

Abstract
The mzQuantML standard from the HUPO Proteomics Standards Initiative has recently been released, capturing quantitative data about peptides and proteins, following analysis of MS data. We present a Java application programming interface (API) for mzQuantML called jmzQuantML. The API provides robust bridges between Java classes and elements in mzQuantML files and allows random access to any part of the file. The API provides read and write capabilities, and is designed to be embedded in other software packages, enabling mzQuantML support to be added to proteomics software tools (http://code.google.com/p/jmzquantml/). The mzQuantML standard is designed around a multilevel validation system to ensure that files are structurally and semantically correct for different proteomics quantitative techniques. In this article, we also describe a Java software tool (http://code.google.com/p/mzquantml-validator/) for validating mzQuantML files, which is a formal part of the data standard.
Keywords: Bioinformatics, Java API, mzQuantML, Proteomics Standards Initiative, XML
The two most common tasks performed in proteomic research are the identification or quantification of proteins or peptides, using MS followed by data analysis. The Proteomics Standards Initiative (PSI) has been working for a number of years to develop data standards to assist data sharing, software development, and database submissions for the different data types produced in these typical workflows. The PSI has released mzML for raw MS data or peak lists [1], mzIdentML for peptide and protein identification, for example exported from a search engine [2], TraML [3] for encoding transition lists and associated metadata, and, recently, mzQuantML for quantitative data [4]. The model is developed as an Extensible Markup Language (XML) Schema Definition file, accompanied by controlled vocabulary (CV) terms and definitions as part of the PSI-MS CV [5], also used in mzML, mzIdentML, and TraML. The use of correct CV terms within the schema is governed by a mapping file, defining the terms that MUST, SHOULD, or MAY (formal keywords) used at particular locations within a file.
Four quantitative proteomics techniques are currently supported in mzQuantML version 1.0: (i) intensity-based (MS1) label free, (ii) MS1 label-based (such as SILAC or N15), (iii) MS2 tag-based (iTRAQ or tandem mass tag), and (iv) spectral counting. A review on software support for quantitative proteomics can be found in [6]. All the latest example files can be found in http://code.google.com/p/mzquantml/source/browse/#svn/trunk/examples/version1.0. The support for SRM is in progress and will be soon submitted as an update to the current specifications. Each of these techniques is represented using the same core mzQuantML structures, but the types of structures that must be used for each technique are governed by a set of “semantic rules.” The semantic rules are written in natural language and define the elements that MUST, SHOULD, or MAY appear in the file for each technique along with some conditions.
The jmzQuantML application programming interface (API) is developed by following a similar design as jmzML [7] and jmzIdentML [8]. It is developed in Java, thus making it platform independent, using open source frameworks such as Java architecture for XML binding (JAXB), Maven 2, Log4j, and JUnit. The API is copyrighted under the Apache Software License 2.0 (http://www.apache.org/licenses/LICENSE-2.0.html). The two recommended ways for using jmzQuantML are by downloading the Java Archive (JAR) file and manually importing the JAR into a new Java project, or by exploiting Maven's dependency mechanism (details can be found in http://code.google.com/p/jmzquantml/). Full Java documentation of the API is available from http://jmzquantml.googlecode.com/svn/trunk/src/docs/api/index.html.
Proteomic data represented in mzQuantML can produce large files, since the format is able to capture quantitative data about proteins or protein groups, peptides, and features (regions of 2D LC-MS space), as well as all references between these elements to ensure a full trace of the steps performed by analysis software is retrievable. As such, if software packages attempted to load complete mzQuantML files into memory, they would run into memory overload problems. A technique called the xxIndex (http://code.google.com/p/pride-toolsuite/wiki/XXIndex) is employed in jmzQuantML to allow random access to any XML element in an mzQuantML file via its XPath (the hierarchy and location of an element within an XML file). With this technique, which is also used in jmzML and jmzIdentML, jmzQuantML API can extract individual elements via the xxIndexer (an index recording both start and end byte positions of each element) from large files stored on disk quickly, and use a low memory overhead. The same technique can also be used to build large files.
Reference resolving is another important feature of the API. Many elements within mzQuantML files contain references to other objects via a unique identifier. The handling of references within jmzQuantML is controlled by a configuration file. The initial stages of reference resolving are performed at the same time when the xxIndexer scans the file for creating the XPath-based index. Depending on the user preference stored in the configuration file, the API decides whether to resolve a reference by creating a full Java object of the referenced element (called autoRefResolving), or store only the unique (String) identifier of the referenced element (called idMapped). There is a default configuration file in the API where the reference resolving is enabled for all the objects by default. It may be desirable for memory saving purpose to turn off the reference resolving mechanism for elements expected to contain a large number of references to another object, such as <PeptideConsensus> elements (representing peptides quantified across replicates) referencing <Feature> elements (representing 2D regions of LC-MS space that have been quantified).
We have generated some example code to demonstrate how to use jmzQuantML for marshalling and unmarshalling mzQuantML files, which is available from http://code.google.com/p/jmzquantml/w/list. The API implements two main functions for mzQuantML files: marshalling and unmarshalling. Marshalling is the process of writing Java objects as XML fragments to an mzQuantML file stored on the disk, whereas unmarshalling is the reverse—reading XML fragments from an mzQuantML file into memory as Java objects. These functions are provided as two main classes: MzQuantMLMarshaller and MzQuantMLUnmarshaller. These classes provide more than one way of performing marshalling and unmarshalling operations in order to give more flexibility to users. Figure 1 illustrates the work-flow of marshalling and unmarshalling. Marshalling an mzQuantML object to a file is more straightforward than unmarshalling. The user first creates an MzQuantMLMarshaller object, then must follow one of two different paths: via the MzQuantML object (Path A in the Marshall flowchart in Fig. 1) or FileWriter (Path B in Fig. 1). Path A requires the user to create an empty MzQuantML object as the root element to which all other element objects are attached. The element objects are generated by the JAXB compiler directly from the mzQuantML schema definition file. Each element object provides setter and getter methods for populating data into correct and valid attribute types. The relationship between the mzQuantML elements and the Java objects is illustrated in Fig. 2. All the required element objects are added to the root object before marshalling the whole object to an mzQuantML file. This method is only recommended for marshalling small files, as the entire object tree becomes loaded into memory. For large file, API users are recommended to follow the FileWriter method (Path B). By using a FileWriter, no memory issues occur and files of almost any size can be produced. The marshalling process in Path B functions via writing each required blocks of elements direct to file, for example the process starts by writing a start tag (MzQuantMLMarshaller.createMzQuantMLStartTag) and finishes by writing closing tag (MzQuantMLMarshaller.createMzQuantMLClosingTag). This method for file writing requires API users to be aware of the order of elements that must be written to create a valid mzQuantML file. The recommended order can be found inspecting the mzQuantML XML Schema. For jmzQuantML users following Path A, the order of element writing is not important.
Figure 1.
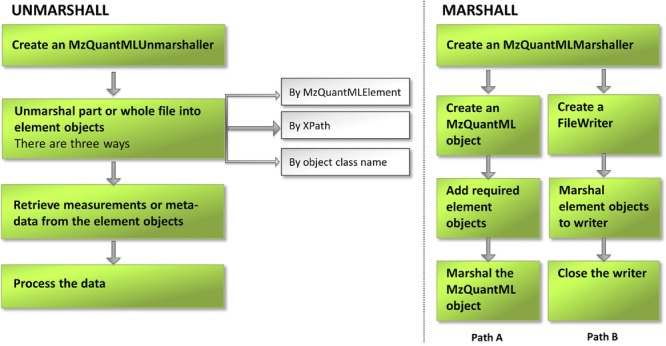
Diagram of the steps for marshalling and unmarshalling mzQuantML files using the jmzQuantML API, 250 × 129 mm (300 × 300 DPI).
Figure 2.

An example illustrating the relationship between the <ProteinList> element in the XML Schema Definition and the ProteinList object class in jmzQuantML, 99 × 70 mm (600 × 600 DPI).
The unmarshalling workflow is shown in left panel of Fig. 1. An MzQuantMLUnmarshaller object must be created to read from the given file. The class equips the user with some useful functions to explore an mzQuantML file (e.g. getMzQuantMLId, getMzQuantMLName, and getMzQuantMLVersion). The unmarshal method can be invoked from one of three parameter types: (i) via a named object type within the MzQuantMLElement class (e.g. MzQuantMLElement.AuditCollection), an XPath string describing the location of the element in the file (e.g. /MzQuantML/AnalysisSummary), or the class name of an element object (e.g. uk.ac.liv.jmzqml.model.mzqml.CvList.class), and it returns a Java object wrapping the element type and attributes from the values in the file. For objects that appear in multiple occurrences in a file, jmzQuantML provides a method (unmarshalCollectionFromXpath) for retrieving a list of objects that can be iterated over. Through any of these options, jmzQuantML processes the data in the file, returning Java objects to the user of the API for onward processing in the local application.
Although mzQuantML version 1.0 has only been a PSI standard since March 2013, there are already several software packages supporting the format, which use jmzQuantML internally, such as ProteoSuite (http://www.proteosuite.org/), x-Tracker (http://www.x-tracker.info/) and the Progenesis Post-Processor [9]. A tutorial describing how to develop software using PSI standards can be found in [10].
There is no guarantee that an mzQuantML file created by the jmzQuantML API is semantically valid, as the schema is designed with flexibility and future extensibility in mind and mzQuantML requires multilevel validation, which is outside the scope of a typical API. Specifically, as part of the formal mzQuantML standard, we require formal validation software to check three levels of validation: (i) a file is valid against the XML Schema, (ii) correct CV terms are used in the correct locations of the file, and (iii) the semantic rules are correctly fulfilled.
As an example of the CV validation requirement, the <AnalysisSummary> element in each file must contain a CV term specifying the quantitative technique used for the experiment, e.g. “LC-MS label-free quantitation analysis” (MS:1001834) or “MS1 label-based analysis” (MS:1002018). These CV mapping rules are captured in an XML file as a formal part of the standard, defining which CV terms MUST, SHOULD, or MAY be present at each location in the file that has the potential to be parameterized. As an example of a semantic rule—for an MS2 tagging-style file (e.g. iTRAQ data)—“If PeptideConsensusList is present there MUST be a FeatureList present and there MUST be an MS2AssayQuantlayer present.” The rule states the general mzQuantML structures that must be present in an iTRAQ-style file and if they are absent a fatal error will result.
We have developed an mzQuantML validator (http://code.cgoogle.om/p/mzquantml-validator/) to ensure that software packages export data in a consistent way. The validator checks if an mzQuantML file is syntactically and semantically valid and has used appropriate CV terms. The validator is implemented using jmzQuantML and the PSI validator framework [11]. The validator comes with a basic graphical interface, as it is intended as a tool for developers rather than laboratory scientists. When a user loads a file, the validator will process the file against the different rules, and export messages at different levels of severity. Since XML schema validation (using any known method) can be slow for very large mzQuantML files, the validator allows the user to skip schema validation and process semantic validation rules only if desired. We have tested the validator with file sizes up to 150 MB, which takes approximately 1 min on a standard desktop PC without schema validation and several hours with schema validation. The validator is particularly relevant for software developers adding mzQuantML export capabilities to data analysis software; since the mzQuantML specifications are large and complex and without robust validation software, there is a danger of producing invalid files. We also produced valid and invalid test files in our repository for users to test. The header of each file contains information about the expected errors. We encourage users to try the validator on these files and see how the validator reports the errors encountered.
We present two Java-based open source resources, named the jmzQuantML API and the mzQuantML validator, to support the new mzQuantML standard from the HUPO-PSI. The jmzQuantML API follows the successful design pattern employed in APIs for other PSI standards that are now in common use in a variety of software toolkits, enabling rapid development of import and export capabilities. The mzQuantML validator will be an essential resource for all developers wishing to add mzQuantML export capabilities to their software packages.
Acknowledgments
This work was funded by BBSRC grants to A.R.J. (BB/I00095X/1, BB/H024654/1, and BB/K004123/1) and the EU FP7 project “ProteomeXchange” (grant number 260558). We also thank the help from Jun Fan, Gerhard Mayer, and Faviel Gonzalez for testing the API and reporting bugs.
Glossary
- API
application programming interface
- CV
controlled vocabulary
- PSI
Proteomics Standards Initiative
- XML
Extensible Markup Language
The authors have declared no conflict of interest.
References
- [1].Martens L, Chambers M, Sturm M, Kessner D, et al. mzML—a community standard for mass spectrometry data. Mol. Cell. Proteomics. 2011;10:R110.000133. doi: 10.1074/mcp.R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jones AR, Eisenacher M, Mayer G, Kohlbacher O, et al. The mzIdentML data standard for mass spectrometry-based proteomics results. Mol. Cell. Proteomics. 2012;11:M111.014381. doi: 10.1074/mcp.M111.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Deutsch EW, Chambers M, Neumann S, Levander F, et al. TraML–a standard format for exchange of selected reaction monitoring transition lists. Mol. Cell. Proteomics. 2012;11:R111 015040. doi: 10.1074/mcp.R111.015040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Walzer M, Qi D, Mayer G, Uszkoreit J, et al. The mzQuantML data standard for mass spectrometry-based quantitative studies in proteomics. Mol. Cell. Proteomics. 2013;12:2332–2340. doi: 10.1074/mcp.O113.028506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mayer G, Montecchi-Palazzi L, Ovelleiro D, Jones AR, et al. The HUPO proteomics standards initiative-mass spectrometry controlled vocabulary. Database. 2013;2013:bat009. doi: 10.1093/database/bat009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gonzalez-Galarza FF, Lawless C, Hubbard SJ, Fan J, et al. A critical appraisal of techniques, software packages, and standards for quantitative proteomic analysis. Omics. 2012;16:431–442. doi: 10.1089/omi.2012.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cote RG, Reisinger F, Martens L. jmzML, an open-source Java API for mzML, the PSI standard for MS data. Proteomics. 2010;10:1332–1335. doi: 10.1002/pmic.200900719. [DOI] [PubMed] [Google Scholar]
- [8].Reisinger F, Krishna R, Ghali F, Rios D, et al. jmzIdentML API: a Java interface to the mzIdentML standard for peptide and protein identification data. Proteomics. 2012;12:790–794. doi: 10.1002/pmic.201100577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Qi D, Brownridge P, Xia D, Mackay K, et al. A software toolkit and interface for performing stable isotope labeling and top3 quantification using Progenesis LC-MS. Omics. 2012;16:489–495. doi: 10.1089/omi.2012.0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gonzalez-Galarza FF, Qi D, Fan J, Bessant C, Jones AR. A tutorial for software development in quantitative proteomics using PSI standard formats. Biochimica et Biophysica Acta. 2014;1844:88–97. doi: 10.1016/j.bbapap.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Montecchi-Palazzi L, Kerrien S, Reisinger F, Aranda B, et al. The PSI semantic validator: a framework to check MIAPE compliance of proteomics data. Proteomics. 2009;9:5112–5119. doi: 10.1002/pmic.200900189. [DOI] [PubMed] [Google Scholar]