

Abstract
Fas death pathway is important for lymphocyte homeostasis, but the role of Fas pathway in T cell memory development is not clear. We show that whereas the expansion and contraction of CD8+ T cell response against Listeria monocytogenes were similar for wild-type (WT) and Fas ligand (FasL) mutant mice, the majority of memory CD8+ T cells in FasL mutant mice displayed an effector memory phenotype in the long-term in comparison with the mainly central memory phenotype displayed by memory CD8+ T cells in WT mice. Memory CD8+ T cells in FasL mutant mice expressed reduced levels of IFN-γ and displayed poor homeostatic and Ag-induced proliferation. Impairment in CD8+ T cell memory in FasL mutant hosts was not due to defective programming or the expression of mutant FasL on CD8+ T cells, but was caused by perturbed cytokine environment in FasL mutant mice. Although adoptively transferred WT memory CD8+ T cells mediated protection against L. monocytogenes in either the WT or FasL mutant hosts, FasL mutant memory CD8+ T cells failed to mediate protection even in WT hosts. Thus, in individuals with mutation in Fas pathway, impairment in the function of the memory CD8+ T cells may increase their susceptibility to recurrent/latent infections.
The importance of T cell memory for the control of intracellular pathogens is well documented (1–3); however, the mechanisms that govern the induction and maintenance of memory T cells are not clear. During T cell priming, CD8+ T cells undergo massive proliferation (>1000-fold), which is followed by a contraction phase wherein the majority (>90%) of activated cells die by apoptosis (4). Engagement of Fas, with its ligand (FasL),3 is thought to play a key role in the regulation of T cells (5, 6) because the interaction between activated T cells expressing Fas and FasL provides a mechanism for deleting T cells. This view is supported by the observation that lpr mice that bear mutation in Fas gene develop lymphadenopathy (7), and T cells from lpr mice are resistant to activation-induced cell death (8–10). Furthermore, T cell tolerance induction to superantigen is impaired or delayed in lpr mice (11–14). The role of Fas in regulating CD8+ T cell homeostasis has been controversial because autoreactive CD8+ T cells induced by cross-presentation of self Ags are deleted by a Fas-dependent mechanism (15), whereas the expansion and contraction of Ag-specific CD8+ T cell response induced after viral infection in vivo have been shown to occur by Fas-independent mechanisms (16, 17).
We evaluated the development and maintenance of OVA-specific CD8+ T cells in normal and FasL mutant mice using recombinant intracellular bacterium, Listeria monocytogenes (LM)-expressing OVA as an immunogen. Our results reveal that although the expansion and contraction of CD8+ T cell response proceed normally in FasL mutant mice, memory CD8+ T cells in FasL mutant mice display a skewed phenotype and poor homeostatic proliferation and fail to mediate effective protection against pathogen rechallenge.
Materials and Methods
Mice
Female C57BL/6J wild-type (WT), B6.Faslpr/lpr, and B6.FasLgld/gld mice, 3–4 wk of age, were obtained from The Jackson Laboratory. Mice were maintained in the animal facility at Institute for Biological Sciences (National Research Council of Canada) in accordance with the guidelines of the Canadian Council on Animal Care.
Intracellular bacteria and infections
OVA-expressing LM (LM-OVA) were generated, as described previously (18). Mice were inoculated with 1 × 104 LM-OVA suspended in 200 μl of 0.9% NaCl, via the lateral tail vein (i.v.). In some experiments, mice were infected with 106 LM-OVA, followed by daily injection with ampicillin (10 mg/mouse) from days 2 to 7. For enumeration of bacterial burden in organs, single-cell suspensions from infected mice were made, and CFUs were determined by plating 100-μl aliquots of serial 10-fold dilutions in 0.9% saline. Plates were incubated for 24 h, and colonies were counted visually.
CD8+ T cell purification
Single-cell suspensions were prepared by tweezing the pooled spleens (n = 2–3) between the frosted ends of two sterile glass slides in RPMI 1640. Cells were subsequently passed through Falcon 2360 cell strainers (BD Discovery Labware), centrifuged, and resuspended (20 × 106/ml) in 0.5–1 ml of RPMI 1640 containing 8% FBS (HyClone) and 50 μg/ml gentamicin (Invitrogen Life Technologies), hereafter refer to as R8-A. CELLection Biotin Binder Dynabeads, precoated per manufacturer’s instructions (Dynal Biotech), with biotin-conjugated rat anti-mouse CD8β.2 mAb (53.5.8; BD Biosciences), were added to the resuspended cell pellet at a ratio of 5 beads/cell, and incubated for 15–20 min at 4°C in a rotating platform. CD8β + T cells were separated by magnetic isolation. Dynabead detachment was done using the CELLection Biotin Binder kit releasing buffer (DNase; 188 U/108 Dynabeads) in 37°C shaker for 15 min, followed by two to three rounds of washing/magnetic separation. This protocol resulted in >95% pure CD8+ T cells, as determined by follow-up analysis with PE-conjugated rat anti-mouse CD8α (BD Biosciences). Analysis was performed using EPICS XL flow cytometer and EXPO software (Beckman Coulter).
Flow cytometry
At various time intervals after infection, aliquots (10 × 106) of spleen cells were incubated in 200 μl of PBS plus 1% BSA (PBS-BSA) with anti-CD16/32 at 4°C. After 10 min, cells were stained with PE-H-2Kb OVA257–264 tetramer and various Abs (anti-CD8, anti-CD62L, anti-IL-7Rα, anti-PD-1, and anti-CD44) for 30 min at room temperature. This was followed by amplification of the tetramer staining using the PE-Faser kit (Miltenyi Biotec). All Abs were obtained from BD Biosciences. PE-H-2KbOVA257–264 tetramer was obtained from Beckman Coulter. Cells were washed with PBS, fixed in 0.5% formaldehyde, and acquired on BD Biosciences FACS Canto analyzer.
Assessment of IFN-γ production
Aliquots of spleen cells (10 × 106/ml) were stained with anti-CD8 Ab and H-2KbOVA257–264 tetramer for 30 min, as described above. Cells were then washed, reconstituted in R8 medium (RPMI 1640 plus 8% FBS), plated into 96-well plates (2 × 106/well), and stimulated with OVA257–264 peptide (1 μg/ml) in the presence of Golgi-stop (BD Biosciences). After 1 h, cells were harvested, washed, permeabilized, and stained for intracellular IFN-γ using the IFN-γ staining kit (obtained from BD Biosciences). Cells were acquired on BD Biosciences FACS Canto analyzer. For measurement of IFN-γ production in vitro, CD8+ T cells were purified and incubated (105) with spleen cells (5 × 105) from uninfected mice in the presence of various concentrations of OVA257–264. Supernatant was collected at 48 h, and the amount of IFN-γ produced was measured by ELISA.
Assessment of cell cycling
Cell cycling was evaluated according to the protocol of Tough and Sprent (19) using the BD Biosciences BrdU staining kit. Three days before the harvesting of spleens from infected mice, BrdU was provided (0.08%) in the drinking water every day. Spleens were harvested, single-cell suspensions were prepared, and aliquots of spleen cells (10 × 106/ml) were stained with anti-CD8 Ab and H-2KbOVA257–264 tetramer for 30 min, as described above. After staining, cells were washed, permeabilized, and incubated with DNase for 30 min at 37°C. Cells were then stained with anti-BrdU Ab on ice for 30 min, washed, fixed in 0.5% formaldehyde, and acquired on BD Biosciences FACS Canto analyzer.
Assessment of commitment to apoptosis
Aliquots of cells (10 × 106/ml) were incubated in RPMI 1640 plus 8% FBS for 3–6 h. Cells were then surface stained with anti-CD8 Ab and OVA tetramers, as described above. Two different intracellular staining kits (TUNEL and caspase 3) were used to evaluate the apoptotic cells. Cells were permeabilized and incubated with Ab against active caspase 3. Cells were also fixed, permeabilized, and incubated with the ApoD enzyme and FITC dUTP, per the BD Biosciences protocol. Cells were finally fixed and analyzed on BD FACS Canto flow cytometer.
ELISPOT assay
Enumeration of IFN-γ -secreting cells was done by ELISPOT assay (18). Briefly, spleen cells were incubated in anti-IFN-γ Ab-coated ELISPOT plates, varying the number of spleen cells from immunized mice to achieve a final cell density of 5 × 105/well using feeder cells from unimmunized mice. These cultures were established in RPMI 1640 plus 8% FBS with or without OVA257–264 (10 μg/ml), supplemented with IL-2 (0.01 ng/ml), and incubated for 48 h at 37°C, 8% CO2. The cells were then lysed with H2O, and the plates were washed (PBS-T) and incubated with the biotinylated secondary Ab (4°C, overnight), followed by avidin-peroxidase conjugate (room temperature, 2 h). Spots were revealed using diaminobenzidine.
Cytotoxicity assays
Single-cell suspensions from spleens of immunized mice were prepared. A total of 30 × 106 spleen cells from immunized mice was incubated with 5 × 105 irradiated (10,000 rad) Ag-bearing target cells (EG7 cells) in 10 ml of RPMI 1640 plus 8% FBS. In some cases, purified CD8+ T cells from infected mice were incubated (1 × 106) with syngeneic feeder cells from unimmunized mice (29 × 106) and EG7 cells, as described above. Cultures contained 0.1 ng/ml IL-2 and were placed in 25-cm2 tissue culture flasks (Falcon; BD Biosciences), kept upright. After 5 days (37°C, 8% CO2), cells were harvested from the flasks, washed, counted, and used as effectors in a standard 51Cr release CTL assay. For preparation of targets for CTL assay, EL4 cells were incubated with medium or with OVA257–264 for 1 h. Cells were then labeled with 50 μl of 51Cr (100 μCi) in the presence of 25 μl of RPMI 1640 plus 8% FBS medium. After 45 min, targets were washed twice, various ratios of effectors and targets were cocultured for 4 h in 96-well round-bottom tissue culture plates, the supernatants were collected, and radioactivity was detected by gamma counting. The percentage of cytotoxicity was calculated using the following formula: 100 × ((cpm experimental − cpm spontaneous)/(cpm total − cpm spontaneous)).
Assessment of cytokine expression by quantitative RT-PCR
Harvested spleens were snap frozen in a dry-ice/100% ethanol bath. Total RNA was extracted using the Qiagen RNeasy mini kit, according to the manufacturer’s instructions, along with rapid mechanical lysis. The whole spleen was cut into pieces, and each piece was lysed in 1 ml of lysis buffer in a miniBeadbeater 3110BX (BioSpec Products) with glass beads (ϕ = 0.5 mm and ϕ = 0.1 mm) (BioSpec Products). Total RNA from homogenates was extracted according to the manufacturer’s instructions and treated with RNase-free DNase I (Roche Applied Science) for 30 min at 37°C. DNase was removed using the Qiagen RNeasy mini kit, according to the manufacturer’s instructions. A quantity amounting to 5 μg of total RNA was taken for cDNA synthesis. cDNA was synthesized using AncT primers purchased from Sigma-Aldrich. RNA was made linear at 65°C for 5 min, and cDNA was synthesized in a 40-μl reaction volume containing 1.5 μl AncT primers (100 pmol/μl), 8 μl of 5× first strand buffer, 4 μl of DTT (100 mM), 5 μl of dNTPs (5 mM), 1 μl of RNase OUT (40 U/μl), 2 μl of Superscript II (200 U/μl) (Invitrogen Life Technologies), and 15 μl of RNA template. Reverse transcription was performed in a Thermo Cycler 9700 (Applied Biosystems) at 42°C for 15 min and 45°C for 2 h. Identical samples not treated with Superscript II were also prepared as controls to measure DNA contamination. Remaining RNA template was hydrolyzed with 1 M NaOH at 65°C for 5 min and neutralized with 1 M HCl. cDNA was purified using Microcon YM-30 centrifugal filter units (Millipore). The number of amplicons was measured by quantitative real-time PCR using gene-specific primers and quantitative PCR SYBR Green supermix (ABgene). Primers were designed using Primer Express 2.0. β-actin was used as an internal reference control. Ten-fold dilutions of cDNA were used as template to generate the standard curve for each primer-template set (1×, 1/10×, 1/100×, and 1/1000×). This standard curve was run together with triplicate reactions of the uncharacterized samples. PCR was performed in sealed tubes in a 96-well microtiter plate in an ABI Prism 7000 thermocycler (Applied Biosystems). The 25-μl reaction consisted of 12.5 μl of quantitative PCR SYBR Green supermix, 2.5 μl of primer mix (1.5 pmol/μl each), and 10 μl of template. Thermal conditions were as follows: activation at 95°C for 15 min, followed by 40 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 1 min, and extension at 72°C for 1 min. Fluorescence was measured during the annealing step and plotted against the amplification cycle. Relative quantitative analysis of the data was extrapolated from the standard curve. Primer efficiencies were between 100 and 98%.
Results
Impaired cytolytic CD8+ T cell response in FasL mutant mice
We evaluated memory CD8+ T cell responses against OVA257–264 in mice with mutation/deficiency in various molecules (indicated in Fig. 1A). Mice were infected with LM-OVA, and cytolytic CD8+ T cell response was evaluated at day 60. As is evident from Fig. 1, mice generated OVA-specific CD8+ T cell responses in the absence of inducible NO synthase-2, IFN-γ, TNF-Rαβ, and IL-12. In contrast, the CD8+ T cell response to OVA in FasL mutant mice was considerably reduced. Similar results were obtained when the frequency of OVA257–264-specific CD8+ T cells was evaluated by ELISPOT assay (data not shown). We then evaluated the cytolytic CD8+ T cell response in FasL mutant mice at various time intervals. At days 7 and 30 after LM-OVA infection, similar cytolytic CD8+ T cell response was noted in control (WT) and FasL mutant mice (Fig. 1B). However, at day 60, the CD8+ T cell response was considerably reduced in FasL mutant mice.
FIGURE 1.

Impaired CD8+ T cell memory in FasL mutant mice. C57BL/6J WT mice, or mice with mutation/deficiency in inducible NO synthase-2, IFN-γ, TNFR-αβ, FasLgld/gld, IL-12, and CD40L were infected with rLM-OVA (1 × 104 i.v.). Spleen cells were restimulated (30 × 106 cells) on days 7 and 30 (B) or day 60 (A and B) with irradiated OVA-expressing EG-7 cells (5 × 105) and IL-2 (0.1 ng/ml). Five days later, cells were harvested and evaluated for cytolytic activity against EL4 targets in the presence or absence of the CTL epitope OVA257–264. Percentage of cytotoxicity in the absence of OVA257–264 ranged from 0 to 10%. Figure is representative of three such experiments conducted.
Impaired cytolytic CD8+ T cell response in Fas and FasL mutant mice
Because mice with a mutation in Fas as well as FasL display lymphoproliferative disease, we determined whether the impairment in cytolytic CD8+ T cell response also occurs in mice with a mutation in Fas. Although WT mice generated a potent OVA257–264-specific cytolytic CD8+ T cell response, mice with a mutation in Fas or FasL displayed very little cytolytic CD8+ T cell response (Fig. 2A). Because the cytolytic CD8+ T cell response was detected after restimulation of cells with OVA-expressing targets for 5 days in vitro, we evaluated the possibility that the detection of the CD8+ T cell response in mutant mice was impaired by the presence of some inhibitory activity in the spleen cells of such mice. We therefore purified CD8+ T cells from LM-OVA-infected WT mice and stimulated them in vitro with WT or FasL mutant spleen cells. Spleen cells from FasL mutant mice did not inhibit cytolytic response of WT memory CD8+ T cells (Fig. 2B). Furthermore, purified memory CD8+ T cells from FasL mutant mice did not display any cytolytic response upon culture with WT spleen cells (Fig. 2C). Thus, the impairment in cytolytic CD8+ T cell response in FasL mutant mice was attributable directly to a defect in the CD8+ T cells generated in FasL mutant mice.
FIGURE 2.
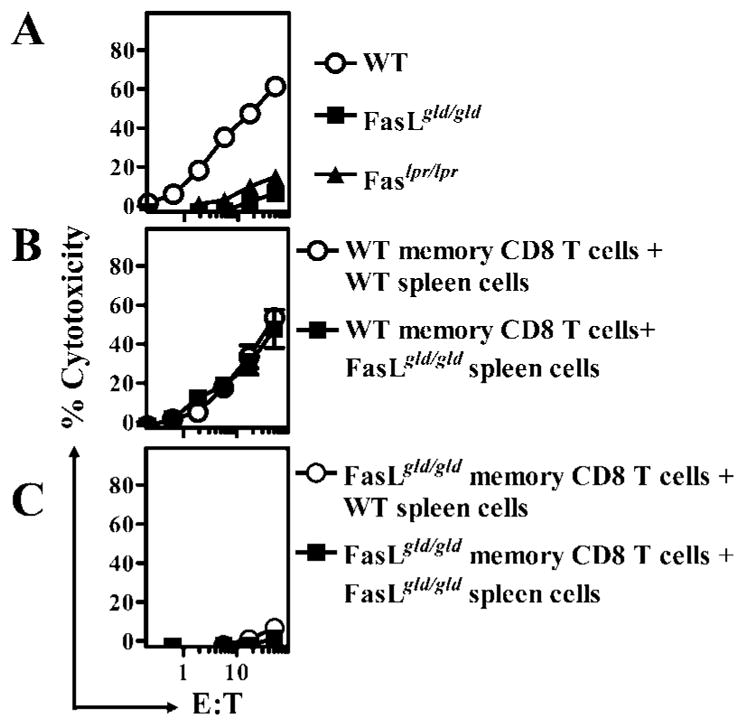
Impaired CD8+ T cell memory in both Fas and FasL mutant mice. C57BL/6J WT, B6.Faslpr/lpr, and B6.FasLgld/gld mice were infected with rLM-OVA (1 × 104 i.v.). On day 60, spleen cells were removed and restimulated (30 × 106 cells) with irradiated OVA-expressing EG-7 cells (5 × 105) and IL-2 (0.1 ng/ml) (A). CD8+ T cells were purified from the spleens of LM-OVA-infected WT (B) and FasLgld/gld (C) mice and restimulated (1 × 106 pure CD8+ T cells) with EG-7 cells (5 × 105 cells) in the presence of splenocytes from WT or FasLgld/gld mice (29 × 106 cells) and IL-2 (0.1 ng/ml). In vitro stimulation was conducted for 5 days, after which the cells were harvested and tested for cytolytic activity against EL4 targets in the presence or absence of the CTL epitope OVA257–264. Percentage of cytotoxicity in the absence of OVA257–264 ranged from 0 to 10%.
Normal expansion and contraction of CD8+ T cell response in FasL mutant mice
We tested the possibility that the impairment in the cytolytic CD8+ T cell response in FasL mutant mice was due to a reduced frequency of OVA-specific CD8+ T cells. We first measured the relative LM-OVA burden in WT vs FasL mutant mice at various time intervals after infection. LM-OVA infection peaked in both groups of mice ~day 3. At day 5, WT mice had cleared LM-OVA from spleens, whereas low-level burden was still detectable in the spleens of some FasL mutant mice. By day 7 of infection, LM-OVA was cleared from the spleens of all FasL mutant mice (Fig. 3A). Similar results were obtained in the livers and lungs of infected mice (data not shown). We measured the frequency of OVA-specific CD8+ T cells by ELISPOT assay (Fig. 3B) and by staining with H-2Kb-OVA tetramers (Fig. 3C). OVA-specific CD8+ T cell response peaked at day 7 after LM-OVA infection, which was followed by a rapid phase of contraction, and the two groups of mice did not display any difference in the timing or extent of this response. However, in the longer term (~day 60 onward), slightly reduced numbers of OVA-specific CD8+ T cells were noted in FasL mutant mice.
FIGURE 3.
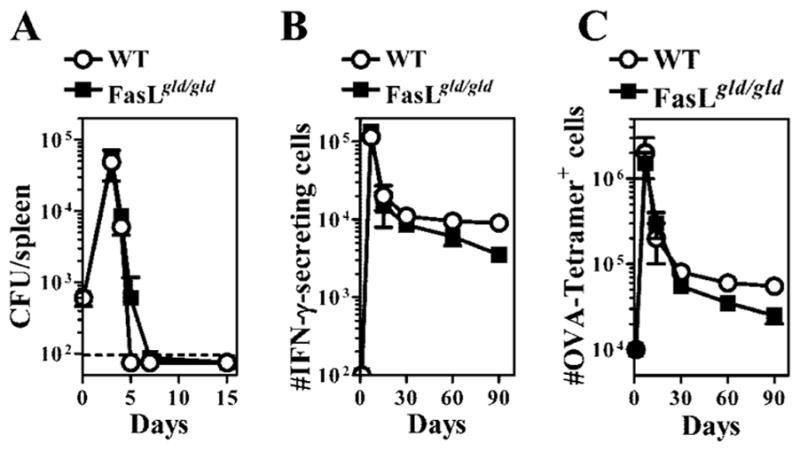
Normal expansion and contraction of CD8+ T cell response in FasL mutant mice. WT or FasL mutant mice were infected with 104 LM-OVA (A and B) or 106 LM-OVA, followed by daily injection with ampicillin (10 mg/mouse) from days 2 to 7 (C). The burden of LM-OVA was evaluated in the spleens of infected mice at various time intervals after infection (A). The frequency of OVA257–264-specific CD8+ T cells/spleen was evaluated by ELISPOT assay (B), and by staining with H-2KbOVA257–264 tetramers, as described in Materials and Methods (C).
Expression of mutant Fas or FasL on CD8+ T cells is not the reason behind the impaired cytolytic CD8+ T cell response in these mice
It is possible that the expansion and contraction of the CD8+ T cell response may proceed normally in FasL mutant mice, but the programming of CD8+ T cell memory development is defective in CD8+ T cells expressing a mutant FasL. We therefore infected WT, FasL, or Fas mutant mice with LM-OVA and purified memory CD8+ T cells from these mice at day 30. Purified CD8+ T cells from the various groups of mice were adoptively transferred into naive WT, FasL, or Fas mutant mice. Cells were transferred at day 30 because we wanted to transfer memory CD8+ T cells at a time point when their numbers remained relatively stable and they had not been subject to immune modulation for too long in the FasL mutant environment. Sixty days after the cell transfer, the fate of the transferred cells was then determined by evaluating OVA-specific cytolytic CD8+ T cell response in recipients. WT memory CD8+ T cells displayed potent response upon transfer into WT hosts, but not when transferred into Fas or FasL mutant hosts (Fig. 4). Memory CD8+ T cells from Fas or FasL mutant mice displayed potent cytolytic response upon transfer into WT hosts. These results indicate that the impairment in cytolytic CD8+ T cell response that occurs in Fas or FasL mutant mice is not related to the expression of mutant Fas or FasL on CD8+ T cells, and that the programming of CD8+ T cell memory proceeds normally in mice with a dysfunctional Fas pathway.
FIGURE 4.

Normal programming of CD8+ T cell memory in Fas or FasL mutant mice. WT, Fas mutant, and FasL mutant mice were infected with 104 LM-OVA. Thirty days after infection, CD8+ T cells were purified from spleens of infected mice and adoptively transferred (1 × 106/mouse) into naive WT, Fas, and FasL mutant mice. Sixty days after cell transfer, spleens were removed from recipient mice and tested for cytolytic activity against OVA257–264, as described in Fig. 1.
We then addressed whether memory CD8+ T cells that are purified from LM-OVA-infected FasL mutant hosts at a very late time period (day 90) would display normal response after transfer into WT hosts. To this end, we purified CD8+ T cells from LM-OVA-infected WT and FasL mutant hosts at day 90, and transferred these cells (same number of OVA-specific cells) into naive WT and FasL mutant hosts. After an additional 60 days, the cytolytic CD8+ T cell response was then evaluated. As is evident in Fig. 5, FasL mutant CD8+ T cells that are transferred at a very late time period (day 90) do not display significant cytolytic response even after transfer into WT hosts. These results indicate that the progressive dysfunction in memory CD8+ T cells that occurs in FasL mutant hosts is not reversible.
FIGURE 5.
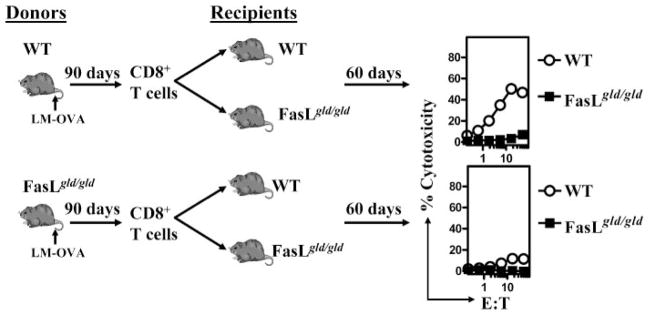
Impaired maintenance of CD8+ T cell memory in FasL mutant mice. WT and FasL mutant mice were infected with 104 LM-OVA. Ninety days after infection, CD8+ T cells were purified from spleens of infected mice and adoptively transferred (1–5 × 106/mouse) into naive WT and FasL mutant mice. Cell numbers were adjusted to inject exactly the same numbers of OVA-specific CD8+ T cells. Sixty days after cell transfer, spleens were removed from recipient mice and tested for cytolytic activity against OVA257–264, as described in Fig. 1.
Majority of memory CD8+ T cells in FasL mutant mice display an effector memory phenotype
Because the phenotype of memory cells gives clues about the activation status and function of memory cells, we measured the relative expression of IL-7Rα, CD62L, and PD-1 on CD8+ T cells that are OVA specific (Fig. 6, left panels) and on CD8+ T cells that are not OVA specific (Fig. 6, right panels). After day 7 of infection, OVA-specific CD8+ T cells in WT mice displayed progressive increase in IL-7Rα and CD62L expression, and a decrease in PD-1 expression, indicative of gradual differentiation into the central memory phenotype. In contrast, in FasL mutant mice, reduced numbers of OVA-specific CD8+ T cells expressed high levels of IL-7Rα and CD62L, indicating a significant phenotypic difference in memory CD8+ T cells in WT vs FasL mutant hosts. Because FasL mutant mice display increased cellularity in lymph nodes and spleens, implying that these mice must be responding to self or environmental Ags, we evaluated the phenotype of CD8+ T cells that were not OVA specific. The majority of these CD8+ T cells in FasL mutant mice were CD62LlowIL-7Rα low and PD-1high, implying that these CD8+ T cells are in a highly activated state.
FIGURE 6.
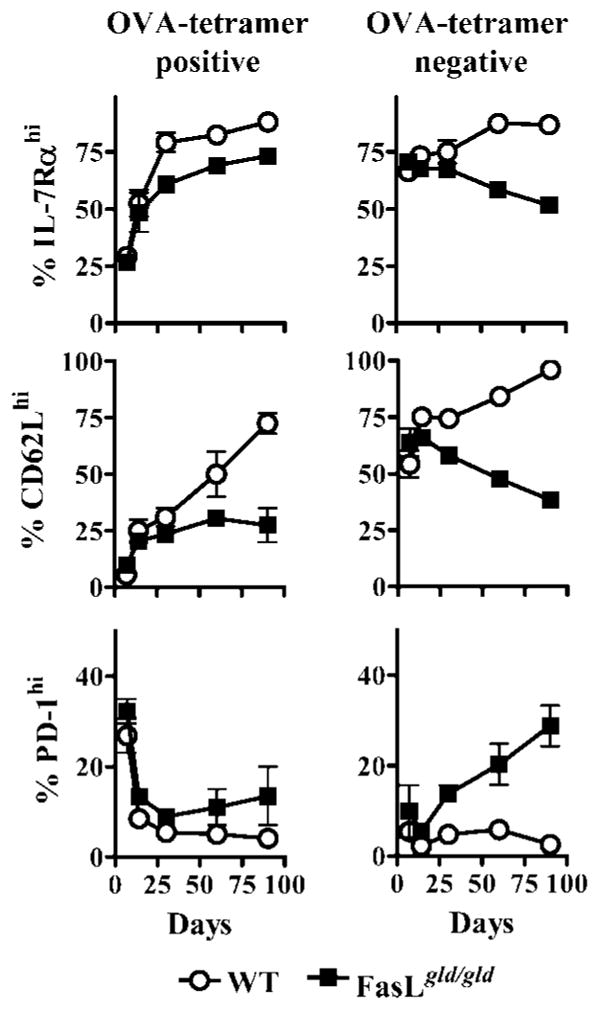
Memory CD8+ T cells in FasL mutant mice display a different phenotype. WT or FasL mutant mice were infected with 106 LM-OVA, followed by daily injection with ampicillin (10 mg/mouse) from days 2 to 7. At various time intervals, the expression of IL-7Rα, CD62L, and PD-1 was evaluated on OVA257–264 tetramer+ (left panels) and tetramer− (right panels) CD8+ T cells.
Memory CD8+ T cells in FasL mutant mice are apoptotic
Because OVA-specific CD8+ T cell numbers decreased progressively in FasL mutant hosts and these cells displayed an effector memory status, we evaluated the relative commitment of these cells to apoptosis. Apoptosis was detected at day 75 after LM-OVA infection by intracellular staining of OVA tetramer+ cells for active caspase 3 and TUNEL. OVA-specific CD8+ T cells in FasL mutant hosts displayed increased commitment to apoptosis (Fig. 7).
FIGURE 7.
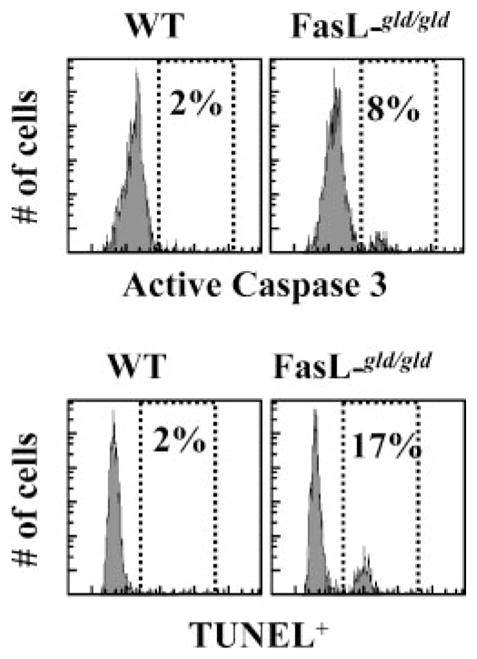
Increased apoptotic commitment of CD8+ T cells in FasL mutant mice. WT or FasL mutant mice were infected i.v. with 106 LM-OVA, followed by daily injection with ampicillin (10 mg/mouse) from days 2 to 7. At day 75, spleens were removed from infected mice, cultured in vitro for 3–6 h in RPMI 1640 plus 8% FBS. Cells were then stained with anti-CD8 Abs and OVA tetramers, followed by intracellular staining for detection of active caspase 3+ and TUNEL+ cells. Expression on gated OVA tetramer+ cells is indicated.
Defective homeostatic proliferation of memory CD8+ T cells in FasL mutant hosts
Based on the phenotypic analysis of OVA-specific memory CD8+ T cells in FasL mutant hosts, we expect impairment in their proliferative ability. We tested this by measuring the relative proliferation of OVA-specific memory CD8+ T cells in the absence of antigenic stimulation at day 70 after LM-OVA infection. In WT controls, ~10% of OVA-specific CD8+ T cells were cycling in comparison with FasL mutant mice, in which only ~5% of cells were cycling (Fig. 8A). At day 7 after LM-OVA infection, when the proliferation of OVA-specific CD8+ T cells was at peak, no difference was noted for proliferation of OVA-specific CD8+ T cells from WT vs FasL mutant mice (data not shown). We also labeled the cells with CFSE and evaluated their homeostatic proliferation in vitro in response to IL-7 and IL-15. CD8+ T cells from FasL mutant mice displayed poor homeostatic proliferation in vitro (Fig. 8B). We also purified CD8+ T cells from LM-OVA-infected WT and FasL mutant mice, labeled them with CFSE, and transferred them to naive RAG-1-deficient hosts. Four days later, spleens were isolated from RAG-1-deficient hosts, and the reduction in the CFSE intensity of OVA tetramer-gated CD8+ T cells was evaluated. Although OVA-specific CD8+ T cells from WT mice displayed good Ag-independent proliferation, OVA-specific CD8+ T cells from FasL mutant mice proliferated poorly (Fig. 8C).
FIGURE 8.
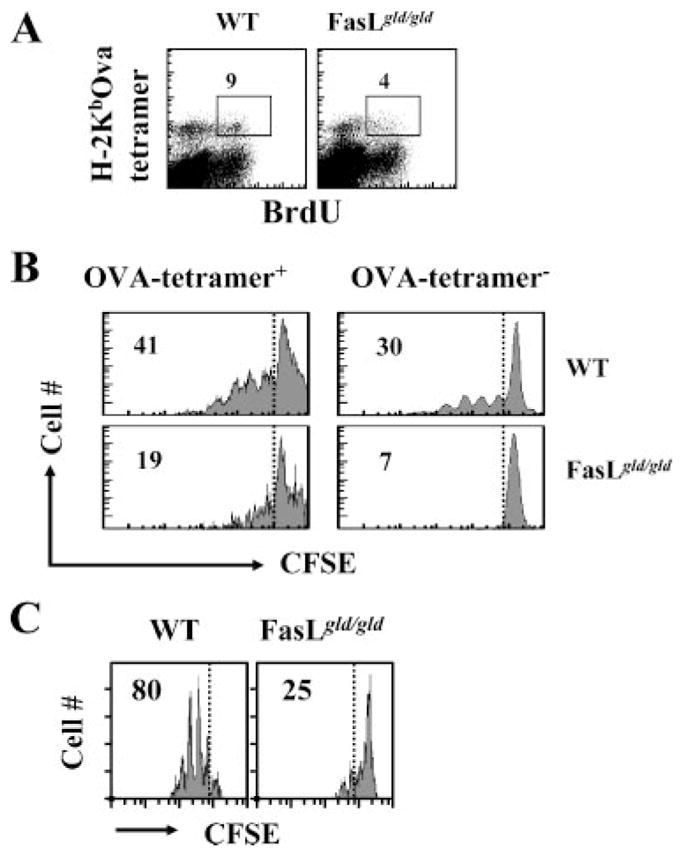
Impaired homeostatic proliferation of memory CD8+ T cells in FasL mutant mice. WT or FasL mutant mice were infected with LM-OVA. At day 70, BrdU (0.8 mg/ml) was incorporated into the drinking water of mice, which was changed daily. At day 73, spleens were removed and spleen cells were stained with anti-CD8 Ab and OVA tetramers, followed by intracellular staining for BrdU. Numbers in the panels indicate the percentage of BrdU+ CD8+ T cells among the tetramer+ cells (A). Spleen cells were also stained with CFSE (0.5 μM) and cultured (1 × 106 cells/ml) in RPMI 1640 plus 8% FBS in the presence of IL-7 and IL-15 (1 ng/ml each). After 5 days, the cells were washed and stained with anti-CD8 Ab and OVA tetramers, and the down-regulation of CFSE expression was evaluated by flow cytometry (B). Purified CD8+ T cells from WT and FasL mutant mice were also labeled with CFSE and transferred (5 × 106/mouse) i.v. into RAG-1−/− mice. Four days later, spleens were removed from RAG-1−/− mice and stained with OVA tetramer and anti-CD8 Ab, and the relative reduction in the expression of CFSE was evaluated by flow cytometry (C).
Impaired function of memory CD8+ T cells in FasL mutant mice
Because the synthesis of cytokines and proliferation in response to Ag encounter in vivo are two important functions of CD8+ T cells, we measured these functions at day 70 after LM-OVA infection of WT and FasL mutant mice. Intracellular staining for IFN-γ was done after cells were stained first with OVA tetramers, followed by in vitro stimulation with OVA peptide for 1 h. Slightly reduced expression of IFN-γ was noted in OVA-specific CD8+ T cells from FasL mutant mice (Fig. 9A). Upon in vitro stimulation for 48 h with OVA-peptide and syngeneic dendritic cells, purified CD8+ T cells from WT mice produced higher levels of IFN-γ in comparison with the CD8+ T cells from FasL mutant mice (Fig. 9B). Furthermore, OVA-specific CD8+ T cells from WT mice expressed higher levels of IFN-γ at a 10,000-fold reduced peptide concentration in comparison with OVA-specific CD8+ T cells from FasL mutant mice. In contrast, when cells were stimulated with plate-bound anti-CD3 Abs, CD8+ T cells from FasL mutant mice produced higher levels of IFN-γ (Fig. 9C).
FIGURE 9.

Impairment in the function of memory CD8+ T cells in FasL mutant mice. WT and FasL mutant mice were infected with LM-OVA. At day 70, spleens were removed, and the numbers of IFN-γ -secreting tetramer+ CD8+ T cells were evaluated (A) after stimulation of cells for 1 h with OVA257–264. Numbers in the panels indicate the percentage of IFN-γ -secreting CD8+ T cells among the tetramer+ cells. CD8+ T cells were also purified from WT and FasL mutant mice at day 70, and stimulated (1 × 105/well) in vitro with different concentrations of OVA peptide (B) and syngeneic dendritic cells as APCs (2 × 105/well). Purified CD8+ T cells were also stimulated with plate-bound anti-CD3 Abs in the absence of dendritic cells (C). Supernatants were collected at 48 h, and the production of IFN-γ was measured by ELISA (B). The relative increase in the numbers of OVA-specific CD8+ T cells after pathogen rechallenge (day 70) was evaluated by measuring the numbers of OVA tetramer+ CD8+ T cells 4 days after rechallenge of mice with 105 LM-OVA (D).
We also measured the ability of memory CD8+ T cells to proliferate in response to pathogen rechallenge. LM-OVA-infected WT and FasL mutant mice were rechallenged with LM-OVA on day 70, and the relative increase in the numbers of OVA tetramer+ CD8+ T cells was enumerated 4 days later. Although OVA-specific CD8+ T cells from WT mice underwent ~10-fold expansion in 4 days, only 2-fold expansion in OVA-specific CD8+ T cells was noted in FasL mutant mice (Fig. 9D).
Compromised protection by CD8+ T cells from FasL mutant mice
Because protection against a lethal dose of LM or LM-OVA requires memory CD8+ T cells (18, 20, 21), we reasoned that an impairment of CD8+ T cell memory may consequently impair the long-term protection against pathogen rechallenge. As expected, WT mice that were preimmunized with LM demonstrated massive protection against a rechallenge with a lethal dose of LM, indicating the development of potent and protective T cell memory (Fig. 10). FasL mutant mice in contrast demonstrated little, if any, protection in the spleen or lungs. It could be argued that the absence of protection in FasL mutant mice may not be due to impairment of CD8+ T cell memory, but could be attributable to other immune defects. We, therefore, purified CD8+ T cells from naive or immune WT and FasL mutant mice and transferred the cells to naive WT or FasL hosts. Four days after the cell transfer, mice were challenged with a lethal dose of LM-OVA, and the influence on bacterial burden was measured 3 days later. As is evident in Fig. 11, transferred memory CD8+ T cells from WT mice mediated reduction in bacterial burden in WT as well as FasL mutant recipients. In contrast, transfer of memory CD8+ T cells from FasL mutant mice did not mediate effective reduction of LM-OVA even in WT hosts. These results clearly indicate that CD8+ T cell memory is compromised in FasL mutant mice. When memory CD8+ T cells from FasL mutant mice were transferred at early time intervals after LM-OVA infection (e.g., day 30), then the CD8+ memory T cells mediated potent protection (our unpublished observations). These results indicate that memory CD8+ T cells may be induced normally in FasL mutant mice, but over time their function is impaired due to prolonged exposure to the FasL environment.
FIGURE 10.
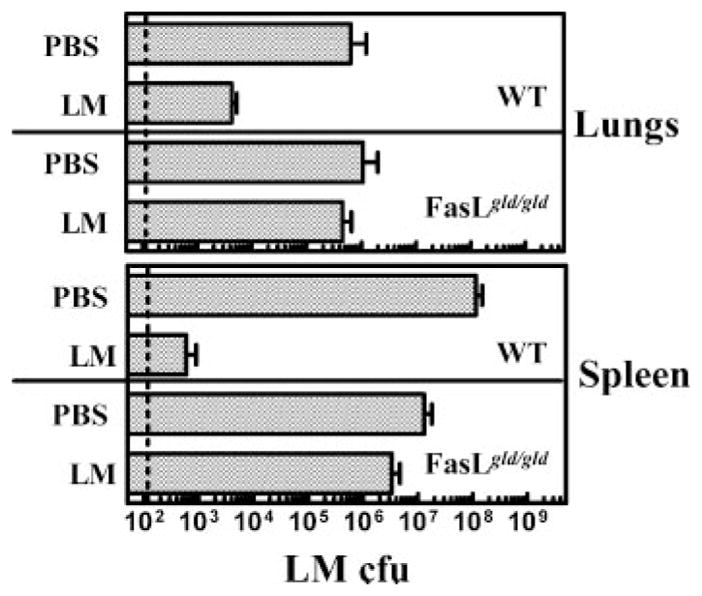
Long-term protection in response to rechallenge of mice with a lethal dose of LM is lost in FasL mutant mice. WT and FasL mutant mice were infected with LM (1 × 103, i.v.). On day 80, mice were rechallenged with LM (1 × 105, i.v.), and the burden of LM was evaluated in lungs and spleen 3 days post rechallenge.
FIGURE 11.

Memory CD8+ T cells from FasL mutant mice fail to confer protection. WT and FasL mutant mice were infected with LM (1 × 103, i.v.). On day 80, CD8+ T cells were purified from naive and LM-OVA-infected mice. Purified CD8+ T cells from WT or FasL mutant mice were transferred (2 × 106) into naive WT or FasL mutant recipients. Four days after the transfer, recipients were challenged with a lethal dose (3 × 105) of LM-OVA. LM-OVA burden in the spleens was evaluated after 3 days.
Impaired cytokine expression in FasL mutant mice
Because our results indicated that the environment that develops in FasL mutant mice, rather than the expression of mutant FasL on CD8+ T cells, was responsible for impairment of CD8+ T cell memory, we sought to determine the relative levels of inflammatory cytokines (IL-12, IFN-α, IFN-γ, TNF-α, IL-18), suppressive cytokines (IL-10, TGF-β), and memory-promoting cytokines (IL-15 and IL-7) in WT vs FasL mutant spleens at day 75 after LM-OVA infection. This was achieved by performing quantitative RT-PCR on spleen samples, and values were normalized to β-actin. Of the various cytokines evaluated, splenic expression of IFN-γ was significantly higher in FasL mutant mice (Fig. 12). Furthermore, compared with the WT spleens, IL-10 levels were higher and IL-7 levels were lower in the spleens of FasL mutant mice, although these differences were not statistically significant.
FIGURE 12.
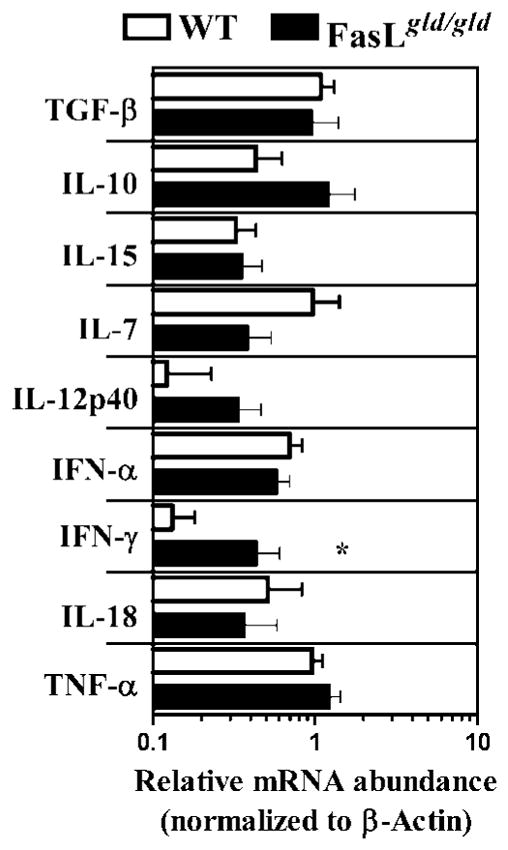
Impaired cytokine expression in FasL mutant mice. WT and FasL mutant mice were infected with LM-OVA. At day 75, spleens were removed and snap frozen in dry-ice ethanol bath. Total RNA was extracted, and 5 μg of RNA was taken to make cDNA. The amount of RNA for various cytokines and β-actin was measured using the SYBR Green method of quantification, as outlined in Materials and Methods. Values represent the levels of cytokine mRNA relative to β-actin mRNA. Although differences in several cytokines were noted between WT vs FasL mutant spleens, only the difference in the relative expression of IFN-γ achieved significance (*, p < 0.05) by Mann-Whitney U test.
Discussion
The role of Fas pathway in the regulation of immune responses after infection has been unclear. On one hand, disruption of the Fas pathway results in generalized, chronic, lymphadenopathy, splenomegaly, and lymphoproliferative disorder in mice (5), and auto-immune lymphoproliferative syndrome in humans (22), which also mirrors the clinical manifestations of the chronic autoimmune disorder, systemic lupus erythematosus (SLE) (7, 23). In contrast, CD8+ T cell response to lymphocytic choriomeningitis virus (LCMV) infection does not appear to be influenced by a mutation of the Fas pathway (16, 17). We have therefore evaluated the role of the Fas pathway in the development and function of CD8+ T cell memory in a murine intracellular bacterial infection model. Our results indicate that whereas the priming, expansion, and contraction of CD8+ T cell response proceed normally in mice with a defective Fas pathway, the phenotype and function of memory CD8+ T cells are compromised in the long-term.
Using P14 CD8 TCR-lpr/lpr transgenic mice, it was shown that the mutation in Fas (lpr) did not affect LCMV gp33 peptide Ag-induced deletion or clonal down-regulation of LCMV gp33-specific CD8+ T cells after a viral infection in vivo (16, 17). However, the functionality of CD8+ T cells was not assessed. Similarly, using female HY-TCR-lpr mice, it was reported that the deletion of HY-TCR transgenic T cells was not influenced by the mutation of Fas (24). Later studies have, however, indicated that Fas-dependent mechanisms appear to be important for controlling autoreactive CD8+ T cells (15). Taken together, these reports suggest that autoreactive CD8+ T cells that are induced in the absence of inflammation are controlled by Fas-dependent mechanisms, whereas the down-regulation of T cells at the end of an immune response to foreign Ags that occurs in the presence of inflammation is controlled by mechanisms other than Fas. Our results are compatible with this interpretation because we did not notice any role of Fas in the expansion or contraction of OVA-specific CD8+ T cells after infection with LM-OVA. However, our results indicate remarkable phenotypic differences in CD8+ T cells in FasL mutant mice. The differences in CD62L expression in OVA-specific CD8+ T cells in WT vs FasL mutant mice emerged only after day 30 of LM-OVA infection. This indicates that the problem with CD8+ T cell memory in FasL mutant mice may be due to mechanisms that develop later in FasL mutant mice.
The impairment of CD8+ T cell memory in FasL mutant hosts depended on the chronic exposure of memory CD8+ T cells in such hosts. When newly formed memory CD8+ T cells (day 30) from Fas or FasL mutant hosts were transferred into WT hosts, no functional impairment in memory CD8+ T cells was noted (Fig. 4). When newly formed memory CD8+ T cells from WT mice were transferred into Fas mutant hosts and kept in that environment for 60 days, functional impairment of memory was noted (Fig. 4). These results indicate that the mutation of Fas or FasL on CD8+ T cells is not the reason for impairment of CD8+ T cell memory; rather, it is the environment that develops in such mice that causes memory impairment. It has been previously reported that IL-7 is beneficial (25) and IFN-γ is detrimental (26) for CD8+ T cell homeostasis. Our results indicate that the relative expression of IFN-γ is significantly enhanced in the spleens of FasL mutant mice. Spleens of FasL mutant mice have a 10-fold reduction in IL-7:IFN-γ ratio in comparison with WT spleens. It is conceivable that chronic activation of self-reactive CD4+ and CD8+ T cells in FasL mutant mice results in increased levels of cytokines such as IFN-γ. CD8+ T cells from FasL mutant mice produced higher levels of IFN-γ in response to anti-CD3 stimulation. CD4−CD8− TCR+ cells accumulate in FasL mutant mice (5, 7), secrete IFN-γ, and suppress CD8+ T cell activation (27). Such regulatory mechanisms could impair the homeostasis of memory CD8+ T cells against non-self Ags.
Impairment in maintenance, but not programming, of CD8+ T cell memory was shown to occur in mice that were deficient in MHC class II or CD4+ T cells (28), although it is not clear how CD4+ T cells promote the maintenance of memory CD8+ T cells. It is therefore not clear whether similar mechanisms induce the impairment of CD8+ T cell memory in CD4+ T cell-deficient mice vs FasL mutant mice, although memory CD8+ T cells in both situations express reduced levels of IL-7Rα. It is important to note that the impairment of memory in FasL mutant mice appears to be related to the phenotype of cells rather than due to the number of cells, because FasL mutant have only marginally reduced numbers of memory cells compared with WT controls.
It has been previously shown that memory, but not naive, CD4+ T cells are costimulated to proliferate by Fas engagement (29). Similarly, the proliferation of peripheral CD8+ T cells is enhanced by Fas engagement (30). Caspases appear to be required for T cell proliferation because anti-CD3-induced proliferation and IL-2 production by human T cells were blocked by inhibitors of caspase activity (31). Furthermore, T cells deficient in Fas-associated death domain protein (which is recruited after Fas engagement and causes activation of caspases) were defective in proliferation (32). It is therefore conceivable that FasL mutant memory CD8+ T cells fail to proliferate efficiently in response to antigenic encounter (30). However, our results indicate that the impairment of memory CD8+ T cells in FasL mutant mice is related to the phenotype of cells rather than due to the expression of mutant FasL on CD8+ T cells.
The gradual, exacerbated T cell expansion and accumulation that occur in mice with a nonfunctional Fas pathway suggest that the T cells may be specific to some self/environmental Ags. Indeed, the majority of the CD8+ T cells in FasL mutant mice that are not OVA-specific progressively became CD62LlowIL-7Rα low, indicating the highly activated state of these cells. In the face of such enormous accumulation of highly activated, self-reactive T cells, resident memory CD8+ T cells against non-self Ags may be under competitive homeostatic pressure. Indeed, it has been previously shown that homeostatic proliferation of T cells can be inhibited by large numbers of nonspecific T cells (33–35), and may involve competition for common resources, such as IL-7 and IL-15 (25, 36, 37). Memory CD8+ T cells need to undergo homeostatic proliferation to survive, and our results do indicate that in the longer term, FasL mutant mice have reduced numbers of OVA-specific CD8+ T cells. However, a mere 2- to 3-fold difference in the number of memory CD8+ T cells in a host does not translate to a significant difference in protective efficacy in response to pathogen rechallenge. The massive difference in protective efficacy that we noted in CD8+ T cells from WT vs FasL mutant mice could be attributed to their difference in phenotype and consequent proliferation in response to antigenic encounter. It has been previously reported that effector memory CD8+ T cells undergo less proliferation in comparison with central memory cells (38). Thus, memory CD8+ T cells from WT mice (mainly central memory phenotype) display a superior protective efficacy in comparison with the mainly effector memory CD8+ T cells in FasL mutant mice. Impairment in the proliferation of memory CD8+ T cells in response to Ag could present a major problem during pathogen re-encounter, in which the memory CD8+ T cells have to proliferate rapidly to counteract the proliferating pathogen (38).
Inactivation of the Fas pathway induces an autoimmune lymphoproliferative syndrome in humans (22), and has been considered to induce a state that is similar to a systemic autoimmune disorder (e.g., SLE) (5, 7). In such individuals, chronic Ag presentation and T cell expansion would force excessive competition among lymphocytes. Any previously generated memory T cells to foreign Ags in such individuals would become susceptible to dysfunction. This puts the individuals at a greater risk because the memory T cells that have been accumulated over a lifetime would be gradually rendered nonfunctional. Support for this hypothesis comes from clinical data in which it has been reported that the herpes zoster infection gets reactivated (shingles) in SLE patients (39) even before the use of immunosuppressants. Similarly, patients with SLE have a difficulty in controlling latent EBV infection due to altered T cell responses against EBV (40). Thus, our study reveals that despite the persistence of memory T cells, their function can get significantly modulated in hosts with defective lymphocyte homeostasis. Identification of the key mechanisms that impair the function of memory T cells in such hosts can pave the way for novel therapeutic approaches.
Footnotes
This work was supported by a grant from the Canadian Institutes of Health Research.
Abbreviations used in this paper: FasL, Fas ligand; LCMV, lymphocytic choriomeningitis virus; LM, Listeria monocytogenes; SLE, systemic lupus erythematosus.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Dutton RW, Bradley LM, Swain SL. T cell memory. Annu Rev Immunol. 1998;16:201–223. doi: 10.1146/annurev.immunol.16.1.201. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- 3.Sprent J, Surh CD. Generation and maintenance of memory T cells. Curr Opin Immunol. 2001;13:248–254. doi: 10.1016/s0952-7915(00)00211-9. [DOI] [PubMed] [Google Scholar]
- 4.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 5.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 6.Henkart PA. Lymphocyte-mediated cytotoxicity: two pathways and multiple effector molecules. Immunity. 1994;1:343–346. doi: 10.1016/1074-7613(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 8.Russell JH, Rush B, Weaver C, Wang R. Mature T cells of autoimmune lpr/lpr mice have a defect in antigen-stimulated suicide. Proc Natl Acad Sci USA. 1993;90:4409–4413. doi: 10.1073/pnas.90.10.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bossu P, Singer GG, Andres P, Ettinger R, Marshak-Rothstein A, Abbas AK. Mature CD4+ T lymphocytes from MRL/lpr mice are resistant to receptor-mediated tolerance and apoptosis. J Immunol. 1993;151:7233–7239. [PubMed] [Google Scholar]
- 10.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumor necrosis factor. Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 11.Scott DE, Kisch WJ, Steinberg AD. Studies of T cell deletion and T cell anergy following in vivo administration of SEB to normal and lupus-prone mice. J Immunol. 1993;150:664–672. [PubMed] [Google Scholar]
- 12.Van PL, Ibraghimov A, Abbas AK. The roles of costimulation and Fas in T cell apoptosis and peripheral tolerance. Immunity. 1996;4:321–328. doi: 10.1016/s1074-7613(00)80440-9. [DOI] [PubMed] [Google Scholar]
- 13.Papiernik M, Pontoux C, Golstein P. Non-exclusive Fas control and age dependence of viral superantigen-induced clonal deletion in lupus-prone mice. Eur J Immunol. 1995;25:1517–1523. doi: 10.1002/eji.1830250607. [DOI] [PubMed] [Google Scholar]
- 14.Renno T, Hahne M, Tschopp J, MacDonald HR. Peripheral T cells undergoing superantigen-induced apoptosis in vivo express B220 and up-regulate Fas and Fas ligand. J Exp Med. 1996;183:431–437. doi: 10.1084/jem.183.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurts C, Heath WR, Kosaka H, Miller JF, Carbone FR. The peripheral deletion of autoreactive CD8+ T cells induced by cross-presentation of self-antigens involves signaling through CD95 (Fas, Apo-1) J Exp Med. 1998;188:415–420. doi: 10.1084/jem.188.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmermann C, Rawiel M, Blaser C, Kaufmann M, Pircher H. Homeostatic regulation of CD8+ T cells after antigen challenge in the absence of Fas (CD95) Eur J Immunol. 1996;26:2903–2910. doi: 10.1002/eji.1830261215. [DOI] [PubMed] [Google Scholar]
- 17.Reich A, Korner H, Sedgwick JD, Pircher H. Immune down-regulation and peripheral deletion of CD8 T cells does not require TNF receptor-ligand interactions nor CD95 (Fas, APO-1) Eur J Immunol. 2000;30:678–682. doi: 10.1002/1521-4141(200002)30:2<678::AID-IMMU678>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 18.Dudani R, Chapdelaine Y, Faassen HH, Smith DK, Shen H, Krishnan L, Sad S. Multiple mechanisms compensate to enhance tumor-protective CD8+ T cell response in the long-term despite poor CD8+ T cell priming initially: comparison between an acute versus a chronic intracellular bacterium expressing a model antigen. J Immunol. 2002;168:5737–5745. doi: 10.4049/jimmunol.168.11.5737. [DOI] [PubMed] [Google Scholar]
- 19.Tough DF, Sprent J. Turnover of naive- and memory-phenotype T cells. J Exp Med. 1994;179:1127–1135. doi: 10.1084/jem.179.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong P, Pamer EG. CD8 T cell responses to infectious pathogens. Annu Rev Immunol. 2003;21:29–70. doi: 10.1146/annurev.immunol.21.120601.141114. [DOI] [PubMed] [Google Scholar]
- 21.Harty JT, Bevan MJ. CD8+ T cells specific for a single nonamer epitope of Listeria monocytogenes are protective in vivo. J Exp Med. 1992;175:1531–1538. doi: 10.1084/jem.175.6.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bidere N, Su HC, Lenardo MJ. Genetic disorders of programmed cell death in the immune system. Annu Rev Immunol. 2006;24:321–352. doi: 10.1146/annurev.immunol.24.021605.090513. [DOI] [PubMed] [Google Scholar]
- 23.Lynch DH, Watson ML, Alderson MR, Baum PR, Miller RE, Tough T, Gibson M, Davis-Smith T, Smith CA, Hunter K. The mouse Fas-ligand gene is mutated in gld mice and is part of a TNF family gene cluster. Immunity. 1994;1:131–136. doi: 10.1016/1074-7613(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 24.Teh SJ, Dutz JP, Motyka B, Teh HS. Fas (CD95)-independent regulation of immune responses by antigen-specific CD4−CD8+ T cells. Int Immunol. 1996;8:675–681. doi: 10.1093/intimm/8.5.675. [DOI] [PubMed] [Google Scholar]
- 25.Schluns KS, Kieper WC, Jameson SC, Lefrancois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 26.Badovinac VP, Tvinnereim AR, Harty JT. Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-γ. Science. 2000;290:1354–1358. doi: 10.1126/science.290.5495.1354. [DOI] [PubMed] [Google Scholar]
- 27.Priatel JJ, Utting O, Teh HS. TCR/self-antigen interactions drive double-negative T cell peripheral expansion and differentiation into suppressor cells. J Immunol. 2001;167:6188–6194. doi: 10.4049/jimmunol.167.11.6188. [DOI] [PubMed] [Google Scholar]
- 28.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. 2004;5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desbarats J, Wade T, Wade WF, Newell MK. Dichotomy between naive and memory CD4+ T cell responses to Fas engagement. Proc Natl Acad Sci USA. 1999;96:8104–8109. doi: 10.1073/pnas.96.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki I, Martin S, Boursalian TE, Beers C, Fink PJ. Fas ligand costimulates the in vivo proliferation of CD8+ T cells. J Immunol. 2000;165:5537–5543. doi: 10.4049/jimmunol.165.10.5537. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy NJ, Kataoka T, Tschopp J, Budd RC. Caspase activation is required for T cell proliferation. J Exp Med. 1999;190:1891–1896. doi: 10.1084/jem.190.12.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, Cado D, Chen A, Kabra NH, Winoto A. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature. 1998;392:296–300. doi: 10.1038/32681. [DOI] [PubMed] [Google Scholar]
- 33.Ernst B, Lee DS, Chang JM, Sprent J, Surh CD. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity. 1999;11:173–181. doi: 10.1016/s1074-7613(00)80092-8. [DOI] [PubMed] [Google Scholar]
- 34.Rocha B, Dautigny N, Pereira P. Peripheral T lymphocytes: expansion potential and homeostatic regulation of pool sizes and CD4/CD8 ratios in vivo. Eur J Immunol. 1989;19:905–911. doi: 10.1002/eji.1830190518. [DOI] [PubMed] [Google Scholar]
- 35.Dummer W, Ernst B, Leroy E, Lee D, Surh C. Autologous regulation of naive T cell homeostasis within the T cell compartment. J Immunol. 2001;166:2460–2468. doi: 10.4049/jimmunol.166.4.2460. [DOI] [PubMed] [Google Scholar]
- 36.Tan JT, Ernst B, Kieper WC, Leroy E, Sprent J, Surh CD. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J Exp Med. 2002;195:1523–1532. doi: 10.1084/jem.20020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldrath AW, Sivakumar PV, Glaccum M, Kennedy MK, Bevan MJ, Benoist C, Mathis D, Butz EA. Cytokine requirements for acute and basal homeostatic proliferation of naive and memory CD8+ T cells. J Exp Med. 2002;195:1515–1522. doi: 10.1084/jem.20020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 39.Pope JE, Krizova A, Ouimet JM, Goodwin JL, Lankin M. Close association of herpes zoster reactivation and systemic lupus erythematosus (SLE) diagnosis: case-control study of patients with SLE or noninflammatory musculoskeletal disorders. J Rheumatol. 2004;31:274–279. [PubMed] [Google Scholar]
- 40.Kang I, Quan T, Nolasco H, Park SH, Hong MS, Crouch J, Pamer EG, Howe JG, Craft J. Defective control of latent Epstein-Barr virus infection in systemic lupus erythematosus. J Immunol. 2004;172:1287–1294. doi: 10.4049/jimmunol.172.2.1287. [DOI] [PubMed] [Google Scholar]