

Abstract
Summary: Next-generation sequencing technologies produce short reads that are either de novo assembled or mapped to a reference genome. Genotypes and/or single-nucleotide polymorphisms are then determined from the read composition at each site, which become the basis for many downstream analyses. However, for low sequencing depths, e.g. 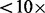 , there is considerable statistical uncertainty in the assignment of genotypes because of random sampling of homologous base pairs in heterozygotes and sequencing or alignment errors. Recently, several probabilistic methods have been proposed to account for this uncertainty and make accurate inferences from low quality and/or coverage sequencing data.
, there is considerable statistical uncertainty in the assignment of genotypes because of random sampling of homologous base pairs in heterozygotes and sequencing or alignment errors. Recently, several probabilistic methods have been proposed to account for this uncertainty and make accurate inferences from low quality and/or coverage sequencing data.
We present ngsTools, a collection of programs to perform population genetics analyses from next-generation sequencing data. The methods implemented in these programs do not rely on single-nucleotide polymorphism or genotype calling and are particularly suitable for low sequencing depth data.
Availability: Programs included in ngsTools are implemented in C/C++ and are freely available for noncommercial use at https://github.com/mfumagalli/ngsTools.
Contact: mfumagalli82@gmail.com
Supplementary Information: Supplementary materials are available at Bioinformatics online.
1 INTRODUCTION
Next-generation sequencing (NGS) technologies have revolutionized population genetics research by enabling unparalleled data collection from genomes or subsets of genomes from many individuals. Current technologies produce short fragments of sequenced DNA called ‘reads’ that are either de novo assembled or mapped to a pre-existing reference genome. This leads to chromosomal positions being sequenced a variable number of times across the genome, usually referred to as the sequencing depth. Individual genotypes are then inferred from the proportion of nucleotide bases covering each site after the reads have been aligned.
Low sequencing depth, along with high error rates stemming from base calling and mapping errors, causes single-nucleotide polymorphism and genotype calling from NGS data to be associated with considerable statistical uncertainty. Recently, probabilistic models that take these errors into account have been proposed to accurately assign genotypes and estimate allele frequencies (e.g. Nielsen et al., 2012).
We present ngsTools, a collection of programs for population genetics analyses that use methods which account for the statistical uncertainty of NGS data. The implemented methods are specially tailored for low-depth sequencing datasets with multiple individuals and populations, and can incorporate deviations from Hardy–Weinberg equilibrium. The inputs for these programs are the files generated by ANGSD, a software for reading and handling NGS data (popgen.dk/angsd).
2 PROGRAMS
2.1 Data preparation
We assume that sequencing reads have already been mapped to a reference sequence or de novo aligned, and the data are in BAM/SAM format. Data should undergo quality control filtering to remove reads, sites, contigs or individuals with low-quality or unusual features. Mapped reads that pass quality controls are then processed by the program ANGSD to compute genotype likelihoods, which are a function of the observed sequencing reads and their qualities. ANGSD can then be used to calculate genotype posterior probabilities under different priors, as well as per-site sample allele frequency posterior probabilities using a maximum likelihood estimate of the sample site frequency spectrum. Programs in ngsTools read and compute summary statistics from files containing this information.
2.2 ngsPopGen
ngsPopGen contains several programs to perform population genetics analyses from sample allele frequency posterior probabilities. ngsStat calculates several basic population genetics summary statistics. Given a file with sample allele frequency posterior probabilities generated by ANGSD, the number of segregating sites and the expected average heterozygosity can be estimated by ngsStat. If data from two species or populations are provided, ngsStat also outputs the expected number of fixed differences. Results can be reported for each site or as sliding window values. ngsFST provides a set of methods to quantify genetic differentiation between pairs of populations without relying on called genotypes using the methods described in Fumagalli et al. (2013). It specifically calculates indices of the per-site expected genetic variance between and within populations, which facilitates calculation of FST in any desired genomic window. Similarly, ngsCovar approximates the covariance matrix among individuals by accounting for genotype uncertainty from genotype posterior probabilities. Eigen-decomposition of the resulting covariance matrix enables one to perform a principal component analysis. ngs2dSFS implements several methods to estimate the joint site frequency spectrum for two populations, which is useful for demographic and selection inference or as a prior in estimating genetic differentiation (Fumagalli et al., 2013). Several scripts to manipulate and plot results are also provided.
2.3 ngsF
ngsF provides a method to estimate individual inbreeding coefficients from genotype likelihoods using an expectation–maximization algorithm described in Vieira et al. (2013). Inbreeding coefficients provide insights into a population’s mating system and demographic history. More importantly, incorporating inbreeding coefficients into the prior when calculating posterior probabilities of genotypes can lead to improved genotype and single-nucleotide polymorphism calling. The output of this program can be parsed by ANGSD and, consequently, by all other programs mentioned here.
2.4 ngsSim and ngsUtils
ngsTools also offers many other useful tools for population genetics analyses. ngsSim is a simple sequencing read simulator that can generate data for multiple populations with variable levels of depth, error rates, genetic variability and individual inbreeding. ngsUtils includes tools to extract data and merge or manipulate files generated by ANGSD.
3 EXAMPLE OF APPLICATION TO EMPIRICAL DATA
To illustrate the use of these programs for analyzing empirical data, we applied them to a publicly available dataset of wild rice accessions (446 Oryza rufipogon and 11 Oryza meridionalis) from 19 countries, at an effective sequencing coverage of 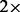 (Huang et al., 2012). We analyzed all 11 O. meridionalis individuals and 150 randomly sampled O. rufipogon accessions.
(Huang et al., 2012). We analyzed all 11 O. meridionalis individuals and 150 randomly sampled O. rufipogon accessions.
We first used ngsF to compute individual inbreeding coefficients for all samples (rice is a partially selfing plant), which were then used to calculate genotype posterior probabilities in ANGSD for each individual at all sites. Because inbreeding is not expected to vary much across chromosomes, we estimated it only on chromosome one. Using the genotypes and their associated probabilities, we estimated a covariance matrix, and the latter was decomposed to produce a principal component analysis plot. We were able to clearly differentiate the two species and highlight fine-scale genetic structure among different O. rufipogon ecotypes (Supplementary Fig. S1).
4 CONCLUSION
Although sequencing costs are decreasing, NGS of large samples is still expensive causing many researchers to focus on low-depth samples. This is particularly true for nonhuman nonmodel organisms for which research funding typically does not provide for deep sequencing of many individuals. Analyses of data from such species are particularly challenging because imputation-based methods used in human genomics are not available and because they may suffer from high levels of inbreeding. This beckons for new and efficient computational methods that directly address the problem of genotyping uncertainty on NGS data.
The methods provided by ngsTools are designed with this problem in mind. ngsTools provides tools to accurately estimate genetic variation in case of low-coverage sequencing data. The individual methods have been previously tested providing extensive documentation of their statistical and computational properties.
We here report on the availability of an integrated open source computer package facilitating access to the methods for the broader research community. ngsTools is available on a public repository for shared development so that additional methods can be developed under this framework and integrated into the software package.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Thorfinn Korneliussen and Anders Albrechtsen for helpful discussions and assistance using ANGSD.
Funding: This work was supported by a NIH (3R01HG03229-07) grant to R.N., an EMBO Long-term Fellowship ALTF 2011-229 to M.F. and an NIH Genomics Training Grant (T32HG000047-13) to T.L.
Conflict of Interest: none declared.
REFERENCES
- Fumagalli M, et al. Quantifying population genetic differentiation from next-generation sequencing data. Genetics. 2013;195:979–992. doi: 10.1534/genetics.113.154740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, et al. A map of rice genome variation reveals the origin of cultivated rice. Nature. 2012;490:497–501. doi: 10.1038/nature11532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, et al. SNP calling, genotype calling, and sample allele frequency estimation from new-generation sequencing data. PLoS One. 2012;7:e37558. doi: 10.1371/journal.pone.0037558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira FG, et al. Estimating inbreeding coefficients from ngs data: impact on genotype calling and allele frequency estimation. Genome Res. 2013;23:1852–1861. doi: 10.1101/gr.157388.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.