

Abstract
Recently, ∼16% of participants in an anti-Aβ passive immunotherapy trial for mild-to-moderate Alzheimer disease (AD) had a negative baseline amyloid positron emission tomography (PET) scan. Whether they have AD or are AD clinical phenocopies remains unknown. We examined the 2005-2013 National Alzheimer's Coordinating Center autopsy database and found that ∼14% of autopsied subjects clinically diagnosed with mild-to-moderate probable AD have no or sparse neuritic plaques, which would expectedly yield a negative amyloid PET scan. More than half of these “Aβ-negative” subjects have low neurofibrillary tangle Braak stages. These findings support the implementation of a positive amyloid biomarker as an inclusion criterion in future anti-Aβ drug trials.
Amyloid positron emission tomography (PET) imaging is a valuable biomarker in clinical trials of anti-Aβ passive immunotherapy in mild-to-moderate Alzheimer disease (AD) patients,1,2 and to diagnose AD in the clinical setting.3 The results of the largest amyloid PET imaging substudy of a clinical trial were recently communicated. 4,5 Remarkably, although all had a clinical diagnosis of mild-to-moderate probable AD, 30 of 184 (16.3%) participants had a negative baseline amyloid PET scan. Interestingly, 22 of 61 (36.1%) APOEε.4 non-carriers were amyloid PET negative, whereas only 8 of 123 (6.5%) APOEε4 carriers were amyloid PET negative, and 1 of these 8 was homozygous for the APOEε4 allele.
Because of its potential adverse impact on the design of ongoing and future clinical trials for AD, it is imperative to recognize and characterize this subset of “amyloid PET-negative mild-to-moderate AD dementia subjects.” To explore the clinical and neuropathologic characteristics of subjects in this category, we examined the 2005-2013 autopsy cohort of the National Alzheimer's Coordinating Center (NACC) database, a large multicenter longitudinal cohort study of aging involving 35 past and present AD centers across the United States.6 We selected all subjects with a clinical diagnosis of probable AD (PRAD) and a last Mini-Mental State Examination (MMSE) score between 16 and 26 within 2 years of death, and excluded subjects with a non-AD primary neuropathological diagnosis. Based on published amyloid PET—postmortem correlations,7,10 we approximated the cutoffs for amyloid PET imaging sensitivity and classified as amyloid positive (Aβ+;) those subjects with moderate or frequent neuritic plaques, and as amyloid negative (Aβ−) those with no or sparse neuritic plaques according to Consortium to Establish a Registry for Alzheimer's Disease (CERAD) score,11 appreciating that the CERAD score is of neuritic plaques and therefore not identical to a determination of amyloid positivity. We investigated the demographic, clinical, and neuropathological characteristics of Aβ+; and Aβ− subjects.
Subjects and Methods
Eligibility Criteria
To approximate the clinical characteristics of anti-Aβ immunotherapy clinical trials, subjects were selected from the 2005–2013 NACC autopsy cohort if they met the following inclusion criteria: (1) age of death ≥ 50 years; (2) last clinical evaluation (including MMSE) within 2 years before death; (3) last MMSE score between 16 and 26, inclusive; (4) clinical diagnosis of PRAD; and (5) if any primary neuropathological diagnosis was present, this had to be AD, although meeting the neuropathological criteria for AD12 was not required. To maximize the correlation between a clinical diagnosis of PRAD and AD neuropathological changes, the following conservative exclusion criteria were implemented: (1) a primary neuropathological diagnosis other than AD (eg, frontotemporal lobar degeneration, dementia with Lewy bodies, hippocampal sclerosis, vascular dementia, prion disease, Parkinson disease, Huntington disease, hypoxia, ischemia, hemorrhage); and (2) cognitive impairment attributable to alcohol use, depression, medication use, or medical illness.
Data Collection
Demographic characteristics included sex, age at death, education, and APOE genotype. Clinical variables included age of onset, disease duration, Unified Parkinson Disease Rating Scale, MMSE score, Clinical Dementia Rating Scale Sum of Boxes (CDR-SOB) score, and part B of the Trail Making Test. Neuropathological data included CERAD score of neuritic plaques,11 Braak stage of neurofibrillary tangles,13 presence of any vascular pathology, presence and severity of cerebral amyloid angiopathy (none, mild, moderate, severe), atherosclerosis (ie, circle of Willis, none, mild, moderate, severe), and arterioloscle-rosis (none, mild, moderate, severe), and presence of ≥1 lacunar infarcts, ≥1 cortical microinfarcts, ≥1 large infarcts, subcortical arteriosclerotic leukoencephalopathy, cortical laminar necrosis, ≥1 brain hemorrhages, hippocampal sclerosis, and incidental Lewy bodies (in any brain region, brainstem, limbic system, neocortex, unspecified).
Statistical Analyses
Statistical analyses were performed with Prism version 5.0 for Mac (GraphPad, La Jolla, CA). Normality of continues variables in the data set was evaluated with the D'Agostino—Pearson omnibus test. For continuous variables, pairwise comparisons between Aβ− and Aβ+; subjects were performed with the Mann-Whitney U test or unpaired Student t test, as appropriate. For categorical variables, comparisons of proportions between Aβ− and Aβ+; subjects were done using Fisher exact test. All the hypothesis tests were 2-sided, and a p value of <0.05 was considered to be statistically significant.
Results
F1 The flowchart in the Fig 1 depicts the selection process based on the eligibility criteria. A total of 161 subjects were eligible for this study. The Table shows the demographic, clinical, and neuropathological characteristics of the subjects. Groups were comparable regarding sex, education, age, and disease duration from symptom onset. Of note, compared to Aβ+ subjects, Aβ− subjects had slightly but significantly better cognitive function, as indicated by antemortem MMSE and CDR-SOB scores. A trend was also observed with part B of the Trail Making Test, a measure of executive function.
Figure 1.
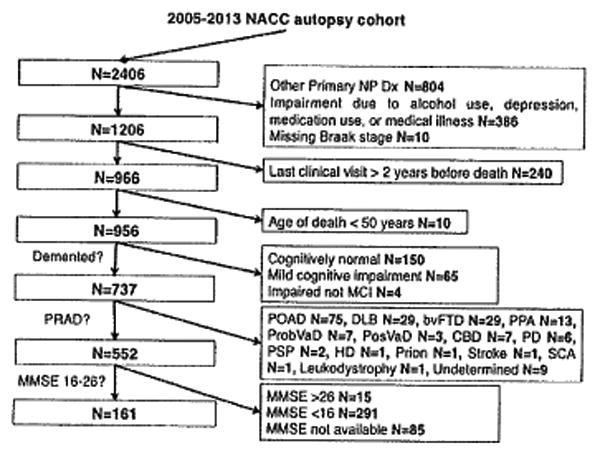
Flowchart of the selection algorithm of the study subjects from the original 2005-2013 National Alzheimer's Coordinating Center (NACC) autopsy cohort. The final sample size of this study is 161. The number of subjects excluded and the reasons for their exclusion are shown on the right side of the flowchart. bvFTD = behavioral variant frontotemporal dementia; CBD = corticobasal degeneration; DLB = dementia with Lewy bodies; HD = Huntington disease; MCI = mild cognitive impairment; MMSE = Mini-Mental State Examination; NP Dx = neuropathological diagnosis; PD = Parkinson disease; POAD = possible Alzheimer disease; PosVaD = possible vascular dementia; PPA = primary progressive aphasia; PRAD = probable Alzheimer disease; Prob-VaD = probable vascular dementia; PSP = progressive supranuclear palsy; SCA = spinocerebellar ataxia.
Table 1. Comparison between Aβ+ and Aβ− Subjects with Mild-to-Moderate (MMSE = 16-26) Probable Alzheimer Disease.
| Characteristic | Aβ+, n = 139 | Aβ−, n = 22 | P |
|---|---|---|---|
| Demographic characteristics | |||
| Sex, No. (%) female | 80 (57.5) | 15 (68.2) | 0.485 |
| Education, yr | 15.3 ±3.2 | 14.4 ±2.9 | 0.272 |
| Age at death, yr | 84.4 ± 7.7 | 86.3 ± 6.7 | 0.267 |
| Age at onset, yr | 76.4 ± 8.6 | 78.7 ±6.4 | 0.273 |
| Disease duration, yr | 8.0 ±3.8 | 7.5 ±3.9 | 0.708 |
| Cognitive performance | |||
| MMSE | 20.0 [17.0–22.0]a | 22.0 [19.7–23.2]a | 0.021a |
| CDR-SOB | 9.0 [6.0–12.0]a | 6.0 [5.0–9.25]a | 0.023a |
| TMT-B, s | 245±71b | 205 ± 86b | 0.074b |
| Motor performance, UPDRS | 11.0 ±7.8 | 10.1 ±9.7 | 0.411 |
| APOE genotype | |||
| APOEε4 carriers, No. (%) | 61/119 (51.3) | 6/18 (33.3) | 0.208 |
| APOEε4 alleles, No. (%) | 69/238 (29.0) | 7/36 (19.4) | 0.318 |
| Neuropathology, No. (%) | |||
| Braak stage 0/I/II | 5 (3.6)a | 13 (59.1)a | <0.0001a |
| Braak stage III/IV | 49 (35.2)a | 5 (22.7)a | |
| Braak stage V/VI | 85 (61.1)a | 4 (18.2)a | |
| Any vascular pathology present | 113 (81.3)b | 14 (63.6)b | 0.088b |
| Moderate/severe CAA | 45/134 (33.6) | 4/22 (18.2) | 0.215 |
| Moderate/severe arteriosclerosis | 37/109 (33.9) | 6/20 (33.3) | 0.802 |
| Moderate/severe atherosclerosis | 61 (43.9) | 8 (36.4) | 0.644 |
| Large artery infarct | 8/130 (5.8) | 2/22 (9.1) | 0.629 |
| Cortical microinfarcts | 23/138 (16.7) | 4/22 (18.2) | 0.768 |
| Lacunar infarcts | 19/138 (13.8) | 4/22 (18.2) | 0.527 |
| Subcortical arteriosclerotic leukoencephalopathy | 15/137 (10.9) | 2/22 (9.1) | 1.000 |
| Cortical laminar necrosis | 1/136 (0.7) | 0/21 (0.0) | 1.000 |
| Hippocampal sclerosis | 13/136 (9.6) | 4/21 (19.0) | 0.249 |
| Hemorrhage | 7 (5.0) | 0 (0.0) | 0.595 |
| Lewy bodies any region | 34 (24.5)a | 1 (4.5)a | 0.048a |
| Lewy bodies neocortex | 8 (5.7) | 0 (0.0) | 0.103 |
| Lewy bodies limbic | 17 (12.2) | 0 (0.0) | |
| Lewy bodies brainstem | 4 (2.9) | 1 (4.5) | |
| Lewy bodies unspecified | 5 (3.6) | 0 (0.0) |
Education, age of death, age of onset, disease duration, and TMT-B are presented as mean ± standard deviation. MMSE and CDR-SOB scores are presented as median [interquartile range]. A higher MMSE score and a lower CDR-SOB score indicate better cognitive performance. Denominators vary due to missing data.
Statistically significant difference (p< 0.05).
Statistical trend (p < 0.1).
Aβ+ = CERAD score of moderate or frequent neuritic plaques; Aβ− = CERAD score of none or sparse neuritic plaques; MMSE = Mini-Mental State Examination; CDR-SOB = Clinical Dementia Rating Scale Sum of Boxes; CERAD = Consortium to Establish a Registry for Alzheimer's Disease; TMT-B = part B of Trail Making Test; UPDRS = Unified Parkinson Disease Rating Scale; CAA = cerebral amyloid angiopathy.
Regarding the neuropathological findings, more than half of the Aβ− subjects (13 of 22, 59.1%) had a Braak stage 0/I/II, indicating that many Aβ− subjects with dementia do not have the neuropathological signs of AD. We reasoned that other concomitant pathologies may account for the cognitive decline of these individuals.14,15 However, Aβ− and Aβ+ subjects did not differ in the frequency of concurrent moderate/severe cerebral amyloid angiopathy (CAA), arteriolosclerosis, or atherosclerosis, nor in the frequency of coexisting lacunar infarcts, large infarcts, cortical microinfarcts, subcortical arteriosclerotic leukoencephalopathy, cortical laminar necrosis, cerebral hemorrhages, or hippocampal sclerosis. If anything, there was a trend toward a significantly higher frequency of “any vascular pathology” in the Aβ+; group. Last, Lewy body pathology was more frequent in Aβ+ subjects than in Aβ− subjects.
The APOE genotype was available for 137 of the 161 eligible subjects. The AP0Eε4 genotype was overre-presented within the Aβ+ group compared to the Aβ−group (see Table). APOEε4 noncarriers were twice as likely to be Aβ− than APOEε4 carriers (12 of 70 [17.1%] vs 6 of 67 [8.9%], odds ratio = 2.1; 95% confidence interval = 0.7–6.0), although these differences did not reach statistical significance in this relatively small autopsy sample.
Discussion
The recent finding of a higher than expected proportion (16%) of amyloid PET-negative subjects who participated in a large multicenter phase 3 anti-Aβ passive immunotherapy trial has raised concerns about the design of this and similar clinical trials.4,5 Do these individuals really have AD? If not, should participation in subsequent trials require demonstration of a positive amyloid biomarker?
We interrogated the 2005–2013 NACC autopsy database and selected a convenience sample of subjects with a clinical diagnosis of PRAD (MMSE = 16–26) excluding those with a primary clinical or neuropathological diagnosis of a disease other than AD and those with cognitive dysfunction attributed to medications, alcohol, medical conditions, or depression. Thus, these were individuals who otherwise would qualify for and may have been enrolled in clinical trials of potentially disease-modifying anti-Aβ drugs. Surprisingly, we discovered that a not negligible proportion (22 of 161, ≈l4%) of these subjects actually have no or only sparse neuritic plaques, which is not sufficient to meet the neuropathological diagnostic criteria for AD.12,16 Had we not excluded individuals with a clinical diagnosis of AD but a non-AD pathological diagnosis, the percentage of individuals without substantial Aβ deposits would have been even higher, similar to an earlier report by Beach et al.14 Importantly, extrapolating the published studies correlating amyloid PET radiotracer uptake and postmortem plaque burden,7-10 these subjects would have had a negative amyloid PET scan in vivo, referred to in this study as Aβ−. Although 2 of these 22 individuals had severe CAA and this could be associated with a positive amyloid PET scan,17 the proportion of Aβ− subjects observed in this study is strikingly similar to the 16% recently observed in clinical trials of anti-Aβ passive immunotherapy4,5 and to that reported in the validation studies of some of the amyloid PET radiotracers.18-20
We next asked whether this singular group of Aβ−subjects could be differentiated from Aβ+; subjects on the basis of demographic, clinical, or neuropathological signatures. We found that: (1) both groups have similar gender, education level, and age of death and symptom onset; (2) APOEε4 carriers were half as likely to be Aβ−as noncarriers; (3) although meeting criteria for mild-to-moderate dementia, the Aβ− subjects were slightly less impaired than Aβ+ subjects; (4) more than half of the Aβ− subjects had a Braak stage 0/I/II of neurofibrillary tangles, which is clearly insufficient to account for their mild-to-moderate dementia; but (5) frequency and severity of concurrent incidental vascular and Lewy body pathologies did not differ between Aβ− and Aβ+; subjects; therefore, these known pathologies—although acknowledging the limitations of NACC scoring—do not appear to account for cognitive loss among the Aβ−subjects.
In summary, our results document that a sizeable number of individuals clinically diagnosed with PRAD at a mild-to-moderate dementia stage in specialized AD centers have insufficient plaques and tangles to meet established neuropathological guidelines for AD. The pathological substrate of these clinical phenocopies of AD remains elusive, and further investigation in other cohorts may help elucidate their distinct clinical or neuropathological features. Our findings support the implementation of a baseline amyloid PET radiotracer uptake above a prespecified cutoff and/or a positive cerebrospinal fluid AD biomarker as inclusion criteria in future clinical trials with anti-Aβ therapies.
Acknowledgments
This work was supported by the Massachusetts Alzheimer's Disease Research Center (NIH grant P50 AQ2 AG0001534; B.T.H.). The NACC database is funded by the NIH National Institute on Aging grant U01 AG016976.
Footnotes
Potential Conflicts of Interest: Nothing to report.
References
- 1.Rinne JO, Brooks DJ, Rossor MN, et al. 11 C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9:363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 2.Ostrowitzki S, Deptula D, Thurfjell L, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantener-umab. Arch Neurol. 2012;69:198–207. doi: 10.1001/archneurol.2011.1538. [DOI] [PubMed] [Google Scholar]
- 3.Johnson KA, Minoshima S, Bohnen Nl, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer's Association. Alzheimers Dement. 2013;9:e-1–e-16. doi: 10.1016/j.jalz.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sperling R, Salloway S, Raskind M, et al. Bapineuzumab phase 3 trials in mild to moderate Alzheimer's disease dementia in apoli-poprotein E ε4 carriers (Study 302) and non-carriers (Study 301): safety and PiB PET amyloid imaging. 2013 Available at: http://AQ4www.ctad.fr/07-download/Congres2012/PressRelease/Final-Sperling-CTAD-Presentation-10-29-12.pdf.
- 5.Vellas B, Carrillo MC, Sampaio C, et al. Designing drug trials for Alzheimer's disease: what we have learned from the release of the phase III antibody trials: a report from the EU/US/CTAD Task Force. Alzheimers Dement. 2013;9:438–444. doi: 10.1016/j.jalz.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer's Coordinating Center (NACC) database: the Uniform Data Set. Alzheimer Dis Assoc Disord. 2007;21:249–258. doi: 10.1097/WAD.0b013e318142774e. [DOI] [PubMed] [Google Scholar]
- 7.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain. 2008;131(pt 6):1630–164S. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikonomovic MD, Abrahamson EE, Price JC, et al. Early AD pathology in a [C-11]PiB-negative case: a PiB-amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathol. 2012;123:433–447. doi: 10.1007/s00401-012-0943-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sojkova J, Driscoll I, Iacono D, et al. In vivo fibrillar beta-amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Arch Neurol. 2011;68:232–240. doi: 10.1001/archneurol.2010.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark CM, Pontecorvo MJ, Beach TG, et al. Cerebral PET with flor-betapir compared with neuropathology at autopsy for detection of neuritic amyloid-β) plaques: a prospective cohort study. Lancet Neurol. 2012;11:669–678. doi: 10.1016/S1474-4422(12)70142-4. [DOI] [PubMed] [Google Scholar]
- 11.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 12.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18(4 suppl):S1–S2. [PubMed] [Google Scholar]
- 13.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 14.Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005-2010. J Neuropathol Exp Neurol. 2012;71:266–273. doi: 10.1097/NEN.0b013e31824b211b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montine TJ, Sonnen JA, Montine KS, et al. Adult Changes in Thought study: dementia is an individually varying convergent syndrome with prevalent clinically silent diseases that may be modified by some commonly used therapeutics. Curr Alzheimer Res. 2012;9:718–723. doi: 10.2174/156720512801322555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bacskai BJ, Frosch MP, Freeman SH, et al. Molecular imaging with Pittsburgh compound B confirmed at autopsy: a case report. Arch Neurol. 2007;64:431–434. doi: 10.1001/archneur.64.3.431. [DOI] [PubMed] [Google Scholar]
- 18.Vandenberghe R, Van Laere K, lvanoiu A, et al. 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Ann Neurol. 2010;68:319–329. doi: 10.1002/ana.22068. [DOI] [PubMed] [Google Scholar]
- 19.Barthel H, Gertz HJ, Dresel S, et al. Cerebral amyloid-β PET with florbetaben (18F) in patients with Alzheimer's disease and healthy controls: a multicentre phase 2 diagnostic study. Lancet Neurol. 2011;10:424–435. doi: 10.1016/S1474-4422(11)70077-1. [DOI] [PubMed] [Google Scholar]
- 20.Johnson KA, Sperling RA, Gidicsin CM, et al. Florbetapir (F18-AV-45) PET to assess amyloid burden in Alzheimer's disease dementia, mild cognitive impairment, and normal aging. Alzheimers Dement. 2013;9(5 suppl):S72–S83. doi: 10.1016/j.jalz.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]