

Abstract
For many years, mitochondria were viewed as semi-autonomous organelles, required only for cellular energetics. This view has been largely supplanted by the concept that mitochondria are fully integrated into the cell and that mitochondrial stresses rapidly activate cytosolic signaling pathways that ultimately alter nuclear gene expression. Remarkably, this coordinated response to mild mitochondrial stress appears to leave the cell less susceptible to subsequent perturbations. This response, termed mitohormesis, is being rapidly dissected in many model organisms. A fuller understanding of mitohormesis promises to provide insight into our susceptibility for disease and potentially provide a unifying hypothesis for why we age.
In 120 BC, King Mithridates V held a lavish banquet. Such feasts were not unusual, since as King of Pontus, a region now in modern day Turkey, Mithridates V was extraordinarily wealthy, a result of his strategic alliance with the neighboring Roman Empire. Midway through this particular banquet, however, the seemingly robust and healthy monarch suddenly died. All signs immediately pointed to poisoning. Suspicion naturally fell on the king’s wife, who by tradition became the caretaker ruler of Pontus, and would remain so, until the time either of the two young sons of Mithridates V would come of age. A short time after his father’s untimely death, the eldest son and potential future king began to notice new, intense abdominal pains after every meal. Convinced his mother was now slowly poisoning him, and determined not to meet the same fate as his recently deceased father, the older son decided to escape. Over the next seven years, while alone in the wild, the young prince attempted to find a way to protect himself against a future assassination. His solution was to regularly ingest small doses of known poisons believing that by doing so he could build up a resistance against larger doses that might someday come his way. After seven years of a self-imposed exile, the son, now Mithridates VI, returned to Pontus. He rapidly retook the throne, whereupon he promptly imprisoned his mother and brother, married his sister and ruled Pontus effectively, albeit ruthlessly, for the next sixty years. It is widely believed that when Mithridates VI returned to Pontus, he carried with him a potion containing 54 known poisonous compounds mixed together in small doses. Ingestion of this potion, thereafter called Antidotum Mithridaticum, was believed to protect the user against any number of lethal assaults. It would be used in one form or another by royals and common folk for the next 1900 years.
However dysfunctional our particular families may be, few of us today have the particular concerns that beset Mithridates VI. Yet surprisingly, many of the notions that he wrestled with during his seven year sabbatical in the Anatolian woods are becoming increasingly rediscovered and explored. In particular, there is growing scientific interest in the idea that a mild sub-lethal stress can protect against larger, subsequent stresses. This concept, termed hormesis, was originally explored in the context of classical 19th century pharmacology, although the term itself was only coined some seventy years ago (Southam and Ehrlich, 1943). In general, hormesis is defined as any adaptive response exhibiting a biphasic dose response, and this general phenomenon has been the subject of many excellent reviews (Calabrese and Baldwin, 2002). Here, we will explore in-depth one particular form of hormesis, termed mitohormesis. In this paradigm, mild mitochondrial stress that can be triggered by any of a variety of insults, results in a broad and diverse cytosolic and nuclear response (Figure 1). Although varied, this response appears to induce a wide-ranging cytoprotective state resulting in long-lasting metabolic and biochemical changes. Remarkably, rather than being harmful, these changes may reduce our susceptibility for disease, as well as potentially determine how long we live.
Figure 1.
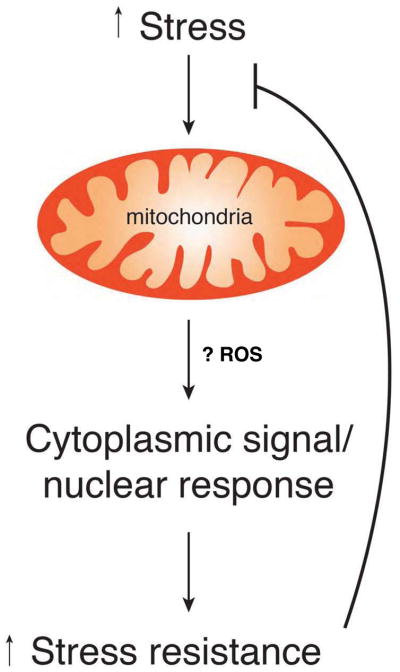
The basis of mitohormesis. Any of a number of endogenous or exogenous stresses can perturb mitochondrial function. These perturbations are relayed to the cytosol through at present, poorly understood mechanisms that may involve mitochondrial ROS as well as other mediators. These cytoplasmic signaling pathways and subsequent nuclear transcriptional changes induce various long lasting cytoprotective pathways. This augmented stress resistance allows for protection from a wide array of subsequent stresses.
Mitochondrial Stress and the Retrograde Response
Mitochondria are entrusted with the difficult task of providing for acute and chronic energetic needs through the generation of ATP, and of producing various biosynthetic intermediates that form part of the TCA cycle and heme biosynthesis. Yet, the genetic complexity of the mitochondria is such that it encodes a mere dozen or so polypeptides, with the other 1000 or so proteins that make up the organelle encoded by the nucleus. This cellular co-dependency suggests that for the cell to grow and divide, any significant deficiency in mitochondrial function might trigger an adaptive nuclear response. How then does the nucleus know that the mitochondria are in trouble? And furthermore, how is this mitochondrial stress signal sent in a retrograde fashion from mitochondria to the nucleus? The first attempt to find answers to these questions came from studying S. cerevisiae, an organism in which mitochondrial DNA (mtDNA) is not required for growth as long as a fermentable carbon source is provided, allowing investigators to analyze the presumed adaptive nuclear response in yeast cells engineered to lack mitochondrial DNA (Parikh et al., 1987). These early studies showed that mitochondrial genetic deficiencies induce a coordinated and complex nuclear response that alters the expression of forty or more genes (Epstein et al., 2001). While the exact response depends in part on the specific mitochondrial perturbation, in general, the transcriptional response results in a reconfiguration of metabolism, allowing for the production of essential intermediates such as glutamate, and increasing glycolytic production of ATP (Epstein et al., 2001). In yeast, the coordination of this response depends on the transcription factors Rtg1 andRtg3, two basic helix-loop-helix/leucine zipper proteins (Rothermel et al., 1995; Rothermel et al., 1997). Rtg1 andRtg3 bind DNA in a sequence specific manner as a heterodimer. The activity of Rtg1/Rtg3 is predominantly regulated by dephosphorylation of Rtg3, resulting in translocation of the complex from the cytoplasm to the nucleus. This process is regulated by a third factor, the cytoplasmic protein Rtg2 (Sekito et al., 2000). Interestingly, the activity of Rgt2 appears to be sensitive to variety of metabolic cues, including the concentration of glutamate, as well as regulation through the important nutrient sensitive kinase, Target of Rapamycin complex I (TORC1) (Komeili et al., 2000; Liu et al., 2001). Presently, it is believed that Rtg2 is the proximal sensor of mitochondrial dysfunction, although the exact signal emitted from damaged or dysfunctional mitochondria remains elusive (Jazwinski and Kriete, 2012). One attractive possibility is mitochondrial reactive oxygen species (ROS), whose levels increase in dysfunctional mitochondria. Alternatively, the activating signal might involve a fall in mitochondrial membrane potential, given that this is also a common phenomenon observed in dysfunctional mitochondria. Unlike ROS, mitochondrial membrane potential is not an emitted signal. Nonetheless, recent evidence in mammalian cells suggest that mitochondrial depolarization can serve as a stimulus to recruit and activate cytosolic factors as observed with the PINK1/Parkin system (Jin et al., 2010). In that particular case, loss of mitochondrial membrane potential leads to the accumulation of the mitochondrial targeted serine/threonine kinase PINK1, which in turn, triggers the recruitment of the cytosolic E3 ubiquitin ligase Parkin. Thus, it’s conceivable that a fall in membrane potential could similarly recruit Rtg2 to the mitochondrial membrane and allow assembly of a signaling complex required for the retrograde response.
Although the studies discussed above established a way, at least in yeast, that perturbations in mitochondrial function could be sensed and compensated for, the interest in this retrograde pathway was enhanced when it was shown that activating this response could extend the replicative lifespan of yeast(Kirchman et al., 1999). Subsequent studies have demonstrated that a loss of mitochondrial membrane potential during normal yeast replicative aging and showed that this fall in membrane potential correlated with induction of the retrograde response as these organisms aged(Borghouts et al., 2004). Taken together, these observations suggested that mitochondrial stress may trigger a hormetic response that provided both short term metabolic benefits and the potential for long term benefits in increased stress resistance and longevity.
Unfortunately, there are no clear analogs of the Rtg1/2/3 system in mammals. Nonetheless, mutations in mitochondrial DNA that result in stable perturbations of mammalian mitochondrial function can also elicit a coordinated alteration in nuclear gene expression(Heddi et al., 1999). Again, this nuclear response can be globally viewed as an attempt to compensate for the primary metabolic defect caused by impaired mitochondrial function. Subsequent analysis suggested that following treatment with various respiratory chain inhibitors or after mtDNA deletion, the disruption of mammalian mitochondrial function elicited a signaling response involving calcineurin-dependent activation of NF-κB (Biswas et al., 1999; Biswas et al., 2003). Recent systems biology approaches have suggested that the retrograde response in mammals may involve a host of factors including the Retinoid X receptor α (RXRA), the peroxisome proliferator-activated receptor γ coactivator-1 (PGC1α), the signaling kinase c-Jun N-terminal kinase (JNK) and ROS generation (Chae et al., 2013).
Signaling Mitochondrial Stress
Thus far we have discussed various mechanisms by which stressed mitochondria may signal outward to the cytosol and the nucleus. These include altering mitochondrial membrane potential to allow recruitment and assembly of signaling molecules or the production of ROS. These are however, not the only available elements contained within the mitochondrial repertoire. For instance, calcium released from the ER is taken up via the mitochondrial calcium uniporter (MCU) through a recently identified inner mitochondrial protein (Baughman et al., 2011; De Stefani et al., 2011). In these and other ways, mitochondria have the capacity to shape cytosolic calcium transients and ultimately to modulate a host of calcium-dependent signaling events. Further, mitochondrial activity plays a central role in determining the levels of metabolic intermediates including NAD+/NADH and Acetyl-CoA, which act as obligate cofactors for modifying enzymes in the sirtuin, PARP and histone acetyltransferase families. Although it remains uncertain to what degree enzymatic activity is normally regulated by cofactor availability, there is accumulating evidence that under various stresses, availability of these intermediates shapes biological outcomes (Dolle et al., 2013; Houtkooper and Auwerx, 2012). Besides these energetic intermediates, mitochondria can influence the cell by their underlying structure and distribution. For instance, there is increasing interest in the role of mitochondrial fusion and fission in physiology. Recent evidence suggests that tubular versus fragmented mitochondria differ in their metabolic capacity, ROS production and ability to induce cell death(Picard et al., 2013). Furthermore, other recent studies have determined that mitochondrial distribution within the cell, particularly mitochondrial distance to the nucleus, can alter the ultimate transcriptional response (Al-Mehdi et al., 2012).
The mitochondrial unfolded protein response
In many of the initial studies exploring the cellular response to mitochondrial perturbations, the nuclear response was in general precipitated by altering the integrity of mtDNA or by acutely poisoning the electron transport chain to interrupt ATP generation. However, a distinctly different nuclear response is induced when the mitochondrial stress is the accumulation of misfolded proteins within the mitochondrial matrix. In one early example, cells were engineered to express a mutant form of the mitochondrial matrix protein ornithine transcarbamylase (OTC) (Zhao et al., 2002). This mutant OTC molecule could not fold correctly and elicited a stress response that was similar, in some respects, to the better characterized ER stress response. In particular, the accumulation of misfolded mitochondrial OTC induced a nuclear transcriptional response resulting in the induction of a number of mitochondrial-specific protein chaperones. This mitochondrial unfolded protein response pathway (UPRmt) is found in lower organisms as well, and recent elegant dissection of this pathway has been achieved in C. elegans (Haynes et al., 2010; Nargund et al., 2012; Yoneda et al., 2004). In particular activation of this response in worms requires a unique transcription factor termed Activating Transcription Factor associated with Stress-1 (ATFS-1) (Haynes et al., 2010; Nargund et al., 2012). ATFS-1 is unique in the sense that it has both a mitochondrial targeting sequence and a nuclear localization signal. In the absence of mitochondrial stress, ATFS-1 is imported into the mitochondria and rapidly degraded by resident proteases. In some ways, this is analogous to factors such as nuclear p53 or cytosolic Hif-1α, both of which are subjected to proteasomal degradation in the absence of their respective stresses, DNA damage and hypoxia. In response to mitochondrial dysfunction, mitochondrial import of ATFS-1 is reduced causing cytosolic accumulation, followed by nuclear translocation. Once in the nucleus, ATFS-1 promotes a transcriptional response that attempts to restore homeostasis by upregulating a host of mitochondrial chaperones and proteases, as well as genes involved in mitochondrial protein importation and ROS metabolism.
The above experimental paradigms represent situations of relatively severe mitochondrial damage. Evidence suggests that milder forms of mitochondrial stress are also sensed and can also elicit a signaling cascade. In yeast, the ATP-binding cassette (ABC) transporter protein Mdl1 exports peptides across the inner mitochondrial membrane (Young et al., 2001). Proteins degraded by specific proteases within the mitochondrial matrix generate peptides of various sizes (600–2100 kD) that can exit into the cytosol through Mdl1. In worms, the analogous protein HAF-1 appears to be important in mediating the response to mild mitochondrial dysfunction (Haynes et al., 2010). Like Mdl1, HAF-1 is an inner mitochondrial membrane protein that belongs to the ABC transporter family, and, like Mdl1, HAF-1 appears capable of exporting matrix generated peptides into the cytosol. Through yet undefined mechanisms, peptide release from the mitochondria triggers a transcriptional response that involves several proteins including the ubiquitin-like protein UBL-5, the homeobox containing transcription factor DVE-1 and ATFS-1(Haynes et al., 2007; Haynes et al., 2010). While much remains to be learned, the use of released peptides from the mitochondrial matrix to trigger a response is vaguely reminiscent of another biological phenomenon, namely quorum sensing in gram-positive bacteria(Battersby and Richter, 2013). In this situation, oligopeptides are released into the environment through an ABC transporter located on the surface of the bacteria (Henke and Bassler, 2004). Release of these peptides into the extracellular environment triggers the activation of a histidine kinase sensor located on the bacterial surface of other gram-positive bacteria. This histidine kinase is part of a bacterial two-component response system that ultimately regulates gene expression. In this fashion, the behavior and biology of multiple gram positive bacteria can be coordinated and these multiple dispersed genomes can communicate with each other. Given that most cells contain hundreds, if not thousands of mitochondria, and given the endosymbiotic theory of mitochondrial origin, it is intriguing to consider that an analogous system to quorum sensing may be in place in order to allow mitochondria within the cell to communicate with each other and with the nucleus (Figure 2). Interestingly, for bacteria, the peptides released often have unique side-chain modification including isoprenyl groups or thio-lactone rings. Whether similar modifications occur for mitochondrial secreted peptides remains unknown.
Figure 2.

Potential parallels between the mitochondrial unfolded protein response and quorum sensing in gram positive bacteria. In the C. elegans UPRmt response, mitochondrial proteins (indicated by blue swirls) are degraded by matrix proteases and the oligopeptides that are generated are then exported through the ABC transporter family member HAF-1. Once in the cytosol, these peptides can influence the subcellular localization of the transcription factor ATFS-1. Nuclear ATFS-1 is capable of orchestrating a broad transcriptional response to mitochondrial stress. As such, this pathway establishes a method for mitochondrial and nuclear genomes to communicate. In some gram positive bacteria, intracellularly generated peptides can be similarly exported through an ABC transporter protein. These peptides can be detected in the environment by a membrane-bound histidine kinases (HK) sensor. The activation of the HK sensor leads to phosphorylation of a response regulator (RR) protein that, in turn, can alter gene expression. This program allows communication between dispersed gram positive bacteria and thus coordinated behavior of widely dispersed bacterial genomes.
There are a number of tantalizing clues that activation of the UPRmt might provide biologically important short and long term adaptation. In particular, there is a growing body of evidence suggesting that activation of the UPRmt might be an important determinant of lifespan. For instance, using C. elegans as a model, recent work has suggested that NAD+ levels decline as worms age (Mouchiroud et al., 2013). Restoration of NAD+ levels extended longevity through a pathway that required activation of the UPRmt response. Interestingly, this study further demonstrated that overexpression of the NAD-dependent deactylase sir-2.1 in C. elegans also extended lifespan in a UPRmt dependent fashion. In another recent study, strains of inbred mice that differ in their longevity were analyzed to identify potential genes that regulate longevity. This analysis identified the mitochondrial ribosomal protein S5 (Mrps5) as a candidate gene regulating mouse lifespan (Houtkooper et al., 2013). As we will discuss later, mitochondrial ribosomal proteins are involved in the translation of those specific electron transport components that are encoded by mtDNA. In worms, it was subsequently demonstrated that knockdown of Mrps5, as well as related mitochondrial ribosomal proteins, could extend lifespan (Houtkooper et al., 2013). When a transgenic worm was used so that the expression of mitochondrial chaperone proteins could be easily visualized, it could be demonstrated that knockdown of mrps-5 triggered the constitutive activation of the UPRmt response. Further, genetic inhibition of the UPRmt response abrogated the ability of mrps-5 to modulate lifespan. Interestingly, the antibiotic doxycycline, which works therapeutically by blocking bacterial translation but also interferes with mitochondrial translation, could also activate UPRmt and extend the life of worms. These observations may help explain the results of previous unbiased screens for longevity genes in C. elegans. For instance, in one such screen, roughly 15% of the genes identified that extended lifespan were functionally linked to the mitochondria or cellular metabolism (Lee et al., 2003). This frequency of mitochondrial and metabolic ‘hits’ represents an approximate 10-fold enrichment of this functional class of gene products. While these earlier large scale unbiased screens did not directly assess the UPRmt response, it remains possible that any variety of genetic, environmental or pharmacological stresses that impair mitochondrial protein quality could trigger a hormetic response that at least in model organisms, appears to extend lifespan.
Mitokines for stresses near and far
So far we have discussed cell autonomous responses in which perturbation of mitochondrial function caused by altering mtDNA, respiratory function or mitochondrial protein quality, triggered a cytoplasmic and nuclear response within the affected cell. Recent evidence suggests however that cell non-autonomous signals can also be generated by mitochondrial stress. As mentioned above, in various systems including yeast(Kirchman et al., 1999), worms(Feng et al., 2001; Lee et al., 2003), flies (Copeland et al., 2009) and mice(Lapointe et al., 2009; Liu et al., 2005) inhibition of elements of the electron transport chain result in lifespan extension. How does altering mitochondrial function regulate longevity? One potential clue came from a recent study in worms in which the UPRmt response was activated and lifespan extended by knocking down a component of the nuclear-encoded cytochrome c oxidase (cco-1) complex (Durieux et al., 2011). Further analysis revealed that tissue specific knockdown of cco-1 in the brain or intestine was sufficient to increase lifespan. Moreover, and quite remarkably, knockdown of cco-1 in the brain could trigger the activation of the UPRmt response in the intestine. The author postulated that stressed mitochondria in the brain might release soluble factors they termed mitokines that somehow govern both the induction of the UPRmt response in distal tissues, and lifespan of the entire organism. Recent evidence suggests that cell non-autonomous signaling stimulated by proteotoxic stress may actually extend beyond the UPRmt response (van Oosten-Hawle et al., 2013).
Why would stressed mitochondria in the brain of a worm need to communicate this stress to the rest of the soma? At present, there are no definitive answers. One possibility is that mitochondria in the nervous system of C. elegans might be tuned to act as sensors of overall metabolic status. Disruption of energy generation in this tissue would generate a signal that could modulate the energy demand or overall stress defenses of other tissues. In many ways, this would function in a fashion not too dissimilar to the control of body temperature. In that case, the stress of cold temperature activates the neuronal release of catecholamines that in turn, increase mitochondrial activity in tissues such as brown fat in order to generate heat. Certainly, it maybe beneficial that disruption of mitochondrial capacity not only triggers a response within the cell with impaired mitochondria but also signals to the rest of the body that a metabolic crisis is at hand. Perhaps this metabolic danger signal could in turn reduce overall energetic demand by altering or delaying local signals that regulate proliferation or other cell fate decisions that require increased energy consumption.
Although intriguing, it remains uncertain what mitokines are and whether they exist in other species. However, there are some hints to suggest that factors secreted from the mitochondria may indeed have systemic biological effects in higher species. One such candidate is humanin, a peptide of approximately 24 amino acids believed to derive from a short, cryptic open reading frame within the mitochondrial 16S rRNA gene. Originally isolated in the context of a protective factor for Alzheimer’s disease (Hashimoto et al., 2001), it is now clear that humanin circulates in plasma and has in various experimental paradigms cytoprotective effects, as well as affording potentially beneficial metabolic protection (Lee et al., 2013). Humanin was proposed to be a founding member of a class of mitochondrially-derived peptides causing systemic effects in mammals (Lee et al., 2013). Exactly how and where humanin is synthesized within cells remains unclear, but there is a growing appreciation that mitochondrial transcription, once thought to be composed of a few relatively bland polycistronic transcripts, in fact has a surprisingly complex and varied transcriptome (Mercer et al., 2011). Whether other mitochondrial peptides and regulatory noncoding RNAs exist remains to be determined.
Very recently yet another stress generated in the mitochondria has been shown to trigger a coordinated nuclear response. These studies centered on the biological effects of actinonin, an antibiotic that inhibits mitochondrial translation. As noted earlier, mitochondria synthesize 13 polypeptides that function as components of the electron transport chain, as well as two ribosomal RNAs and 22 mitochondria-specific tRNAs. Actinonin specifically blocks the translation of the mitochondrial polypeptides. It does so by inhibiting a unique peptide deformylase that catalyzes the removal of a formyl group from the initial N-terminal methionine. Interestingly, actinonin and related compounds are being developed as a new class of anticancer agents (Lee et al., 2004). Indeed, certain tumors appear uniquely sensitive to inhibition of mitochondrial translation (Skrtic et al., 2011). A recent study demonstrated that treating fibroblasts with actinonin causes stalling of mitochondrial ribosomes (Richter et al., 2013). The stalling triggers subsequent depletion and decay of mitochondrial ribosomal subunits and mitochondrial mRNAs. Furthermore, actinonin induced a block in cell proliferation, which may represent part of a mitochondrial ribosome quality control mechanism. This proliferative block appears to precede any fall in cellular energetics and may involve, at least based on gene expression studies, the activation of the tumor suppressor p53 (Richter et al., 2013).
Thus a number of mitochondrial perturbations - including inhibiting mitochondrial function, incorrect folding of mitochondrial proteins, or stalling of mitochondrial ribosomes - elicit a nuclear response (Figure 3). In addition, mitochondrial number is regulated by the balance between mitochondrial biogenesis and mitophagy. The mechanisms underlying biogenesis have been reviewed elsewhere (Scarpulla et al., 2012) with a number of studies demonstrating a central role for the PGC-1 family of coactivators. The activity of PGC-1α is modulated by energy availability as sensed by factors including AMPK and Sirt1 that modify PGC-1α by either phosphorylation or acetylation to regulate the coactivator’s activity (Jeninga et al., 2010; Nemoto et al., 2005; Rodgers et al., 2005). Therefore, mitochondrial biogenesis triggered by energetic stress represents another hormetic response, especially since besides increasing mitochondrial number, PGC-1α can also induce a potent antioxidant stress resistance program (St-Pierre et al., 2006). Interestingly, starvation and energetic stress can also result in the biogenesis of new lysosomes. This response appears to be orchestrated not by PGC-1α, but rather by transcription factor EB (TFEB). Outside their biogenesis, mitochondria and lysosomes share the property that both organelles can trigger cell death when their outer membranes become permeable. For mitochondria, this occurs through the classical mitochondrial outer membrane permeabilization (MOMP) pathway, leading to caspase activation, whereas for lysosomes, permeabilization leads to the release of various cathepsins and other enzymes that can induce cell death through various means (Boya and Kroemer, 2008). As discussed above, mitochondria also share some similarity with the endoplasmic reticulum as the UPRmt response and the classic ER stress response share numerous features. As such, it is likely that each organelle within the cell is capable of perceiving stress signals and communicating stress to the nucleus. Furthermore, for each organelle, unresolved stress can be a potent inducer of cell death.
Figure 3.

The complexity of mitochondrial stresses and responses. A wide array of extrinsic and intrinsic mitochondrial perturbations can elicit cellular responses. As detailed in the text, genetic or pharmacological disruption of electron transport, incorrect folding of mitochondrial proteins, stalled mitochondrial ribosomes, alterations in signaling pathways or exposure to toxins, all appear to elicit specific cytoprotective programs within the cell. These adaptive responses include increased mitochondrial number (biogenesis), alterations in metabolism, increased antioxidant defenses and augmented protein chaperone expression. The cumulative effect of these adaptive mechanisms might be an extension of lifespan and a decreased incidence of age-related pathologies.
Mitochondria as internal sensors of external threats
The above discussion illustrates stresses generated within the mitochondria that can elicit a cellular response. In many cases, this cellular response involves upregulation of a set of genes that alters cellular metabolism and intrinsic stress resistance. However, mitochondria also play a key role in orchestrating the response to perceived external threats. One well studied example involves the mitochondrial antiviral signaling (MAVS) complex. The MAVS protein is an outer mitochondrial membrane protein that serves as an adaptor and platform orchestrating a response to viral infection(Seth et al., 2005). In particular, MAVS can bind to several members of retinoic-acid-inducible protein I-like receptor (RLR) family of pattern recognition receptors. The RLR family in turn binds to unique RNA structures that are generated during viral infection. Once the RLR family engages dsRNA it is believed to trigger a conformational change in the RLR that enables subsequent interaction with MAVS and the mitochondria. This interaction activates MAVS, and in a prion-type fashion, leads to a self-propagating signal (Hou et al., 2011). The MAVS self-assembled polymer can subsequently recruit additional signaling components including TNF-associated factors (TRAF2 and TRAF6) and RIPK1 to activate an innate immune response(Hou et al., 2011; Seth et al., 2005). Another example in which mitochondria are activated by potential external threats involves activation of the inflammasome. The inflammasome is a large protein complex whose assembly is triggered by a wide range of stimuli including uric acid, potentially released from dying cells or lipopolysaccharide (LPS) stimulation that mimics bacterial infection (Lamkanfi and Dixit, 2012). Activation of the inflammasome results in the enzymatic activation of caspase-1, and the subsequent proteolytic cleavage of interleukin 1β. Mitochondrial ROS plays an important role in inflammasome activation, although the precise target remains unidentified (Bauernfeind et al., 2011; Zhou et al., 2011). Indeed, there are a growing number of examples where the generation of mitochondrial ROS appears to be a tightly regulated process that is essential for tuning the magnitude or effectiveness of the immune response (Bulua et al., 2011; West et al., 2011).
Mitochondrial Oxidants
One of the best described mechanisms for mitochondrial communication with its cellular host is through the release of ROS. We have already discussed several examples in which mitochondrial ROS act as downstream effector molecules. Indeed, oxidants in general (Finkel, 2011), and mitochondrial oxidants in particular (Finkel, 2012), can function in numerous signaling pathways. These include diverse processes such as the regulation of cytosolic stress kinases (Kamata et al., 2005), modulation of hypoxic signaling (Chandel et al., 1998), and activation of macroautophagy (Scherz-Shouval et al., 2007). Several recent examples suggest that mitochondrial oxidants are active participants in mitohormesis. One of the first clear examples came from studies in which glucose metabolism was impaired either pharmacologically by exposing worms to 2-deoxy-D-glucose (2DG), or by simply restricting glucose availability(Schulz et al., 2007). Both maneuvers resulted in an extension of lifespan. A more detailed examination of metabolism in these animals demonstrated that restricting glucose availability resulted in a presumed compensatory increase in mitochondrial respiration, with evidence for increased utilization of fat through β-oxidation. These metabolic changes appeared to require activation of aak-2, the C. elegans homolog of the well characterized energy sensor AMP-dependent kinase (AMPK). Consistent with the observed increase in mitochondrial respiration, treatment of worms with 2DG resulted in an increase in ROS levels. Following this oxidative stress, the level of the hydrogen peroxide scavenging enzyme catalase was elevated approximately one week after 2DG exposure. Worms given 2DG but pre-treated with the antioxidant N-acetylcysteine (NAC) showed no evidence of a rise in ROS levels or the subsequent induction of catalase expression. Remarkably, antioxidant treatment also blocked the extension of lifespan by 2DG treatment. These observations support the notion that a shift away from glucose utilization resulted in a transient state of increased mitochondrial metabolism and subsequent oxidative stress. Mitochondrial oxidants in turn induced a hormetic response that ultimately resulted in increased resistance to oxidative stress and increased overall longevity. Others have come to similar conclusions, although it remains to be determined if the relevant mitochondrial oxidant species is hydrogen peroxide or superoxide anion (Yang and Hekimi, 2010). Recent studies have also implicated the phenomenon of mitohormesis in at least part of the life span increase seen in the classic long-lived C. elegans mutant daf-2, which has alterations in the insulin/IGF-1 pathway (Zarse et al., 2012). In another example, long-lived mutants induced through inhibition of mitochondrial respiration were found to increase the activity of the hypoxia-inducible factor HIF-1 (Lee et al., 2010). In this setting, the activation of HIF-1 was seemingly dependent on the release of mitochondrial oxidants. A similar hormetic response involving mitochondrial ROS dependent activation of HIF-1 was shown to be important in rescuing AMPK-null mutant of C. elegans (Xie and Roy, 2012). Finally, a recent study has demonstrated that a redox-dependent, mitohormetic response can also regulate the lifespan of Drosophila (Owusu-Ansah et al., 2013).
From a strictly metabolic viewpoint these studies appear contradictory as they would suggest that increased mitochondrial respiration (e.g. 2DG) or decrease mitochondrial respiration (e.g. respiratory chain mutants) can both correlate with an increase in an organism’s lifespan. Perhaps the only unifying factor is that both conditions appear to result in an inappropriate release of ROS, thus triggering a hormetic response. Another potential source of confusion is that, as previously discussed, disruption of mitochondrial function was thought to induce lifespan extension through the ROS-independent activation of the UPRmt. Nonetheless, some recent evidence suggests that stimuli that activate the previously describe ATFS-1 pathway, also activate a parallel ROS-dependent signaling network (Baker et al., 2012). It is also important to stress that release of mitochondrial oxidants is likely important in the regulation of multiple pathway. One system that is clearly activated by oxidative stress and participates in the subsequent hormetic induction of increased stress resistance involves the nuclear factor erythroid 2-related factor (Nrf2). This leucine zipper transcription factor is capable of regulating the expression of a host of gene products that help mediate resistance to oxidative stress (Ma, 2013). Furthermore, the worm homolog SKN-1 directly binds mitochondria (Paek et al., 2012), and is required to mediate the longevity benefits associated with mitochondrial oxidant release (Schmeisser et al., 2013b; Zarse et al., 2012).
Although the preceding examples have come largely from studies in C. elegans, other recent elegant studies in yeast have come to similar conclusions regarding the potential hormetic role for mROS. For instance, in yeast, a reduction in target of rapamycin (TOR) signaling results in an extension of chronological lifespan. A careful analysis of such long-lived yeast strains with impaired TOR signaling revealed that reducing TOR signaling led to initial increase in mitochondrial ROS production(Pan et al., 2011). Again, rather than being harmful, this mROS production was required for the increased lifespan observed. Remarkably, in this setting, expression of the antioxidant protein manganese superoxide dismutase reduced lifespan while treatment with the redox-cycling compound menadione extended lifespan. A further dissection of the role of mROS in yeast longevity demonstrated that mROS are sensed by the two kinases, Tel1p and Rad53p (Schroeder et al., 2013). These proteins are known to sense DNA damage and are the yeast homologs of the well known mammalian proteins, ATM and Chk2, respectively. Activation of this pathway results in alterations in epigenetic silencing through a mechanism involving the sirtuin family of deacetylases. Interestingly, recent work in C. elegans suggest that sirtuin overexpression in that organism may extend lifespan through a mechanism involving mitohormesis(Schmeisser et al., 2013a).
Mitohormesis in human biology
What are the implications of mitohormesis for human health? Clearly, in model organisms, the evidence is consistent with the idea that a variety of mild stresses can protect the organism from subsequent larger stresses, and thus translate to a longer lifespan. In this context, antioxidants appear to have a negative effect presumably by preventing the hormetic response. Indeed, in C. elegans, there is significant evidence that an augmented cytoprotective response, be it the induction of chaperones, xenobiotic detoxification or antioxidant defenses, is tightly coupled and required for most, if not all, lifespan extensions (Shore et al., 2012). These observations may provide an explanation for the thoroughly disappointing clinical experience with antioxidant therapies. Numerous randomized studies have, in general, failed to demonstrate a benefit from antioxidant therapy and large meta-analysis studies suggests that in some cases, certain antioxidants may actually increase mortality(Bjelakovic et al., 2007; Lippman et al., 2009; Lonn et al., 2005). There are a number of possible explanations for why antioxidants have in general been ineffective including issues of improper dosing or insufficient localization to the mitochondrial source of ROS. Similarly, it is possible that the hint that antioxidants might increase cancer incidence may be a result of the ability of antioxidants to protect genetically damaged pre-cancerous cells from undergoing apoptosis. Nonetheless, it is also possible that chronic low dose antioxidants inhibit the normal hormetic response and therefore block the induction of a broad array of cytoprotective measures the organism would normally undertake. This is supported by some human experimentation in which physical exercise is viewed as a stress and where the salutary benefits of exercise appeared to be inhibited in those subjects given antioxidant supplements(Gomez-Cabrera et al., 2008; Ristow et al., 2009). In these experiments, whereas exercise by itself increased the levels of antioxidant proteins such as superoxide dismutase and glutathione peroxidase in the patient’s skeletal muscle, this response was not observed in individuals taking a combination of vitamin C and vitamin E (Ristow et al., 2009). Again, returning to the large-scale human trials, Vitamin E appears to be associated with an increased risk of developing heart failure in patients with vascular disease or diabetes (Lonn et al., 2005). Given the evidence presented earlier, is it possible that the ischemic myocardium is generating ROS in a fashion similar to cells exposed to hypoxia (Chandel et al., 1998)and that these beneficial hormetic signals are actually inhibited by Vitamin E?
It may also be interesting to assess how mitohormesis regulates disease susceptibility. For instance, nutritional overload contributes to obesity, and obesity contributes to susceptibility for diabetes. Yet, not all obese individuals develop clinically apparent disease. Increased nutrient supply presumably results in increased mitochondrial respiration and increased mitochondrial ROS production. Indeed, ROS production has been tightly linked mechanistically to diabetes (Houstis et al., 2006). Some have further suggested that individual variation in the mitohormesis response might explain why it is that not all obese individual developed clinically apparent disease (Kolb and Eizirik, 2012). A similar line of reasoning could be applied to a host of diseases where patients have multiple risk factors for the disease but no evidence of the condition. Interestingly, certain naturally occurring plant compounds that appear to have medicinal value appear to induce a hormetic response when ingested. Since these compounds are often synthesized under conditions in which the plants are undergoing some form of environmental stress, this has led to the hypothesis known as xenohormesis (Howitz and Sinclair, 2008). In this scenario, these naturally synthesized, plant small molecules represent a form of cross-species signaling. Their production is stimulated when the plant is stressed, as might happen during a prolonged drought. In turn, when consumed they stimulate a hormetic response in the recipient animals, thereby helping to prepare the animal for the presumed worsening of environmental conditions (e.g. drought for the plant, famine for the animal).
Could other effective man-made drugs represent examples of such a strategy? One potential candidate might be the widely used anti-diabetic drug metformin that is thought to work by inhibiting mitochondrial function (Owen et al., 2000). Could the known beneficial effects of this agent for both diabetes and cancer prevention (Franciosi et al., 2013)be through a hormetic mechanism? Although this mechanism to our knowledge has not been suggested, evidence in both lower organisms and mice suggest that treatment with metformin induces an increase in oxidant defenses as well as an extension of lifespan (Martin-Montalvo et al., 2013; Onken and Driscoll, 2010). Does this agent therefore represent the first example of exploiting hormesis to broadly treat a range of age-related diseases and even aging itself? Will other molecules emerge that gently perturb mitochondrial function and generate a mitohormetic signal that results in abroad therapeutic benefit? Will these molecules represent an entirely new approach for how we treat various diseases and as such, are we now on the verge of a new era of therapy and understanding? Or, perhaps, after 2000 years, we have simply re-discovered the ancient secret of King Mithridates VI.
Acknowledgments
We are grateful for the assistance of I.I. Rovira. This work was supported NIH Intramural Funds.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, Gillespie MN. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal. 2012;5:ra47. doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BM, Nargund AM, Sun T, Haynes CM. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet. 2012;8:e1002760. doi: 10.1371/journal.pgen.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battersby BJ, Richter U. Why translation counts for mitochondria - retrograde signalling links mitochondrial protein synthesis to mitochondrial biogenesis and cell proliferation. J Cell Sci. 2013;126:4331–4338. doi: 10.1242/jcs.131888. [DOI] [PubMed] [Google Scholar]
- Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. 2011;187:613–617. doi: 10.4049/jimmunol.1100613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas G, Adebanjo OA, Freedman BD, Anandatheerthavarada HK, Vijayasarathy C, Zaidi M, Kotlikoff M, Avadhani NG. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J. 1999;18:522–533. doi: 10.1093/emboj/18.3.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas G, Anandatheerthavarada HK, Zaidi M, Avadhani NG. Mitochondria to nucleus stress signaling: a distinctive mechanism of NFkappaB/Rel activation through calcineurin-mediated inactivation of IkappaBbeta. J Cell Biol. 2003;161:507–519. doi: 10.1083/jcb.200211104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. 2007;297:842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- Borghouts C, Benguria A, Wawryn J, Jazwinski SM. Rtg2 protein links metabolism and genome stability in yeast longevity. Genetics. 2004;166:765–777. doi: 10.1534/genetics.166.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–6451. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, Sack MN, Kastner DL, Siegel RM. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS) J Exp Med. 2011;208:519–533. doi: 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese EJ, Baldwin LA. Defining hormesis. Hum Exp Toxicol. 2002;21:91–97. doi: 10.1191/0960327102ht217oa. [DOI] [PubMed] [Google Scholar]
- Chae S, Ahn BY, Byun K, Cho YM, Yu MH, Lee B, Hwang D, Park KS. A systems approach for decoding mitochondrial retrograde signaling pathways. Sci Signal. 2013;6:rs4. doi: 10.1126/scisignal.2003266. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland JM, Cho J, Lo T, Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol. 2009;19:1591–1598. doi: 10.1016/j.cub.2009.08.016. [DOI] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolle C, Rack JG, Ziegler M. NAD and ADP-ribose metabolism in mitochondria. FEBS J. 2013;280:3530–3541. doi: 10.1111/febs.12304. [DOI] [PubMed] [Google Scholar]
- Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein CB, Waddle JA, Hale Wt, Dave V, Thornton J, Macatee TL, Garner HR, Butow RA. Genome-wide responses to mitochondrial dysfunction. Mol Biol Cell. 2001;12:297–308. doi: 10.1091/mbc.12.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Bussiere F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell. 2001;1:633–644. doi: 10.1016/s1534-5807(01)00071-5. [DOI] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287:4434–4440. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franciosi M, Lucisano G, Lapice E, Strippoli GF, Pellegrini F, Nicolucci A. Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PLoS One. 2013;8:e71583. doi: 10.1371/journal.pone.0071583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Cabrera MC, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo FV, Sastre J, Vina J. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am J Clin Nutr. 2008;87:142–149. doi: 10.1093/ajcn/87.1.142. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta. Proc Natl Acad Sci U S A. 2001;98:6336–6341. doi: 10.1073/pnas.101133498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13:467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heddi A, Stepien G, Benke PJ, Wallace DC. Coordinate induction of energy gene expression in tissues of mitochondrial disease patients. J Biol Chem. 1999;274:22968–22976. doi: 10.1074/jbc.274.33.22968. [DOI] [PubMed] [Google Scholar]
- Henke JM, Bassler BL. Bacterial social engagements. Trends Cell Biol. 2004;14:648–656. doi: 10.1016/j.tcb.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Auwerx J. Exploring the therapeutic space around NAD+ J Cell Biol. 2012;199:205–209. doi: 10.1083/jcb.201207019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howitz KT, Sinclair DA. Xenohormesis: sensing the chemical cues of other species. Cell. 2008;133:387–391. doi: 10.1016/j.cell.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazwinski SM, Kriete A. The yeast retrograde response as a model of intracellular signaling of mitochondrial dysfunction. Front Physiol. 2012;3:139. doi: 10.3389/fphys.2012.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeninga EH, Schoonjans K, Auwerx J. Reversible acetylation of PGC-1: connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene. 2010;29:4617–4624. doi: 10.1038/onc.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- Kirchman PA, Kim S, Lai CY, Jazwinski SM. Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics. 1999;152:179–190. doi: 10.1093/genetics/152.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H, Eizirik DL. Resistance to type 2 diabetes mellitus: a matter of hormesis? Nat Rev Endocrinol. 2012;8:183–192. doi: 10.1038/nrendo.2011.158. [DOI] [PubMed] [Google Scholar]
- Komeili A, Wedaman KP, O’Shea EK, Powers T. Mechanism of metabolic control. Target of rapamycin signaling links nitrogen quality to the activity of the Rtg1 and Rtg3 transcription factors. J Cell Biol. 2000;151:863–878. doi: 10.1083/jcb.151.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–161. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- Lapointe J, Stepanyan Z, Bigras E, Hekimi S. Reversal of the mitochondrial phenotype and slow development of oxidative biomarkers of aging in long-lived Mclk1+/− mice. J Biol Chem. 2009;284:20364–20374. doi: 10.1074/jbc.M109.006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Yen K, Cohen P. Humanin: a harbinger of mitochondrial-derived peptides? Trends Endocrinol Metab. 2013;24:222–228. doi: 10.1016/j.tem.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MD, She Y, Soskis MJ, Borella CP, Gardner JR, Hayes PA, Dy BM, Heaney ML, Philips MR, Bornmann WG, et al. Human mitochondrial peptide deformylase, a new anticancer target of actinonin-based antibiotics. J Clin Invest. 2004;114:1107–1116. doi: 10.1172/JCI22269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20:2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, Parnes HL, Minasian LM, Gaziano JM, Hartline JA, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2009;301:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005;19:2424–2434. doi: 10.1101/gad.1352905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Sekito T, Epstein CB, Butow RA. RTG-dependent mitochondria to nucleus signaling is negatively regulated by the seven WD-repeat protein Lst8p. EMBO J. 2001;20:7209–7219. doi: 10.1093/emboj/20.24.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonn E, Bosch J, Yusuf S, Sheridan P, Pogue J, Arnold JM, Ross C, Arnold A, Sleight P, Probstfield J, et al. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA. 2005;293:1338–1347. doi: 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood AM, Haugen E, Bracken CP, Rackham O, Stamatoyannopoulos JA, et al. The human mitochondrial transcriptome. Cell. 2011;146:645–658. doi: 10.1016/j.cell.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- Onken B, Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS One. 2010;5:e8758. doi: 10.1371/journal.pone.0008758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–614. [PMC free article] [PubMed] [Google Scholar]
- Owusu-Ansah E, Song W, Perrimon N. Muscle Mitohormesis Promotes Longevity via Systemic Repression of Insulin Signaling. Cell. 2013;155:699–712. doi: 10.1016/j.cell.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paek J, Lo JY, Narasimhan SD, Nguyen TN, Glover-Cutter K, Robida-Stubbs S, Suzuki T, Yamamoto M, Blackwell TK, Curran SP. Mitochondrial SKN-1/Nrf mediates a conserved starvation response. Cell Metab. 2012;16:526–537. doi: 10.1016/j.cmet.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Schroeder EA, Ocampo A, Barrientos A, Shadel GS. Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab. 2011;13:668–678. doi: 10.1016/j.cmet.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh VS, Morgan MM, Scott R, Clements LS, Butow RA. The mitochondrial genotype can influence nuclear gene expression in yeast. Science. 1987;235:576–580. doi: 10.1126/science.3027892. [DOI] [PubMed] [Google Scholar]
- Picard M, Shirihai OS, Gentil BJ, Burelle Y. Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am J Physiol Regul Integr Comp Physiol. 2013;304:R393–406. doi: 10.1152/ajpregu.00584.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter U, Lahtinen T, Marttinen P, Myohanen M, Greco D, Cannino G, Jacobs HT, Lietzen N, Nyman TA, Battersby BJ. A mitochondrial ribosomal and RNA decay pathway blocks cell proliferation. Curr Biol. 2013;23:535–541. doi: 10.1016/j.cub.2013.02.019. [DOI] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Bluher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Rothermel BA, Shyjan AW, Etheredge JL, Butow RA. Transactivation by Rtg1p, a basic helix-loop-helix protein that functions in communication between mitochondria and the nucleus in yeast. J Biol Chem. 1995;270:29476–29482. doi: 10.1074/jbc.270.49.29476. [DOI] [PubMed] [Google Scholar]
- Rothermel BA, Thornton JL, Butow RA. Rtg3p, a basic helix-loop-helix/leucine zipper protein that functions in mitochondrial-induced changes in gene expression, contains independent activation domains. J Biol Chem. 1997;272:19801–19807. doi: 10.1074/jbc.272.32.19801. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. 2012;23:459–466. doi: 10.1016/j.tem.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser K, Mansfeld J, Kuhlow D, Weimer S, Priebe S, Heiland I, Birringer M, Groth M, Segref A, Kanfi Y, et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat Chem Biol. 2013a;9:693–700. doi: 10.1038/nchembio.1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser S, Schmeisser K, Weimer S, Groth M, Priebe S, Fazius E, Kuhlow D, Pick D, Einax JW, Guthke R, et al. Mitochondrial hormesis links low-dose arsenite exposure to lifespan extension. Aging Cell. 2013b;12:508–517. doi: 10.1111/acel.12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder EA, Raimundo N, Shadel GS. Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab. 2013;17:954–964. doi: 10.1016/j.cmet.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Sekito T, Thornton J, Butow RA. Mitochondria-to-nuclear signaling is regulated by the subcellular localization of the transcription factors Rtg1p and Rtg3p. Mol Biol Cell. 2000;11:2103–2115. doi: 10.1091/mbc.11.6.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Shore DE, Carr CE, Ruvkun G. Induction of cytoprotective pathways is central to the extension of lifespan conferred by multiple longevity pathways. PLoS Genet. 2012;8:e1002792. doi: 10.1371/journal.pgen.1002792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20:674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southam CM, Ehrlich J. Effects of extract of western red-cedar heartwood on certain wood-decaying fungi in culture. Phytopathology. 1943;33:517–524. [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- van Oosten-Hawle P, Porter RS, Morimoto RI. Regulation of organismal proteostasis by transcellular chaperone signaling. Cell. 2013;153:1366–1378. doi: 10.1016/j.cell.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Roy R. Increased levels of hydrogen peroxide induce a HIF-1-dependent modification of lipid metabolism in AMPK compromised C. elegans dauer larvae. Cell Metab. 2012;16:322–335. doi: 10.1016/j.cmet.2012.07.016. [DOI] [PubMed] [Google Scholar]
- Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;117:4055–4066. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- Young L, Leonhard K, Tatsuta T, Trowsdale J, Langer T. Role of the ABC transporter Mdl1 in peptide export from mitochondria. Science. 2001;291:2135–2138. doi: 10.1126/science.1056957. [DOI] [PubMed] [Google Scholar]
- Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, Guthke R, Platzer M, Kahn CR, Ristow M. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012;15:451–465. doi: 10.1016/j.cmet.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]