

Abstract
Hyperventilation, and reduced cerebral blood flow velocity can occur in postural tachycardia syndrome (POTS). We studied orthostatically intolerant patients, with suspected POTS, with a chief complaint of upright dyspnea. Based on our observations of an immediate reduction of cerebral blood flow velocity with orthostasis, we hypothesize that the resulting ischemic hypoxia of the carotid body causes chemoreflex activation, hypocapnic hyperpnea, sympathetic activation, and increased heart rate and blood pressure in this subset of POTS. We compared 11 dyspneic POTS subjects to 10 healthy controls during a 70° head-up tilt. In POTS subjects during initial orthostasis prior to BP recovery; central blood volume and mean arterial pressure were reduced (P<0.025) resulting in a significant (P<0.001) decrease in cerebral blood flow velocity, which temporally preceded (17±6 s; P<0.025) a progressive increase in minute ventilation and decrease in end tidal CO2 (P<0.05), compared to controls. Sympathoexcitation, measured by muscle sympathetic nerve activity, was increased in POTS, (P<0.01), and inversely proportional to end tidal CO2 and resulted in an increase in heart rate, (P<0.001), total peripheral resistance (P<0.025), and a decrease in cardiac output (P<0.025). The decrease in cerebral blood flow velocity and mean arterial pressure during initial orthostasis was greater (P<0.025) in POTS. Our data suggest that exaggerated initial central hypovolemia during initial orthostatic hypotension in POTS results in reduced cerebral blood flow velocity and postural hypocapnic hyperpnea that perpetuates cerebral ischemia. We hypothesize that sustained hypocapnia and cerebral ischemia produce sympathoexcitation, tachycardia and a statistically significant increase in blood pressure.
Keywords: Orthostatic Intolerance, Autonomic nervous system, Muscle Sympathetic Nerve Activity
Introduction
Orthostatic intolerance (OI), the inability to tolerate upright posture, relieved by recumbancy1, can have lightheadedness, cognitive deficits, abnormal blood pressure (BP), and heart rate (HR) regulation2, 3. Some with OI complain of dyspnea associated with hyperventilation and hypocapnia4-6. Thus, neurogenic orthostatic hypotension and vasovagal syncope are frequently associated with hyperventilation. Additionally, induced hypotension of healthy controls results in hyperventilation and hypocapnia7, 8. Hypotension also evokes hyperventilation via the ventilatory baroreflex9.
Hyperventilation and hypocapnia occur in postural tachycardia syndrome (POTS)10, defined as OI with excessive upright tachycardia without hypotension11. POTS patients are often categorized as neuropathic or hyperadrenergic. Neuropathic POTS has regional adrenergic denervation resulting in a redistributive central hypovolemia and associated reflex tachycardia12. In hyperadrenergic POTS widespread sympathoexcitation drives the tachycardia13.
We associate hyperventilation with neuropathic POTS3, 14, especially in those with splanchnic pooling following orthostasis14. We previously attributed hyperventilation to splanchnic hyperemia resulting in thoracic hypovolemia, baroreflex unloading and consequent sympathoexcitation and activation of respiratory centers3. In addition, hyperventilation is facilitated by enhanced hypoxic and reduced hypercapnic ventilatory responses in POTS due to altered peripheral and central chemoreceptor sensitivity6. Hyperventilation produces large reductions in cerebral blood flow (CBF)15 and neuronal ischemia.
We evaluated POTS patients with the chief complaint of shortness of breath and large reductions of cerebral blood flow velocity (CBFv) followed by hyperventilation upon change from supine to upright. Based on this response, we hypothesize a paradigm for hyperventilation sequentially comprising a large immediate reduction in CBFv upon orthostasis, ischemic [stagnant] hypoxia- driven carotid chemoreflex activation, followed by hypocapnic hyperpnea, sympathetic activation, increased BP and tachycardia. This is a CBF-driven form of POTS. To test this hypothesis we compared POTS subjects with immediate postural hyperpnea to “healthy” subjects during orthostatic challenge.
Methods
Subjects
Twenty-one subjects (11 POTS; 8 female, 3 male: 10 healthy control subjects; 7 female, 3 male) were tested supine and during 70° upright tilt (HUT). All were referred for suspected POTS with OI lasting longer than six months; comprising excessive tachycardia, lightheadedness, exercise intolerance, headache, fatigue, neurocognitive deficits, palpitations, nausea or abdominal pain, blurred or altered vision, shortness of breath, or sensation of heat while upright, with no other medical explanation for the symptoms. We confirmed POTS by HUT which required symptoms of OI with an increase in HR exceeding 35 beats per minute (bpm) in patients >21 years old, or exceeding 40 bpm in patients <21 years old during the first 10 minutes of HUT16, 17. POTS subjects were only enrolled if they had a chief complaint of shortness of breath. All had normal pulmonary function tests and no cardiopulmonary or systemic illness. Thus, these patients represent a subgroup, as not all POTS patients present with the chief complaint of shortness of breath.
All subjects were non-smokers and normotensive. Healthy control subjects had no previously known medical conditions, no OI, and were free of systemic illness, with normal physical exam, electrocardiogram, and echocardiogram. Subjects were not taking any medications, or ceased their medications for a minimum of two weeks before study. All stopped food and liquid intake for 12 hrs before study, abstained from caffeine and/or xanthine containing products, and did not exercise for 24 hr before testing. All protocols were approved by the New York Medical College IRB, and conformed to the Declaration of Helsinki. All participants signed an informed consent.
Protocol
Testing began at 10 AM using an electronic motorized tilt table (Colin Medical Instruments Corp., San Antonio, TX). Measurements were made, supine, after a 30 min resting period using methods as described.
Participants were instrumented for electrocardiography, respiratory plethysmography using a RespiTrace device (NIMS, Inc., North Bay Village, FL) calibrated against pneumotachography (Hans Rudolph Inc., Shawnee, KS) via facemask, SaO2 by pulse oxymetry and combined capnography (Smith Medical PM Inc., Waukesha, WI) to measure end tidal carbon dioxide (ETCO2). Transcranial Doppler (TCD) (Neurovision; Multigon, Yonkers, NY) measured CBFv of the left middle cerebral artery (MCA) using a 2 MHz probe.
Occlusion cuffs were placed around the mid-thigh above a mercury in silastic strain gauge (D. E. Hokanson, Inc., Bellevue, WA) placed at mid-calf to measure supine calf blood flow in response to venous occlusion plethysmography (VOP). Impedance plethysmography (IPG) detected internal volume shifts during orthostatic stress and Tetrapolar High Resolution Impedance Monitor (THRIM) digital impedance plethysmography (model 2994D, UFI, Inc., Morro Bay, CA)18 to estimate the changes in thoracic and splanchnic blood volume (BV).
Continuous electrocardiography measured HR (Finapress Medical Systems, Amsterdam, Netherlands). Beat-to-beat BP was recorded using finger photo-plethysmography (Finapress Medical Systems, Amsterdam, Netherlands) calibrated every 5 min by automated brachial sphygmomanometer (Colin Medical Instruments Corp., San Antonio TX).
Muscle sympathetic nerve activity (MSNA) was recorded using a tungsten microelectrode (FHC, Corp. Bowdoin, ME) inserted into the common peroneal nerve near the leg’s fibular head as previously reported19.
All measurements were sampled at 200 Hz. All recordings were obtained and analyzed using custom data acquisition software. All data were analyzed by the same trained scientist.
After 30 min of supine acclimatization, there was an additional 30 min rest period where ETCO2 was obtained by capnography. Mean ETCO2 over the last 5 min of the rest period was defined as baseline isocapnia for the particular subject throughout the protocol. Beat-to-beat BP, TCD, HR, impedance measurements, respirations and MSNA were recorded to obtain ten min baseline data. Following baseline measurements all participants had HUT to 70° for 10 min while measurements continued.
Data Analysis
Baseline data was recorded for 10 min prior to HUT. The first minute of active standing or HUT often includes a period of hemodynamic instability known as “initial orthostatic hypotension” (IOH) characterized by translocation of blood from cephalic towards the caudal portion of the body causing a fall in central BV and BP and concomitant rise in HR attributed to a lag in compensatory adrenergic vasoconstriction20, 21. Often the first minute of tilting is not used for analysis. However, we retained all of these physiological measurements because early non-equilibrium changes in hemodynamics are critical to understanding the initial hyperventilatory response. Thus, we tabulated the lowest value for the decreased BP, CO, central BV and the highest value for the increased HR and TPR during the initial tilt. Thereafter upright measurements were divided into four additional stages 1-2, 2-4, 4-6, and 8-10 min from the onset of HUT. Data was time averaged during these time periods.
Beat-to-beat changes in systolic (SAP), diastolic (DAP), and mean arterial (MAP) pressures and relative changes in CO were determined from Finometer data using ModelFlow® software22. Total peripheral resistance (TPR) was obtained by dividing MAP by CO and HR derived from the electrocardiogram. MSNA was compared to DAP after correcting for a 1.3 s lag time from a triggering R-wave. Bursts of MSNA activity were utilized if they had a > 3:1 burst to noise ratio. Burst frequency (bursts per minute) and total MSNA (bursts per minute × area underneath the bursts) were obtained. Total activity was normalized to the largest single burst occurring during the baseline period assigned a value of 1000. We successfully recorded MSNA in 6/11 POTS and 6/10 controls supine and during all time stages.
Normalized respiratory impedance data was used to calculate respiratory rate, and estimate tidal volume and expiratory minute volume (VE). Data were detrended to remove artifact and tidal volume was obtained as peak to trough volume per breath. VE was determined by averaging tidal volume over one min and multiplying by respiratory rate (RR). Changes in electrical impedance were measured continuously from baseline, throughout, and following HUT.
Statistics
SPSS 16 (SPSS Inc. Chicago, IL) was used for statistical calculations. SBP, DBP, MAP, HR, CO, TPR, ETCO2, RR, VE, CBFv, and MSNA were analyzed using a repeated measure ANOVA. Bonferoni post-hoc analysis was performed when findings were significant. Because of inter-subject variability in baseline arterial pressures, CBFv, MSNA, and impedance measurements of regional volume changes, we also expressed repeated measure data as percent change from baseline. Changes in impedance and estimated compartmental variance were analyzed using a two-tailed Student’s t-test. All measures were reported as mean ± SEM. Statistical significance was set a priori at P < 0.05 for all tests.
Results
POTS subjects mean age was 19 ± 3 years and controls 23 ± 3 years. POTS subjects’ height was 169 ± 7 cm while controls’ height was 169 ± 5 cm and POTS subjects’ weight was 65 ± 12 kg while controls’ was 71 ± 9 kg; thus body mass indices were the same with 22.9 ± 4.4 kg · m-2 for POTS and 24.4 ± 2.8 kg · m-2 for controls. Of the 11 POTS subjects, 9 were able to complete 10 min of HUT; 2 POTS subjects were unable due to orthostatic precipitation of pre-syncope. All 10 control subjects withstood the complete duration of the HUT.
Supine Hemodynamics
Table 1 shows supine HR was significantly increased for POTS subjects compared to the controls (P < 0.01). While systolic BP (SBP) in POTS was lower than in controls, the difference was not significant. None of the other parameters shown were different, comparing POTS to control.
Table 1.
Supine Hemodynamic and Neurocardiopulmonary Data (Mean ± SEM.)
| Measurements | POTS | Control |
|---|---|---|
| Heart Rate (bpm) | 85 ± 6 * | 60 ± 2 |
| Systolic BP (mmHg) | 114 ± 5 | 122 ± 4 |
| Diastolic BP (mmHg) | 58 ± 3 | 60 ± 3 |
| MAP (mmHg) | 76 ± 12 | 81 ± 11 |
| Pulse Pressure (mmHg) | 56 ± 3 | 61 ± 3 |
| Calf blood Flow (ml/100 ml/min) | 2.7 ± 0.2 | 2.6 ± 0.2 |
| Respiratory Rate (breaths · min-1) | 13 ± 1 | 13 ± 1 |
| VE (L · min-1) | 7.3 ± 0.9 | 6.6 ± 0.8 |
| ETCO2 (Torr) | 41.4 | 43.6 ± 0.8 |
| Cardiac Output (L · min-1) | 5.1 ± 0.4 | 5.5 ± 0.3 |
| TPR (mmHg · L-1 · min-1) | 15.2 ± 0.8 | 14.2 ± 1.0 |
| Mean CBFv (cm · s-1) | 78 ± 4 | 73 ± 4 |
| CBF Conductance Index (CBFv / MAP) | 1.07 ± 0.10 | 0.96 ± 0.05 |
| MSNA Burst Area (AU) | 2055 ± 802 | 1894 ± 242 |
| MSNA Burst Count (Burst · min-1) | 17 ± 4 | 15 ± 2 |
= Different from control, P < 0.05.
bpm = Beats per minute. BP = Blood Pressure. CBFv = Cerebral Blood Flow Velocity. mm Hg = millimeters of mercury. MAP = Mean Arterial Pressure. VE = Minute ventilation. ETCO2 = End tidal carbon dioxide. TPR = Total Peripheral Resistance L = Liters. cm = Centimeters.
Response to Orthostatic Stress
Respiratory Responses
Respiratory data for all subjects are shown in Figure 1. While an increased tidal volume (TV) accounts for most of the VE increase, some also had a significant (P < 0.05) increase in RR at the initial and 1-2 min stages of HUT compared to supine baseline. There was an obvious and significant (P < 0.001) decrease in ETCO2 for POTS subjects at each stage of HUT, compared to controls, in which ETCO2 decreased to a lesser extent with time and was not different than baseline. VE increased significantly (P < 0.001) following HUT in POTS for all time points after the 1-2 min period. Controls showed no significant increase in VE. Maximum ventilation in POTS subjects was achieved 2-4 minutes following HUT and was maintained through.
Figure 1.
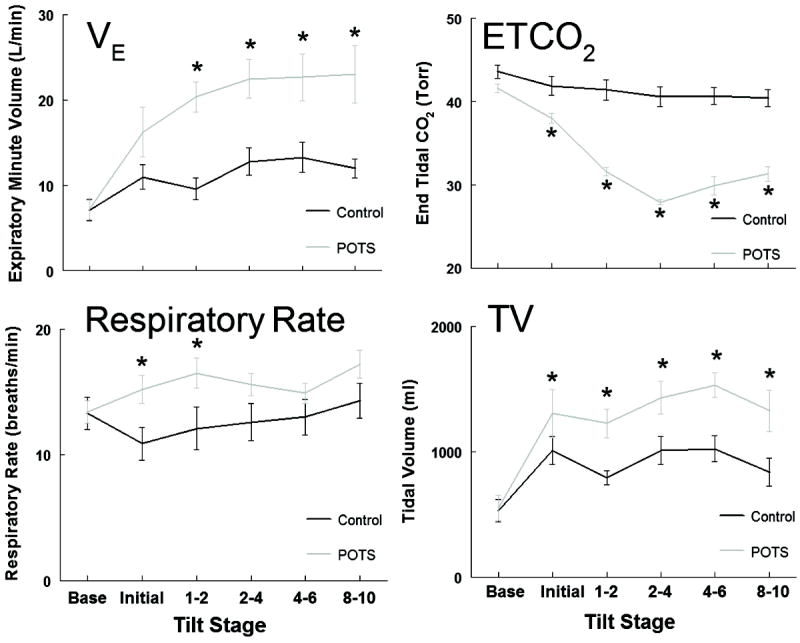
Averaged respiratory data for all participants, (Control - black line, POTS - grey line) before and during a 10 min 70° head-up tilt. Expiratory Minute Volume (VE) is expressed as liters per minute (L · min-1), End tidal carbon dioxide (ETCO2) as Torr, Respiratory Rate (Rate) as breaths per minute (bpm) and Tidal Volume (TV) expressed as milliliters (ml). * indicates different from control, P < 0.05.
Neurocardiopulmonary Responses to Tilt
Figure 2 compares respiratory, hemodynamic and MSNA data for the same representative POTS subject shown in Figure S1 (see http://hyper.ahajournals.org). VE illustrates the differences in timing of the neurocardiopulmonary response to HUT. Total MSNA increased in parallel to VE and was inversely related to ETCO2. CBFv fell immediately upon HUT in synchrony with a brief, abrupt fall in BP comprising IOH23. Notably, the fall in CBFv preceded the increased VE and reciprocal decreased ETCO2. Figure S2 (see http://hyper.ahajournals.org) shows the time delay between CBFv fall and onset of hyperventilation in one representative subject; in all POTS subjects there was a delay of 17 ± 6 s (P<0.025). Hyperventilation was not elicited by HUT in controls. Figure 3, the hemodynamic response of all participants shows HR was significantly increased at baseline in POTS subjects compared to controls (P < 0.001). The increase in HR during HUT in POTS was greater than in controls (P < 0.001) and associated with a decreased CO (P < 0.025) and increased TPR (P < 0.025) compared to control. The short-lived initial decrease in MAP, associated with IOH, was larger in POTS compared to control (P <0.025).
Figure 2.
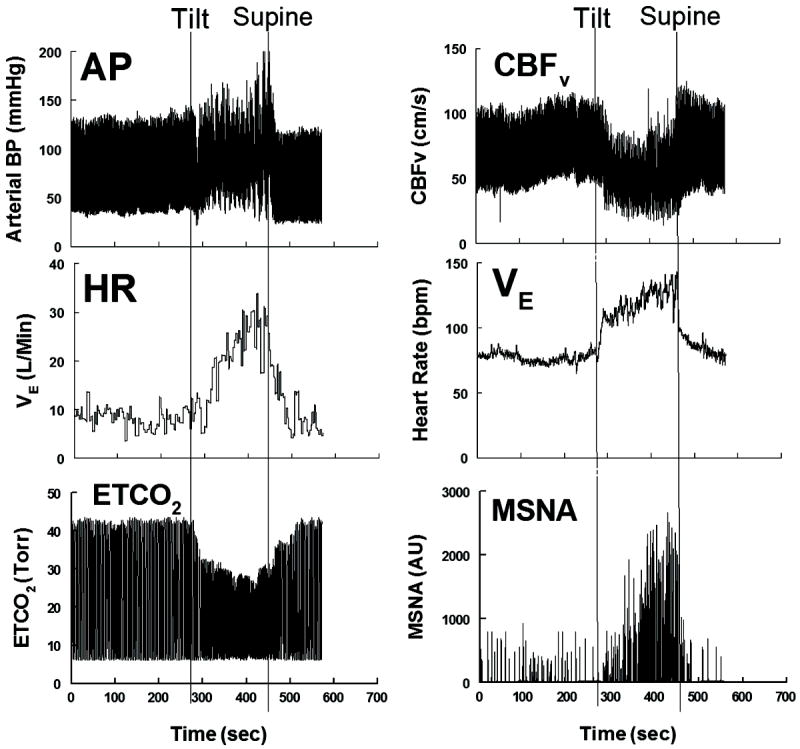
Arterial blood pressure (AP) expressed as millimeters of mercury (mmHg), cerebral blood flow velocity (CBFv) as centimeters per second (cm · s-1), Heart Rate (HR) as beats per minute (bpm), Expiratory Minute Volume (VE) in liters per minute (L · min-1), End Tidal Carbon Dioxide (ETCO2) in Torr and Muscle Sympathetic Nerve Activity (MSNA) expressed as arbitrary units (AU) shown for a representative POTS patient before, during and after a 10 min 70° head-up tilt.
Figure 3.
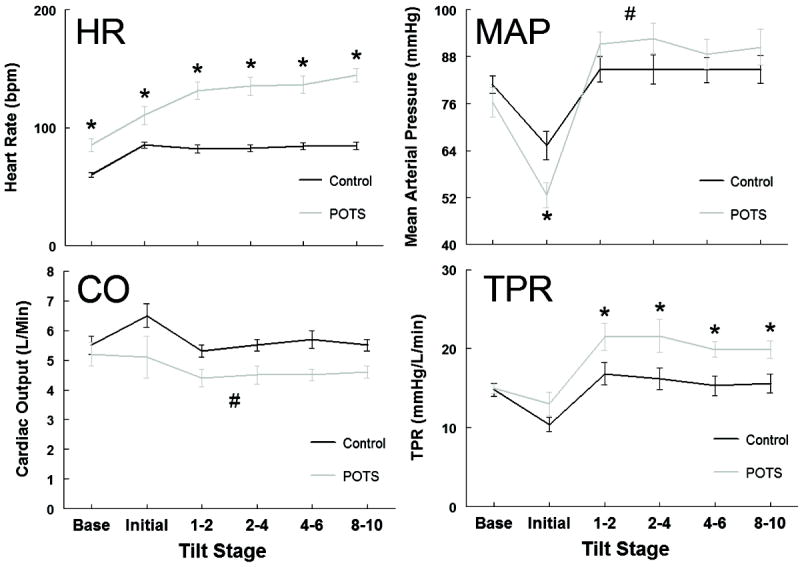
Hemodynamic data for all participants (Control - black line, POTS - grey line) before and during a 10 min 70° head-up tilt. Heart Rate (HR) is expressed as beats per min (bpm), Mean Arterial Pressure (MAP) as millimeters of mercury (mmHg), Cardiac Output (CO) as liters per minute (L · min-1), and Total Peripheral Resistance (TPR) as millimeters of mercury per liter per minute (mm Hg · L · min-1). * indicates difference from control, P < 0.05. # indicates overall effect different from control, P < 0.05.
Because baseline values differed between POTS and controls, Figure 6 shows the percent change from baseline averaged for all subjects within each group. There was a two-fold larger emptying of the thorax [central BV] in POTS compared to controls, with reciprocal filling of the splanchnic reservoir. However, BV redistribution was only transiently different between the two groups during the initial tilt. Initial decreases in CBFv were synchronous with and proportionate to initial changes in central BV (r2 = 0.80) and were significantly greater in POTS, compared to controls (P < 0.01). Once hyperventilation and hypocapnia occurred, CBFv decreased further in POTS subjects.
MSNA, (Figure 4) increased similarly following HUT for POTS and controls. Thereafter, MSNA increased directly with VE and inversely to ETCO2 for POTS subjects while remaining stable for controls. Due to sympathetic activation, MAP and TPR were increased in POTS subjects compared to controls for the remainder of the HUT.
Figure 4.
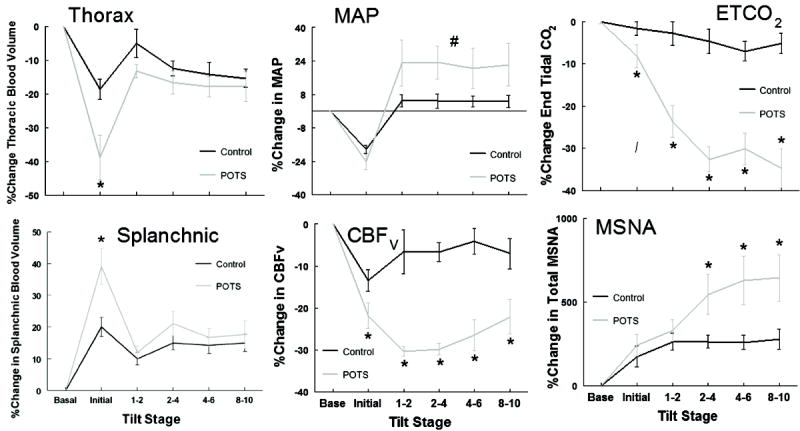
Mean hemodynamic and respiratory data expressed as a percent (%) change from baseline before and during a 10 min 70° head-up tilt for all subjects (Control group - black line, POTS - grey line) showing Thoracic BV (Thorax), Mean Arterial Pressure (MAP), End Tidal Carbon dioxide (ETCO2), changes in splanchnic BV (Splanchnic), changes in cerebral blood flow velocity (CBFv), and changes in total Muscle Sympathetic Nerve Activity (MSNA). * indicates difference from control, P < 0.05. # indicates overall effect different from control, P < 0.05.
Discussion
Changing from supine to standing transfers >500ml of central BV caudally, initially decreasing central BV and increasing BV within the splanchnic vasculature and lower extremites20, 21, 24. There is often a period of IOH during which BP and CBF transiently decrease, sometimes markedly, reaching their nadir 10-20 s after standing25, A reflex tachycardia results, and BP and CBFv are restored within 30-60 s. IOH results from the normal delay of arterial baroreflex detection and autonomic response to gravitational BV redistribution. Thereafter upright HR slows remaining elevated compared to supine; CBFv recovers to somewhat less than supine26; and BP is restored with a slightly higher MAP but reduced pulse pressure due to musculoskeletal mechanisms23, 27, arterial vasoconstriction, elastic recoil of pooled blood in dependent veins, and active venoconstriction of splanchnic veins28. IOH is common, and occurs in healthy individuals who do not develop OI during prolonged postural stress25. Hitherto, studies of POTS subjects have not distinguished differences in IOH from control.
We describe here for the first time, a subset of POTS patients with the chief complaint of dyspnea in which IOH during HUT is exceptional, comprising an exaggerated initial decrease in CBFv that coincides with a notably large fall in central thoracic BV prior to BP restitution. Moreover, the reduced CBFv is immediately followed by hyperpnea and hypocapnia, perpetuating the reduced CBFv. Lastly, sympathetic activity, measured by MSNA, increases in parallel with hyperpnea and hypocapnia at least until orthostatic stress is discontinued. We hypothesize that hyperpnea and hypocapnia increase brain-wide vasoconstriction and hypoxia-ischemia via the Bohr Effect29 resulting in increased cerebral oxygen demand, neuronal excitability and continued sympathetic activation (Figure 4)30.
Posturally increased ventilation can also occur in Control Subjects and in POTS
A modest increase in ventilation and decrease in ETCO2 are observed during transitions from supine to upright31, 32. However, these changes are often exaggerated in POTS subjects compared to controls3, 10, 28, 33. It has been assumed that a reduction in CBF in these POTS patients follows hypocapnia3, 10, 33. However, here we show the reverse may be true; a reduction in CBFv precedes hyperventilation in these POTS subjects.
Hyperventilation in POTS may be a mechanism for compensation of reduced thoracic BV34 and teleologically may be an explanation for hyperpnea and hyperventilation. However, voluntary hyperventilation fails to induce POTS symptoms10 and the respiratory pump (increased intra-abdominal and decreased intra-thoracic pressures) does not improve CO in POTS subjects3. Indeed, in past studies spontaneous hyperventilation in POTS subjects occurs after HUT is well underway. Also, increased ventilation has no effect on reductions of peripheral blood flow in arms, legs and pelvic regions compared to normocapnic POTS subjects and healthy controls3. Moreover, hyperpneic breathing, causing increased abdominal pressure during inspiration and expiration, can restrict venous return to the heart as observed in our POTS patients35.
What causes the exaggerated decrease in CBFv?
Transduction from BV, to arterial BP, to CBF can function as a high pass filter system in which, a rapid decrease in central BV during IOH results in a rapid decrease in AP that drives similar decreases in CBF36. This attenuates slow variations and permits pressure to drive CBF only at higher frequencies (with periods of seconds or smaller). However, central BV and AP can also modulate cerebral autoregulation37. Central BV is linked by the baroreflexes to cerebrovascular regulation via the extrinsic (extracerebral) vascular innervation system and excessive baroreflex unloading causes parasympathetic withdrawal and reduction of nitric oxide-dependent tonic dilation of the extrinsic cerebral vasculature resulting in cerebral vasoconstriction. Our work supports the ability of this extrinsic vasculature to respond well to nitric oxide in humans38. Thus we propose that the exaggerated initial fall in CBFv is directly caused by the initial decrease in BP of IOH and indirectly by reflex loss of NO-dependent dilation of the extracerebral brain vasculature due to parasympathetic withdrawal.
What causes the exaggerated hyperpnea and hypocapnia?
A decreased chemoreflex response to CO2 and increased hypoxic ventilatory response found in POTS subjects6 contributes to hyperventilation. CBF derives largely from carotid artery blood flow. If CBF were reduced, carotid artery blood flow would also be reduced, causing decreased carotid body blood flow and increased chemoreflex sensitivity to hypoxia39. While these data are from rabbits, they are closely paralleled in primates by “stagnant ischemia” or “ischemic hypoxia” in which large reductions in carotid body blood flow is similar to the response to hypoxic hypoxia40. Once engaged, hyperventilation and hypocapnia sustain reduced CBFv. CNS hypocapnia and ischemia result in neuronal excitation and sympathetic activation28.
Limitations
We used TCD which only measures CBFv, however, changes in CBFv accurately reflect changes in CBF during orthostatic stress41. The decrease in arterial BP with initial orthostatic hypotension is larger during standing than during HUT, although present in both20. Nevertheless, these patients uniformly identified their symptoms including dyspnea during tilt with symptoms that occur during “real world” orthostasis.
We did not capture MSNA during all tilts because the electrode position was sometimes lost with HUT. However, the time-dependent responses of respiratory measurements, BP and CBFv were similar in all subjects and recorded successfully during all studies.
Subjects for this study were recruited if they were first referred to our clinic for evaluation of suspected POTS. Thus we have no insight into how this subgroup of POTS patients fits into the larger sphere of patients affected by POTS. Custom data acquisition software is used however, it has been used extensively in previously validate experiments.
Perspectives
Hyperventilation is associated with POTS. Its mechanism was unknown but, based on telelogical arguments, it was assumed to subserve the respiratory-abdominal pump supporting cardiac output and brain blood flow. This study indicates that hyperventilation takes the form of hyperpnea resulting in a decrease in cardiac output and a disproportionate fall in cerebral blood flow in POTS. Hyperpnea seems caused by a profound reduction of central BV early during the initial hypotensive phase of orthostasis. We hypothesize that this results in stagnant ischemic stimulation of the carotid body and consequent hyperpnea, sympathetic activation, tachycardia and an increase in BP.
Supplementary Material
Novelty and Significance.
What is new
To our knowledge this is the first report of decreased cerebral blood flow initiating hypocapic hyperpnea.
What is relevant
Hyperpnea and hypocapnia perpetuate brain wide vasoconstriction and hypoxia-ischemia via the Bohr effect resulting in increased cerebral oxygen demand, neuronal excitability and continued sympathetic activation with tachycardia.
Summary
The ensuing potentiating and sustained effects of hypocapnia on reducing cerebral blood flow and the effects of respiratory alkalosis on oxygen dissociation cause a state of severe hypoxia-ischemia that severely limits brain oxygen and substrate supply while increasing oxygen demand. An ischemia-reperfusion mechanism with oxidative stress could ensue.
Acknowledgments
The authors thank Mr. Zachary Messer and Ms. Seli Dzogbeta for help in data collection and Mrs. Courtney Terilli for help with subject recruitment and screening.
Funding Sources
Funding was provided by the NIH grants RO1 HL 112736 and RO1 HL 074873.
Footnotes
Disclosures
None
References
- 1.Robertson D. The epidemic of orthostatic tachycardia and orthostatic intolerance. Am J Med Sci. 1999;317:75–77. doi: 10.1097/00000441-199902000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Low PA, Opfer-Gehrking TL, McPhee BR, Fealey RD, Benarroch EE, Willner CL, Suarez GA, Proper CJ, Felten JA, Huck CA. Prospective evaluation of clinical characteristics of orthostatic hypotension. Mayo Clin Proc. 1995;70:617–622. doi: 10.4065/70.7.617. [DOI] [PubMed] [Google Scholar]
- 3.Stewart JM, Medow MS, Cherniack NS, Natelson BH. Postural hypocapnic hyperventilation is associated with enhanced peripheral vasoconstriction in postural tachycardia syndrome with normal supine blood flow. Am J Physiol Heart Circ Physiol. 2006;29:H904–H913. doi: 10.1152/ajpheart.01359.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burnum JF, Hickman JB, Stead EA., Jr Hyperventilation in postural hypotension. Circulation. 1954;10:362–365. doi: 10.1161/01.cir.10.3.362. [DOI] [PubMed] [Google Scholar]
- 5.Lagi A, Cencetti S, Corsoni V, Georgiadis D, Bacalli S. Cerebral vasoconstriction in vasovagal syncope: any link with symptoms? A transcranial Doppler study. Circulation. 2001;104:2694–2698. doi: 10.1161/hc6172.099397. [DOI] [PubMed] [Google Scholar]
- 6.Taneja I, Medow MS, Clarke DA, Ocon AJ, Stewart JM. Baroreceptor unloading in postural tachycardia syndrome augments peripheral chemoreceptor sensitivity and decreases central chemoreceptor sensitivity. Am J Physiol Heart Circ Physiol. 2011;301:H173–H179. doi: 10.1152/ajpheart.01211.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Convertino VA, Rickards CA, Lurie KG, Ryan KL. Hyperventilation in response to progressive reduction in central blood volume to near syncope. Aviat Space Environ Med. 2009;80:1012–1017. doi: 10.3357/asem.2598.2009. [DOI] [PubMed] [Google Scholar]
- 8.Lelorier P, Klein GJ, Krahn A, Yee R, Skanes A, Shoemaker JK. Combined head-up tilt and lower body negative pressure as an experimental model of orthostatic syncope. J Cardiovasc Electrophysiol. 2003;14:920–924. doi: 10.1046/j.1540-8167.2003.03065.x. [DOI] [PubMed] [Google Scholar]
- 9.Stewart JM, Rivera E, Clarke DA, Baugham IL, Ocon AJ, Taneja I, Terilli C, Medow MS. Ventilatory baroreflex sensitivity in humans is not modulated by chemoreflex activation. Am J Physiol Heart Circ Physiol. 2011;300:H1492–H1500. doi: 10.1152/ajpheart.01217.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Novak V, Spies JM, Novak P, McPhee BR, Rummans TA, Low PA. Hypocapnia and cerebral hypoperfusion in orthostatic intolerance. Stroke. 1998;29:1876–1881. doi: 10.1161/01.str.29.9.1876. [DOI] [PubMed] [Google Scholar]
- 11.Schondorf R, Low PA. Idiopathic postural orthostatic tachycardia syndrome: an attenuated form of acute pandysautonomia? Neurology. 1993;43:132–137. doi: 10.1212/wnl.43.1_part_1.132. [DOI] [PubMed] [Google Scholar]
- 12.Jacob G, Costa F, Shannon JR, Robertson RM, Wathen M, Stein M, Biaggioni I, Ertl A, Black B, Robertson D. The neuropathic postural tachycardia syndrome. N Engl J Med. 2000;343:1008–1014. doi: 10.1056/NEJM200010053431404. [DOI] [PubMed] [Google Scholar]
- 13.Jordan J, Shannon JR, Diedrich A, Black BK, Robertson D. Increased sympathetic activation in idiopathic orthostatic intolerance: role of systemic adrenoreceptor sensitivity. Hypertension. 2002;39:173–178. doi: 10.1161/hy1201.097202. [DOI] [PubMed] [Google Scholar]
- 14.Stewart JM, Medow MS, Glover JL, Montgomery LD. Persistent splanchnic hyperemia during upright tilt in postural tachycardia syndrome. Am J Physiol Heart Circ Physiol. 2006;290:H665–H673. doi: 10.1152/ajpheart.00784.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito H, Kanno I, Ibaraki M, Hatazawa J, Miura S. Changes in human cerebral blood flow and cerebral blood volume during hypercapnia and hypocapnia measured by positron emission tomography. J Cereb Blood Flow Metab. 2003;23:665–670. doi: 10.1097/01.WCB.0000067721.64998.F5. [DOI] [PubMed] [Google Scholar]
- 16.Plash WB, Diedrich A, Biaggioni I, Garland EM, Paranjape SY, Black BK, Dupont WD, Raj SR. Diagnosing postural tachycardia syndrome: comparison of tilt testing compared with standing haemodynamics. Clin Sci (Lond) 2013;124:109–114. doi: 10.1042/CS20120276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singer W, Sletten DM, Opfer-Gehrking TL, Brands CK, Fischer PR, Low PA. Postural tachycardia in children and adolescents: what is abnormal? J Pediatr. 2012;160:222–226. doi: 10.1016/j.jpeds.2011.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Montgomery LD, Hanish HM, Marker RA. An impedance device for study of multisegment hemodynamic changes during orthostatic stress. Aviat Space Environ Med. 1989;60:1116–1122. [PubMed] [Google Scholar]
- 19.Schwartz CE, Lambert E, Medow MS, Stewart JM. Disruption of phase synchronization between blood pressure and muscle sympathetic nerve activity in postural vasovagal syncope. Am J Physiol Heart Circ Physiol. 2013;305:H1238–H1245. doi: 10.1152/ajpheart.00415.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stewart JM, Clarke D. “He’s dizzy when he stands up”: an introduction to initial orthostatic hypotension. J Pediatr. 2011;158:499–504. doi: 10.1016/j.jpeds.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wieling W, Krediet CT, van Dijk N, Linzer M, Tschakovsky ME. Initial orthostatic hypotension: review of a forgotten condition. Clin Sci (Lond) 2007;112:157–165. doi: 10.1042/CS20060091. [DOI] [PubMed] [Google Scholar]
- 22.Dyson KS, Shoemaker JK, Arbeille P, Hughson RL. Modelflow estimates of cardiac output compared with Doppler ultrasound during acute changes in vascular resistance in women. Exp Physiol. 2010;95:561–568. doi: 10.1113/expphysiol.2009.050815. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Marshall RJ, Shepherd JT. The effect of changes in posture and of graded exercise on stroke volume in man. J Clin Invest. 1960;39:1051–1061. doi: 10.1172/JCI104120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheriff DD, Nadland IH, Toska K. Role of sympathetic responses on the hemodynamic consequences of rapid changes in posture in humans. J Appl Physiol. 2010;108:523–532. doi: 10.1152/japplphysiol.01185.2009. [DOI] [PubMed] [Google Scholar]
- 25.Thomas KN, Cotter JD, Galvin SD, Williams MJ, Willie CK, Ainslie PN. Initial orthostatic hypotension is unrelated to orthostatic tolerance in healthy young subjects. J Appl Physiol. 2009;107:506–517. doi: 10.1152/japplphysiol.91650.2008. [DOI] [PubMed] [Google Scholar]
- 26.Levine BD, Giller CA, Lane LD, Buckey JC, Blomqvist CG. Cerebral versus systemic hemodynamics during graded orthostatic stress in humans. Circulation. 1994;90:298–306. doi: 10.1161/01.cir.90.1.298. [DOI] [PubMed] [Google Scholar]
- 27.Miller JD, Pegelow DF, Jacques AJ, Dempsey JA. Skeletal muscle pump versus respiratory muscle pump: modulation of venous return from the locomotor limb in humans. J Physiol. 2005;563:925–943. doi: 10.1113/jphysiol.2004.076422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tani H, Singer W, McPhee BR, Opfer-Gehrking TL, Haruma K, Kajiyama G, Low PA. Splanchnic-mesenteric capacitance bed in the postural tachycardia syndrome (POTS) Auton Neurosci. 2000;86:107–113. doi: 10.1016/S1566-0702(00)00205-8. [DOI] [PubMed] [Google Scholar]
- 29.Riggs AF. The Bohr effect. Annu Rev Physiol. 1988;50:181–204. doi: 10.1146/annurev.ph.50.030188.001145. [DOI] [PubMed] [Google Scholar]
- 30.Laffey JG, Kavanagh BP. Hypocapnia. N Engl J Med. 2002;347:43–53. doi: 10.1056/NEJMra012457. [DOI] [PubMed] [Google Scholar]
- 31.Anthonisen NR, Bartlett D, Tenney SM. Postural effect on ventilatory control. J Appl Physiol. 1965;20:191–196. [Google Scholar]
- 32.Hughson RL, Edwards MR, O’Leary DD, Shoemaker JK. Critical analysis of cerebrovascular autoregulation during repeated head-up tilt. Stroke. 2001;32:2403–2408. doi: 10.1161/hs1001.097225. [DOI] [PubMed] [Google Scholar]
- 33.Schondorf R, Stein R, Roberts R, Benoit J, Cupples W. Dynamic cerebral autoregulation is preserved in neurally mediated syncope. J Appl Physiol. 2001;91:2493–2502. doi: 10.1152/jappl.2001.91.6.2493. [DOI] [PubMed] [Google Scholar]
- 34.Shepherd JT. The lungs as receptor sites for cardiovascular regulation. Circulation. 1981;63:1–10. doi: 10.1161/01.cir.63.1.1. [DOI] [PubMed] [Google Scholar]
- 35.Takata M, Wise RA, Robotham JL. Effects of abdominal pressure on venous return: abdominal vascular zone conditions. J Appl Physiol. 1990;69:1961–1972. doi: 10.1152/jappl.1990.69.6.1961. [DOI] [PubMed] [Google Scholar]
- 36.Zhang R, Zuckerman JH, Giller CA, Levine BD. Transfer function analysis of dynamic cerebral autoregulation in humans. Am J Physiol. 1998;274:H233–H241. doi: 10.1152/ajpheart.1998.274.1.h233. [DOI] [PubMed] [Google Scholar]
- 37.Van Lieshout JJ, Wieling W, Karemaker JM, Secher NH. Syncope, cerebral perfusion, and oxygenation. J Appl Physiol. 2003;94:833–848. doi: 10.1152/japplphysiol.00260.2002. [DOI] [PubMed] [Google Scholar]
- 38.Stewart JM, Medow MS, DelPozzi A, Messer ZR, Terilli C, Schwartz CE. Middle cerebral O(2) delivery during the modified Oxford maneuver increases with sodium nitroprusside and decreases during phenylephrine. Am J Physiol Heart Circ Physiol. 2013;304:H1576–H1583. doi: 10.1152/ajpheart.00114.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding Y, Li YL, Schultz HD. Role of blood flow in carotid body chemoreflex function in heart failure. J Physiol. 2011;589:245–258. doi: 10.1113/jphysiol.2010.200584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitchell RA. Respiration. Annu Rev Physiol. 1970;32:415–438. doi: 10.1146/annurev.ph.32.030170.002215. [DOI] [PubMed] [Google Scholar]
- 41.Serrador JM, Picot PA, Rutt BK, Shoemaker JK, Bondar RL. MRI measures of middle cerebral artery diameter in conscious humans during simulated orthostasis. Stroke. 2000;31:1672–1678. doi: 10.1161/01.str.31.7.1672. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.