed for 10 min in the incubator chamber. The culture medium from each well was replaced with the same volume of MTT solution. The wells were covered with aluminum foil and then incubated for 1 h. Then the medium was removed carefully, without expiration of the MTT formazan precipitates. The formazan precipitates were solubilized in 200 μL of 91% isopropyl alcohol. Their absorbance was measured at 570 nm in a UV plate reader Multiskan GO, Thermo Scientific. The cell viability (CV) was calculated with respect to the average cell viability of the negative control:
| (3) |
3. Results and discussion
3.1. Optimization of nanocore preparation conditions
3.1.1. Influence of pH
The hydrodynamic diameter of prepared CPT cores changes drastically with the pH used (Fig. 2a). It reaches a few tens of micrometers in the pH range from 4 to 6. In this range the particles have a negative ζ potential, which, however, is not sufficient for colloidal stability of the dispersions. Whereas the core size at pH 7.4 is close to 400 nm as their surface charge goes beyond −20 mV. The cores exhibit the particle size of 140 ±20 nm at pH 3. Despite the CPT nanoparticles are only slightly positively charged at this low pH, stable nanocolloids are formed.
Fig. 2.
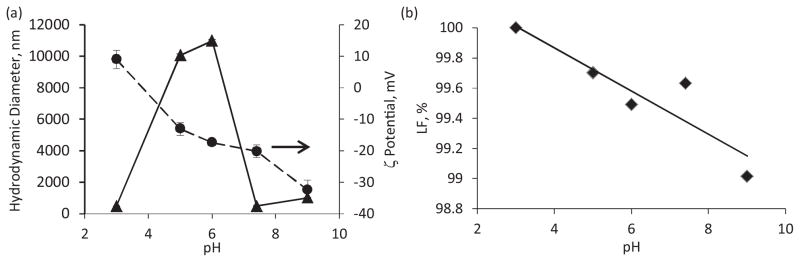
(a) Influence of pH on hydrodynamic diameter and ζ potential of CPT cores. (b) The content of the lactone form in the CPT cores prepared at different pH. CBSA = 0.64 mg/mL, CPVP = 1.44 mg/mL.
The CPT cores were prepared under different pH conditions to evaluate the fraction of the active lactone form of the drug that retains after 20 min of intense sonication in the presence of BSA and PVP. The concentration of the lactone form in the obtained dispersion of nanoparticles reduces gradually as pH increases from 3 to 9 (Fig. 2b). For longer storage this stabilization effect was more profound. The active lactone form of camptothecin and its derivatives is easily hydrolyzed into less active and toxic carboxylic form (Hatefi and Amsden, 2002; McCarron et al., 2008; Sanna et al., 2009; Dora et al., 2006; Hausheer et al., 1998) and its preservation during preparation and storage is of high priority.
Therefore, the preparation of CPT cores at pH 3 is the most reliable method to obtain smaller than 200 nm CPT particles preserving the highest content of its active lactone form.
3.1.2. Amount of BSA in CPT nanocores
The amount of BSA-FITC bound to camptothecin cores coated with a Hep/PLB16-5 bilayer at pH 3.0 is 1.05 ±0.03 mg/mg and remains the same for all concentrations of BSA above 0.64 mg/mL (Table 1). At this concentration, more than 90% of the added BSA-FITC is adsorbed into the core material. If a smaller concentration of BSA is used, the protein is completely bound to the core, but the apparent diameter of nanoparticles increases sufficiently.
Table 1.
Amount of BSA-FITC bound to CPT cores at different concentration of protein.
| C (BSA), mg/mL | C (BSA)/C (CPT), mg/mg | % BSA bound to the core | Hydrodynamic diametera, nm |
|---|---|---|---|
| Coated with (Hep/PLB16-5) shell, pH 3.0 | |||
| 0.35 | 0.62 | 97 ± 3 | 254 ± 15 |
| 0.64 | 1.16 | 91 ± 3 | 167 ± 12 |
| 1.28 | 1.01 | 43 ± 2 | 180 ± 15 |
| 1.90 | 1.13 | 30 ± 2 | 303 ± 14 |
| 2.50 | 4.96 | 100 ± 3 | 1152 ± 56 |
| Coated with a (Hep/PLB16-5)4.5 shell, pH 7.4 | |||
| 0.24 | 0.24 | 64 ± 2 | 245 ± 2 |
| 0.36 | 0.36 | 69 ± 3 | 220 ± 4 |
| 0.64 | 0.69 | 54 ± 2 | 178 ± 2 |
CPVP = 1.44 mg/mL
After changing pH from 3 to 7.4, a partial desorption of BSA-FITC from CPT nanocores takes place (Table 1). At pH 7.4, the charge of BSA (pI = 5 (Zhao and Li, 2008)) is reversed to a negative value affecting adsorption of the protein. However, due to mainly hydrophobic interaction between CPT and BSA (Fleury et al., 1997; Selvi et al., 2008; Mi and Burke, 1994) and variety of possible polyelectrolyte binding sites on the protein (Mattison et al., 1998), a sufficient amount of the protein remains bound to the nanocore and to LbL shell. The concentrations of BSA-FITC higher than 0.64 mg/mL lead to micrometer-sized particles during further LbL assembly at pH 7.4, while the nanocapsules prepared at a 0.64 mg/mL BSA concentration give stable colloids up to 24 h with a nanoparticles diameter of 178 nm. Therefore, these conditions are considered to be the optimum parameters for the core preparation protocol.
3.1.3. Influence of PVP
PVP was used as an excipient for the formulation to improve colloidal stability of the dispersion of CPT nanoparticles. As the concentration of PVP in the core preparation medium increases, the hydrodynamic diameter of CPT cores goes through a maximum at 0.7 mg/mL PVP concentration and then decreases significantly (Fig. 3). It reaches 200 ±20 nm over the 1.4–2.2 mg/mL range, which was further used to get optimal size on CPT nanoparticles while utilizing as lesser amounts of all excipients as possible.
Fig. 3.

Influence of PVP concentration on hydrodynamic diameter of CPT core (5) and amounts of polyelectrolyte needed for recharging their surface upon adsorption: 1 – first step: Hep on BSA-stabilized CPT cores; 2 – second step: PLB16-5 on CPT/Hep nanoparticles; 3 – third step: Hep on CPT/Hep/PLB16-5 nanocapsules, and 4 – fourth step: P16-5 on CPT/(Hep/PLB16-5)1.5 nanocapsules. pH 3.0. CBSA = 0.64 mg/mL.
The feature of the current approach is formation of polyelectrolyte shell on the BSA-stabilized particles directly in the medium used for preparation of CPT cores that is in the presence of polyvinylpyrrolydone. In this series of experiments, CPT cores were produced at different PVP concentration varying from 0 to 2.2 mg/mL. Besides the already mentioned effect of the excipient on core size, we observed some dependence of the amounts of polyelectrolytes needed for ζ-potential reversal on the concentration of PVP (Fig. 3). The light increase of the polyelectrolyte amounts needed for each layer in more concentrated PVP solutions correlates well with smaller apparent size and higher specific surface of the cores. The difference between amounts of Hep needed to complete charge reversal in the first and the third layer (curves 1 and 3) displays the difference in the adsorption conditions on BSA and PLB16-5 layers.
In all further experiments each polyelectrolyte was added in one aliquot in the amount of ~0.7–0.8 mg per 1 mg of CPT that corresponds precisely or somewhat higher than the values in Fig. 3. Therefore, a very small amount of each polyelectrolyte remains in supernatant. As shown previously (Shutava et al., 2012), such excessive polyelectrolytes, if any, can react with alternately charged polymer added on the next stage forming a small fraction of 20–50 nm Hep/PLB16-5 complexes in supernatant. In the case of CPT nanocapsules, formation of polyelectrolyte complexes outside the nanoparticle surface was negligible as confirmed by the measurements of hydrodynamic diameter of nanoparticles after deposition of each layer. When the drug loaded nanocapsules are separated by centrifugation such complexes stay afloat in supernatant.
3.2. LbL assembly
Stable nanocapsules of camptothecin with a 140 nm diameter were prepared at pH 3.0 as describe above. However, to be injectable the dispersion should be reconstituted in a physiologically relevant medium with pH 7 in isotonic PBS buffer or 0.9% sodium chloride solution.
The advantage of LbL assembly of polyelectrolytes is the possibility to form several pairs of layers initially at one pH and further shift pH to another value and finish the coating (Lvov, 2000; Sukhorukov, 2002; De Geest et al., 2009). We utilize this approach and deposited 3.5 Hep/PLb16-5 bilayers at low pH to stabilize the colloids of desired diameter with the highest content of lactone, and further added several layers at pH 7.4.
It was shown that to avoid complications related with material lost and nanoparticles aggregation during centrifugation-based assembly nanoparticles can be coated with polyelectrolytes using the washless LbL technique directly in the presence of uncharged excipients (Shutava et al., 2012). Utilization of block-copolymers of polyamino acids and polyethylene glycol and polysaccharides of low molecular weight allows preserving colloidal stability of the nanoparticles. The polyelectrolytes are added in the amounts that are needed to reverse ζ-potential to an opposite value as high as 20 mV, thus allowing building up of LbL shell.
The camptothecin cores were found to be positively charged at pH 3 (the first point in Fig. 4). Their positive charge is caused by BSA macromolecules on the surface (Zhao and Li, 2008). The minimum amounts of BSA macromolecules which is only sufficient to support colloidal stability of the nanoparticles but leaves almost no free protein in the supernatant on the core preparation stage was used. Thereby, BSA simultaneously works as a surfactant and the first layer in the assembly that anchors the rest of the polyelectrolyte shell.
Fig. 4.

Reversal of ζ-potential value of CPT nanoparticles in the process of washless LbL assembly of polyelectrolyte shell.
The alternation of positive and negative values of ζ-potential confirms the multilayer shell formation on the surface of the nanoparticles (Fig. 4) at pH 3. Three and a half Hep/PLB16-5 bilayers allowed to obtain CPT nanocapsules of about 150 nm in size and well dispersed in PBS at pH 3. Further, 7.0 Hep/PLB16-5 bilayers were deposited in PBS at pH 7.4 to stabilize the colloids at this physiologically relevant condition.
TEM images of CPT nanoparticles coated with a (Hep/PLB16-5)7.0 shell (Fig. 5, top row) show particles with uniform spherical shape. The polyelectrolyte shell material is not distinguishable from the organic core because it has almost similar electron density. For extra PEGylated nanoparticles (Fig. 5, bottom row), a hazy irregular outer periphery of particles indicates the presence of the shells. The hydrophilic PEG5 kDa tails attached to the polyelectrolyte layers makes the shell more distinguishable from the nanoparticle interior.
Fig. 5.

TEM image of CPT nanoparticles with a (Hep/PLB16-5)7.0 (top row) and (Hep/PLB16-5)7.0 /PEG5 kDa (bottom row) shell (stained with ammonium molybdate).
3.3. QCM analysis of capsule wall thickness, enhanced PEGylation and serum protein attachment
The thickness of nanocapsule wall was evaluated by QCM-R technique. The mass of polyelectrolyte film deposited on flat surface of a quartz resonator was recalculated from its frequency decrease using the Sauerbray equation (QCM 200, 2005; Marx, 2003; Voros, 2004). The thickness of 4.5 and 7.5 (PLB16-5/Hep) bilayer films is ca. 5.2 and 7.5 nm (Table 2). The difference between the films with 4.5 and 7.5 bilayers in attachment of PEG upon the treatment with a PEGylator is negligible. Therefore, it is safe to assume that only film outermost amine groups interact with the substances. The mass of mPEG20 kDa attached to the film is ~1.2–1.7 times exceeding that of mPEG5 kDa. This is the PEGylation degree that corresponds to the maximum surface coverage by PEG tails. Despite of the fact that, many amine groups remain unreacted on the surface, the further PEG attachment is hindered by neighboring PEG tails. The mass of these attached PEG corresponds to a layer with apparent thickness of 6–10 nm. The PEGylation significantly decreases the attachment of serum proteins to a (PLB16-5/Hep)n film (Table 3). The ΔF shift which is proportional to the deposited mass is 5–11 Hz for PEGylated surfaces vs. 50 Hz for surfaces without enhanced PEGylation.
Table 2.
PLB16-5/Hep thickness, amount of attached PEG.
| Assembly | Bilayers (n) | dF, Hz | Absorbed mass, μg/cm2 | Thicknessa, nm |
|---|---|---|---|---|
| LbL film | ||||
| (PLB16-5/Hep)n | 7.5 | 51.2 ± 10.7 | 0.9 ± 0.2 | 7.5 |
| 4.5 | 35.0 ± 5.6 | 0.6 ± 0.1 | 5.2 | |
| Attached PEGylator on (PLB16-5/Hep)n, C (mPEG-SVA) = 50 mg/mL | ||||
| mPEG 5 kDa | 7.5 | 41.1 ± 7.3 | 0.7 ± 0.1 | 5.8 |
| mPEG 5 kDa | 4.5 | 49.4 ± 16.1 | 0.9 ± 0.3 | 7.5 |
| mPEG 20 kDa | 7.5 | 70.3 | 1.2 | 10 |
| mPEG 20 kDa | 4.5 | 58.0 | 1.0 | 8.3 |
Using 1.2 g/cm3 film density (Lvov, 2000; Sukhorukov, 2002; De Geest et al., 2009).
Table 3.
Binding of serum proteins on modified polyelectrolyte films.
| Serum protein binding
| |||
|---|---|---|---|
| Assembly on (PLB16-5/Hep)n | Bilayers (n) | dF, Hz | Absorbed mass, μg/cm2 |
| No additional PEGylation | 4.5 | 50.9 | 0.9 |
| mPEG 5 kDa | 7.5 | 5.7 | 0.1 |
| mPEG 5 kDa | 4.5 | 4.7 ± 4.2 | 0.1 |
| mPEG 20 kDa | 7.5 | 11.0 | 0.2 |
| mPEG 20 kDa | 4.5 | 11.0 | 0.2 |
3.4. Colloidal stability of nanocapsules
One of the major parameters of nanocapsule applicability for intravenous administration is their colloidal stability in physiologically relevant buffer and the size remaining <200 nm.
The CPT nanocores coated only with BSA are stable for 2 h in PBS buffer both at pH 3 and 7.4. The samples with a five (Hep/PLB16-5) bilayer shell (positive outermost layer) had a hydrodynamic diameter within 130 ± 10 nm for at least 7 days, while the size of the sample coated with 7 Hep/PLB16-5 bilayers increases to 300 nm (Fig. 6a). The CPT colloids with a shell that have negative Hep layer on the top are more stable than that with positive PLB16-5 (Fig. 6b). One of possible explanations is that due to a strong negative charge Hep macromolecules deposited atop nanoparticles provide better repulsion of nanoparticles in PBS buffer, pH 7.4 than a layer of PLB16-5, PLL part of which is less ionized (pKa = 9.0 (Fasman, 1976)).
Fig. 6.
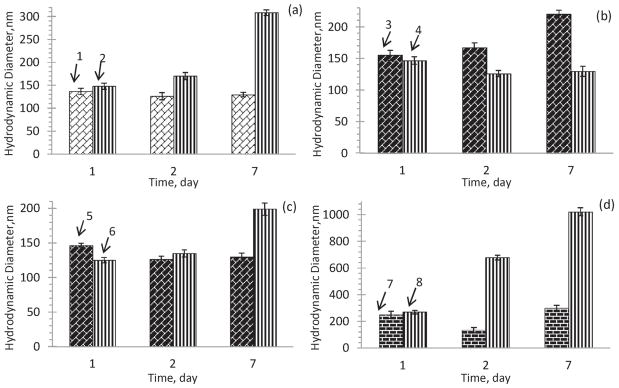
Colloidal stability of nanocapsules in PBS buffer, pH 7.4 with varying: (a) number of bilayers: 1 – (Hep/PLB16-5)5.0 and 2 – (Hep/PLB16-5)7.0 ; (b) different outermost layer: 3 – (Hep/PLB16-5)5.0 and 4 – (Hep/PLB16-5)5.5 ; (c) PEGylation: 5 – (Hep/PLB16-5)5.0 and 6 – (Hep/PLB16-5)5.0 /mPEG5 kDa; (d) different molecular weight of PEGylator on a (Hep/PLB16-5)8.0 shell: 7 – mPEG5 kDa and 8 – mPEG20 kDa.
The nanoparticles with a PLB16-5 terminated shell additionally modified with mPEG5 kDa, also show good colloidal stability. Their hydrodynamic diameter is of 123 ± 2 nm for 48 h, and after 7 days their size increased only by 30% (Fig. 6b).
At the same time, apparent stability of the CPT colloids that are additionally PEGylated with mPEG20 kDa is not as good as that of colloids treated with shorter 5 kDa PEGylator and even worse than the expected stability of PLB16-5 terminated nanoparticles. Low density corona and higher length of grafted PEG chains can favor aggregation of nanoparticles, as it was recently shown for gold and super paramagnetic iron oxide nanoparticles (Cheng and Cao, 2011; Gillich et al., 2013).
3.5. Chemical stability of CPT in nanocapsules at different pH
Camptothecin has two forms, lactone and carboxylate. Lactone is less toxic and more active form of camptothecin for inhibiting Topoisomerase I (Pommier, 2006; Srivastava et al., 2005; Wall et al., 1993). The lactone group of the substance easily hydrolyses to carboxylate at neutral and slightly alkaline conditions.
In PBS, at all studied pH except pH 9 the lactone form of CPT in nanocapsules was preserved at least for 17 h with a light decrease of its content to 97–98% during next 5 h (Fig. 7a). At the same time, free CPT added to the release medium as a solution in DMSO degrades faster at all pH, including pH 3–5 (Fig. 7c). In the presence of 25 mg/mL BSA, lactone in CPT nanocapsules is stable over a wide range of pH including pH 9.0 (Fig. 7b), LF is ~96% after 24 h. Moreover, the lactone form is more preserved as compared to that in PBS alone (Fig. 7a). The improved chemical stability of CPT in LbL capsules, makes such formulation less toxic.
Fig. 7.

Chemical stability of CPT in the (Hep/PLB16-5)5 nanocapsules (a, b) and free CPT (c, d) in PBS, pH 7.4 (a, c) and in PBS with 25 mg/mL BSA (b, d). pH: 1 – 3.0; 2 – 5.0; 3 – 7.4; 4 – 9.0.
3.6. Release kinetics of CPT in PBS and FBS
In PBS buffer supplemented with 2% Polysorbate 80, an initial burst of drug into release medium, lead to at least 25–35% dissolution of nanoparticles for 1.1–1.6 μg/mL of CPT and 60% release for 0.5 μg/mL was observed. It is followed by slowing down drug dissolution during next 3 h (Fig. 8a). After that the released percentage of drug did not change significantly. For the lowest concentration of CPT, 0.5 μg/mL, that is eighteen times lower than the CPT solubility (ca. 9.4 μg/mL), the release reaches the maximum of ~70% after 24 h. No sufficient difference in release profile was observed at pH 5 as compared to pH 7.4 at a 1.6 μg/mL CPT concentration. However, after drug is being released from nanocapsules at pH 7.4, a slow hydrolysis of the lactone form of CPT to the carboxylic one takes place. After 6 h, up to 16% of the drug is converted to the carboxylic form (Fig. 8b). On the other hand, up to 96% of released CPT retains the lactone form after 24 h if released at pH 5.
Fig. 8.
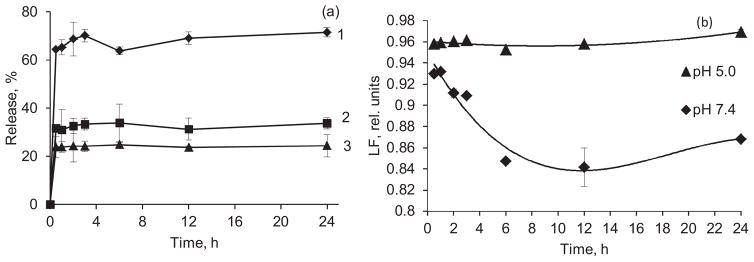
Release of CPT from nanocapsules with a (Hep/PLB16-5)5 shell in PBS with 2% Polysorbate 80, (a) cumulative release of CPT, C (CPT), μg/mL: 1 – 0.5, 2 – 1.1, 3 – 1.6. (b) Lactone fraction remaining in the release medium at pH 7.4 and 5.0 as a function of time. C(CPT) = 1.6 μg/mL.
The release profile of CPT from (Hep/PLB16-5)5 nanocapsules in FBS was quite different from that in PBS (Fig. 9). An initial fast release up to 85% takes place within first 30 min. However, later the concentration of CPT in the release medium decreases and the apparent percent of released drug diminishes to 25–30%. It was apparently due to initial aggregation of non-extra-PEGylated nanoparticles with the serum proteins. It has a lower CPT solubility limit in FBS (6.7 μg/mL) as compared to that in PBS. Similar release profiles were obtained for different concentrations of CPT nanocapsules (SI, Fig. A). We assume that the released CPT molecules interact with proteins from bovine serum. As the time of release increased, a growing volume of the sediment was observed upon separation of nanoparticles from the release medium by centrifugation. A sufficient part of the precipitate was found to be the proteins. The interaction with camptothecin seems to enhance aggregation of the serum proteins or their attachment to the walls of polyelectrolyte capsules.
Fig. 9.

Percentage of free CPT found in FBS (a) and lactone fraction retained in the dissolution medium (b) as a function of time of CPT release from (Hep/PLB16-5)5 (1) and (Hep/PLB16-5)5 /mPEG5 kDa (2) nanocapsules. C(CPT) = 2.27 μg/mL.
The hydrophilic PEG5kDa layer formed on the surface of nanocapsules after treatment with mPEG5kDa-SVA shields them from the FBS proteins, thus minimizing their attachment and stabilizing the colloid in FBS. The initial release of CPT from the nanocapsules with enhanced PEGylation attains only 40% of the drug after 1 h, and then almost a linear increase of CPT concentration in the release medium was found (Fig. 9). After 24 h, the released amount of drug reaches 60%. The apparent thickness of the extra PEG layer is comparable with the thickness of (PLB16-5/Hep)7.5 shell beneath it (Table 2), therefore a lower initial burst and prolonged time of release for the treated capsules are not surprising. An improvement of sustained release performance upon grafting PEG 4000 chains on the surface was previously reported for polyelectrolyte hybrid microspheres consisted of chitosan and magnetite nanoparticles (Zhao et al., 2012).
Apart from the much higher initial CPT release rate in serum, the hydrolysis of the drug in serum is also faster. For nanocapsules without enhanced PEGylation, the highest fraction of the lactone form, ~0.9, was observed in FBS immediately after the initial burst and then it decreases almost to 0.1 (Fig. 9b). The difference in the hydrolysis rates in PBS and serum can be related to the presence of high concentration of albumin that accelerates conversion of lactone to carboxyl (Hatefi and Amsden, 2002; McCarron et al., 2008; Sanna et al., 2009; Dora et al., 2006; Hausheer et al., 1998). For the sample with extra PEGylated surface due to much lower initial CPT release rate, the lactone fraction reaches its maximum after 3 h, indicating that on the earlier stages lactone interacts with serum components and hydrolyses faster than accumulates. Due to the prolonged character of release from the PEGylated nanocapsules, a new portion of lactone CPT is supplied into the supernatant during long period of time, thus supporting its high relative concentration.
3.7. In vitro cell culture studies
The treatment of CRL2303 glioblastoma cells with both free CPT and nanocapsules with extra PEGylated shell causes prominent changes in their morphology that indicate disappearance of cellular processes, while after addition of comparable volume of PBS buffer the cells proliferate (Fig. 10).
Fig. 10.
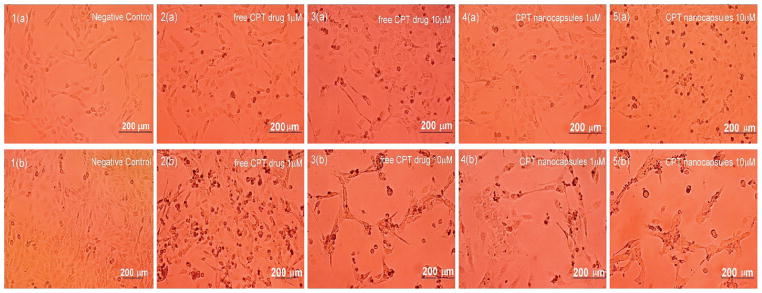
Effect of free CPT (2&3) and CPT nanocapsules with (Hep/PLB16-5)7 /mPEG5 kDa shells (4&5) on Rat brain glioblastoma cells after 16 (upper row, a) and 40 h (lower row, b) treatment. (1) Negative control (PBS buffer), (2, 4) 1.0 μM, (3, 5) 10.0 μM. Images shown are representative of multiple wells (= 6) and multiple platings of cells for each condition tested.
Initially, at 16 h, while glioblastoma cells treated with CPT nanocapsules did show more punctate cells (Fig. 10, panel 5a), cell coverage of the well was still significant compared to controls. However, after 40 h, the treatment with CPT nanocapsules led to clear disruption of cell membranes and necrosis of the cells (Fig. 10, panel 5b), while controls continued to grow and proliferate (Fig. 10, panel 1b). Thus, the cytotoxic effect of CPT nanocapsules showed a delayed effect.
3.8. Effect of CPT nanocapsules on glioblastoma cells viability
Both CPT nanocapsules and free drug inhibit CRL2303 glioblastoma cell growth after 16 h incubation; the decrease of cell viability depends on the applied concentration of CPT (Fig. 11). The prolongation of the incubation up to 40 h increases the effect except for the case of 10 nM free drug that has been apparently hydrolyzed in the course of long cell incubation. The nano-formulation cytotoxic effect is mostly observed in the case of medium drug concentration, 100 nM (circled in Fig. 11). For 100 nM CPT nanocapsules, a clear delayed effect is observed, with a decrease in tumor cell viability down to about 30% by 40 h (Fig. 11). The difference between cell growth inhibition activities of CPT nanocapsules with (Hep/PLB16-5)7 and (Hep/PLB16-5)7/mPEG5 kDa is negligible. The corresponding IC50 values are ca. 103.8 and 103.5 nM (SI, Fig. B). At the same time, free CPT is less active, IC50 is ca. 146.3 nM. We presume that after free CPT is added to cell culture medium, a sufficient part of the drug is quickly hydrolyzed into less active carboxylic form, while the lactone CPT in nanocapsules is preserved and releases slowly in the course of the cell incubation.
Fig. 11.
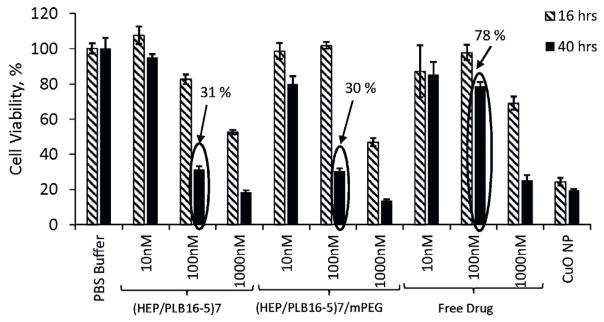
Effect of different concentrations of free CPT, and CPT nanocapsules with (Hep/PLB16-5)7 and (Hep/PLB16-5)7 /mPEG5 kDa shells on CRL2303 glioblastoma cell viability. Data shown are representative of multiple wells (=6) and multiple platings of cells for each condition tested. The percentage values of 100 nM are indicated. Toxic CuO nanoparticles (NP) data are given as positive control.
4. Conclusion
The objectives of using LbL formulation technique to encapsulate camptothecin were to preserve the lactone form of drug; to attain <200 nm mono-dispersed stable colloid dispersion at a high concentration of 0.5 mg/mL; to get a sustained release of the drug for 24 h with good lactone fraction and, finally, to have a better in vitro cancer cell growth inhibition effect than the uncoated CPT drug.
160 nm capsules were formulated with CPT core and BSA covering it, followed by 7–8 bilayer of alternating anionic heparin and cationic PEGylated polylysine (PLB16-5) with total shell thickness of ca. 10 nm. By attaining non-washing LbL assembly at two pH values (starting from lower pH 3 to preserve lactone form and proceeding at pH 7.4), we formulated nanocapsules with loading capacity of approximately 60 wt% and as high as 99% lactone CPT fraction. Colloidal stability of the nanocapsules was improved by incorporating mPEG 5 kDa tails on the outermost layer of PLB16-5. The encapsulation of CPT in the shells of polyelectrolytes modified with PEG reduces the rate of hydrolysis of lactone to carboxylic form at neutral and alkaline media as compared with free drug. The modification of nanocapsule outer surface with additional mPEG 5 kDa tails also increases protein-resistance of the shell, decreases initial drug burst and prolongs release time in biologically relevant media. It allows preserving the active lactone form of CPT in the course of 40 h nanocapsules incubation with cancer cell. As a result, activity of CPT encased into LbL nanocapsules toward CRL2303 glioblastoma cell is improved in three times.
Supplementary Material
Acknowledgments
This work was supported by Award 1R01CA134951 from the National Cancer Institute (NCI). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NCI or National Institute of Health. The authors acknowledge Kirill A. Arapov for assistance with CPT nanocapsules preparation and TEM imaging and Umamaheswari Manoharan for in vitro cell culture studies.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.ijpharm.2014.01.041.
References
- Alarifi S, Ali D, Verma A, Alakhtani S, Ali BA. Cytotoxicity and genotoxicity of copper oxide nanoparticles in human skin keratinocytes cells. Int J Toxicity. 2013;32:296–307. doi: 10.1177/1091581813487563. [DOI] [PubMed] [Google Scholar]
- Bantchev G, Lu Z, Lvov Y. Layer-by-layer nanoshell assembly on colloids through simplified washless process. J Nanosci Nanotechnol. 2009;9:396. doi: 10.1166/jnn.2009.j055. [DOI] [PubMed] [Google Scholar]
- Bulcke F, Thiel K, Dringen R. Uptake and toxicity of copper oxide nanoparticles in cultured primary brain astrocytes. Nanotoxicology. 2013 doi: 10.3109/17435390.2013.829591. http://dx.doi.org/10.3109/17435390.2013.829591. [DOI] [PubMed]
- Chen H, Khemtong C, Yang X, Chang X, Gao J. Nanonization strategies for poorly water-soluble drugs. Drug Discov Today. 2011;16:354. doi: 10.1016/j.drudis.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Cheng L, Cao D. Aggregation of polymer-grafted nanoparticles in good solvents: a hierarchical modeling method. J Chem Phys. 2011;135:124703. doi: 10.1063/1.3638176. [DOI] [PubMed] [Google Scholar]
- Cho YW, Lee J, Lee SC, Huh KM, Park K. Hydrotropic agents for study of in vitro paclitaxel release from polymeric micelles. J Control Release. 2004;97:249–257. doi: 10.1016/j.jconrel.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Chodanowski P, Stoll S. Polyelectrolyte adsorption on charged particles: ionic concentration and particle size effects—a Monte Carlo approach. J Chem Phys. 2001;115:4951. [Google Scholar]
- De Geest B, Sukhorukov G, Möhwald H. The pros and cons of polyelectrolyte capsules in drug delivery. Expert Opin Drug Deliv. 2009;6:613–624. doi: 10.1517/17425240902980162. [DOI] [PubMed] [Google Scholar]
- Dora C, Alvarez-Silva M, Trentin A, de Faria T, Fernandes D, da Costa R, Stimamiglio M, Lemos-Senna E. Evaluation of antimetastatic activity and systemic toxicity of camptothecin-loaded microspheres in mice injected with B16-F10 melanoma cells. J Pharm Pharm Sci. 2006;9:22. [PubMed] [Google Scholar]
- Fasman GD. Handbook of Biochemistry and Molecular Biology. CRC Press; Boca Raton, FL: 1976. p. 464. [Google Scholar]
- Fleury F, Ianoul A, Berjot M, Feofanov V, Alix AJP, Nabiev I. Camptothecin-binding site in human serum albumin and protein transformations induced by the drug binding. FEBS Lett. 1997;411:215–220. doi: 10.1016/s0014-5793(97)00693-5. [DOI] [PubMed] [Google Scholar]
- Gillich T, Acikgöz C, Isa L, Schlüter AD, Spencer ND, Textor M. PEG-stabilized core-shell nanoparticles: impact of linear versus dendritic polymer shell architecture on colloidal properties and the reversibility of temperature-induced aggregation. ACS Nano. 2013;7:316–329. doi: 10.1021/nn304045q. [DOI] [PubMed] [Google Scholar]
- Hatefi A, Amsden B. Biodegradable injectable in situ forming drug delivery systems. Pharm Res. 2002;19:1389. doi: 10.1016/s0168-3659(02)00008-1. [DOI] [PubMed] [Google Scholar]
- Hausheer F, Haridas K, Murali D, Reddy D. Formulations and compositions of poorly water soluble camptothecin derivatives. 5,726,181 US Pat. 1998
- Joshi C, Karumuri B, Newman JJ, DeCoster MA. Cell morphological changes combined with biochemical assays for assessment of apoptosis and apoptosis reversal. In: Mendez-Vilas A, editor. Current Microscopy Contributions to Advances in Science and Technology. Formatex Research Center; Badajoz: 2012. pp. 756–762. [Google Scholar]
- Kahlweit M. Ostwald ripening of precipitates. Adv Colloid Interface Sci. 2008;5:1. [Google Scholar]
- Keck C, Kobierski S, Mauludin R, Muller R. Second generation of drug nanocrystals for delivery of poorly soluble drugs: smart crystal technology. Dosis. 2008;2:124. [Google Scholar]
- Kim S, Kim JY, Huh KM, Acharya G, Park K. Hydrotropic polymer micelles containing acrylic acid moieties for oral delivery of paclitaxel. J Control Release. 2008;132:222. doi: 10.1016/j.jconrel.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J, Lu Z, Lvov Y, Schenkman J, Rusling J. Direct electrochemistry of myoglobin and cytochrome P450cam in alternate layer-by-layer films with DNA and other polyions. J Am Chem Soc. 1998;120:4073. [Google Scholar]
- Lee J, Choi JY, Park CH. Characteristics of polymers enabling nano-comminution of water-insoluble drugs. Int J Pharm. 2008;355:328. doi: 10.1016/j.ijpharm.2007.12.032. [DOI] [PubMed] [Google Scholar]
- Li Y, Xia J, Dubin PL. Complex formation between polyelectrolytes and oppositely charged mixed micelles: static and dynamic light scattering. Study of the effect of polyelectrolyte molecular weight and concentration. Macromolecules. 1994;27:7049. [Google Scholar]
- Lu CY, Liu P. Effect of chitosan multilayers encapsulation on controlled release performance of drug-loaded superparamagnetic alginate nanoparticles. J Mater Sci Mater Med. 2012;23:393–398. doi: 10.1007/s10856-011-4477-2. [DOI] [PubMed] [Google Scholar]
- Lvov Y. Electrostatic layer-by-layer assembly of proteins and polyions. In: Lvov Y, Möhwald H, editors. Protein Architecture: Interfacial Molecular Assembly and Immobilization Biotechnology. Marcel Dekker Publ; New York: 2000. pp. 1–394. [Google Scholar]
- Lvov Y, Pattekari P, Shutava T. Making aqueous nanocolloids from low soluble materials: LbL shells on nanocores. In: Decher G, Schlenoff J, editors. Multilayer Thin Films: Sequential Assembly of Nanocomposite Materials. Chapter 14 Wiley-VCH; NY, London: 2012. pp. 151–170. [Google Scholar]
- Marriott J, Wilson K, Langley C, Belcher D. Pharmaceutical Compounding and Dispensing. Pharmaceutical Press; London, Chicago: 2006. p. 277. [Google Scholar]
- Marx K. Quartz crystal microbalance:? A useful tool for studying thin polymer films and complex biomolecular systems at the solution-surface interface. Biomacromolecules. 2003;4:1099. doi: 10.1021/bm020116i. [DOI] [PubMed] [Google Scholar]
- Mattison KW, Dubin PL, Brittain IJ. Complex formation between bovine serum albumin and strong polyelectrolytes: effect of polymer charge density. J Phys Chem B. 1998;102:3830–3836. [Google Scholar]
- McCarron PA, Marouf WM, Quinn DJ, Fay F, Burden RE, Olwill SA, Scott CJ. Antibody targeting of camptothecin-loaded PLGA nanoparticles to tumor cells. Bioconj Chem. 2008;19:1561–1569. doi: 10.1021/bc800057g. [DOI] [PubMed] [Google Scholar]
- Mi Z, Burke TG. Differential interactions of camptothecin by human serum albumin. Biochemistry. 1994;33:10325–10336. doi: 10.1021/bi00200a013. [DOI] [PubMed] [Google Scholar]
- Mu B, Zhong W, Dong Y, Du PC, Liu P. Encapsulation of drug micro-particles with self-assembled Fe3 O4 /alginate hybrid multilayers for targeted controlled release. J Biomed Mater Res B: Appl Biomater. 2012;100B:825–831. doi: 10.1002/jbm.b.32646. [DOI] [PubMed] [Google Scholar]
- Müller RH, Jacobs C, Kayser O. Nanosuspensions for the formulation of poorly soluble drugs. In: Nielloud F, Marti-Mestres G, editors. Pharmaceutical Emulsions and Suspensions. Marcel Dekker; New York: 2008. pp. 383–407. [Google Scholar]
- Parikh I, Selvaraj U. Composition and method of preparing microparticles of water-insoluble substances. 5,922,355 US Pat. 2008
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- QCM 200. Quartz Crystal Microbalance Digital Controller Operation and Service Manual, Revision 2.1. Stanford Research Systems, Inc; 2005. p. 106. [Google Scholar]
- Sanna N, Chillemi G, Gontrani L, Grandi A, Mancini G, Castelli S, Zagotto G, Zazza C, Barone V, Desideri A. UV–vis spectra of the anticancer camptothecin family drugs in aqueous solution: specific spectroscopic signatures unraveled by a combined computational and experimental study. J Phys Chem B. 2009;113:5369. doi: 10.1021/jp809801y. [DOI] [PubMed] [Google Scholar]
- Schneider G, Decher G. From functional core/shell nanoparticles prepared via layer-by-layer deposition to empty nanospheres. Nano Lett. 2004;4:1833–1839. [Google Scholar]
- Selvi B, Patel S, Savva M. Physicochemical characterization and membrane binding properties of camptothecin. J Pharm Sci. 2008;97:4379–4390. doi: 10.1002/jps.21314. [DOI] [PubMed] [Google Scholar]
- Shutava TG, Pattekari PP, Arapov KA, Torchilin VP, Lvov YM. Architectural layer-by layer assembly of drug nanocapsules with PEGylated polyelectrolytes. Soft Matter. 2012;8:9418–9427. doi: 10.1039/C2SM25683E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivasankar T, Kumar B. Role of nanoparticles in drug delivery system. Int J Res Pharm Biomed Sci. 2010;1:41. [Google Scholar]
- Srivastava V, Negi AS, Kumar JK, Gupta MM, Khanuja SP. Plant-based anticancer molecules: a chemical and biological profile of some important leads. Bioorg Med Chem. 2005;13:5892–5908. doi: 10.1016/j.bmc.2005.05.066. [DOI] [PubMed] [Google Scholar]
- Sukhorukov G. Multilayer Hollow Microspheres. In: Arshady R, Guyot A, editors. Dendtrimers. Vol. 5. Citrus Books; NY: 2002. p. 111. MML Series. [Google Scholar]
- Tsuzuki T. Commercial scale production of inorganic nanoparticles. Int J Nanotechnol. 2009;6:567. [Google Scholar]
- Vertegel AV, Siegel RW, Dordick JS. Silica nanoparticle size influences the structure and enzymatic activity of absorbed lysozyme. Langmuir. 2004;20:6800. doi: 10.1021/la0497200. [DOI] [PubMed] [Google Scholar]
- Voros J. The density and refractive index of adsorbing protein layers. Biophys J. 2004;87:553. doi: 10.1529/biophysj.103.030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall ME, Wani MC, Nicholas AW, Manikumar G, Tele C, Moore L, Truesdale A, Leitner P, Besterman JM. Plant antitumor agents. 30 Synthesis and structure activity of novel camptothecin analogs. J Med Chem. 1993;36:2689–2700. doi: 10.1021/jm00070a013. [DOI] [PubMed] [Google Scholar]
- Wei XH, Nui YP, Xu YY, Du YZ, Hu FQ, Yuan H. Salicylic acid-grafted chitosan oligosaccharide nanoparticle for paclitaxel delivery. J Bioact Compat Polym. 2010;25:319. [Google Scholar]
- Zhao Q, Li B. pH-controlled drug loading and release from biodegradable microcapsules. Nanomed Nanotechnol Biol Med. 2008;4:302–310. doi: 10.1016/j.nano.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XB, Du PC, Liu P. Preparation of aggregation-resistant biocompatible superparamagnetic noncovalent hybrid multilayer hollow microspheres for controlled drug release. Mol Pharm. 2012;9:3330–3339. doi: 10.1021/mp300363g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.