

Abstract
Due to their role in cellular signaling mitogen activated protein (MAP) kinases represent targets of pharmaceutical interest. However, the majority of known MAP kinase inhibitors compete with cellular ATP and target an ATP binding pocket that is highly conserved in the 500 plus representatives of the human protein kinase family. Here we review progress toward the development of non-ATP competitive MAP kinase inhibitors for the extracellular signal regulated kinases (ERK1/2), the c-jun N-terminal kinases (JNK1/2/3) and the p38 MAPKs (α, β, γ, and δ). Special emphasis is placed on the role of computational methods in the drug discovery process for MAP kinases. Topics include recent advances in X-ray crystallography theory that improve the MAP kinase structures essential to structure-based drug discovery, the use of molecular dynamics to understand the conformational heterogeneity of the activation loop and inhibitors discovered by virtual screening. The impact of an advanced polarizable force field such as AMOEBA used in conjunction with sophisticated kinetic and thermodynamic simulation methods is also discussed.
INTRODUCTION
Protein kinases are important drug targets due to their critical roles in cellular signaling. A number of protein kinase inhibitors, such as Iressa (gefitinib), Tarceva (erlotinib), Gleevec (imatinib), and Tykerb (lapatinib) are in clinical use for the treatment of cancer [1, 2]. Imatinib, for example, is approved for the treatment of chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GIST). Each of these inhibitors competes with ATP for binding to the ATP binding site.
In this review we examine progress toward the discovery of drugs that target mitogen activated protein (MAP) kinases, mainly from a computational perspective. MAP kinases are serine/threonine-specific protein kinases that are activated in response to extracellular stimuli [3]. Important MAP kinases include extracellular signal regulated kinases (ERK1/2), the c-jun N-terminal kinases (JNK1/2/3) and p38 MAPKα, β, γ, and δ. MAP kinase signaling pathways are critical targets for a number of human diseases including cancer, diabetes, neurodegenerative and inflammatory diseases [3]. Here we focus on the potential for developing non-ATP competitive inhibitors that target sites such as the MAPK recruiting sites [4]. Such ligands have potential to regulate MAPK activities by blocking the ability of MAPKs to bind upstream activating kinases, scaffold proteins or downstream substrates.
A significant advantage of non-ATP competitive ligands over ATP competitive ligands is that they are not in competition with cellular ATP for binding to active sites. In addition, MAP kinase substrate binding sites may offer unique specificity against other families of kinases. On the other hand, it may be more difficult to design potent non-ATP competitive inhibitors due to protein docking sites being largely solvent exposed and relatively shallow [2]. It is also a challenge to identify inhibitors that are selective within the MAPK family. However, we see increasing evidence, including our own unpublished work that suggests that the substrate/upstream protein-docking sites of MAPKs are drugable. We suggest, that detailed computational studies when combined with NMR and X-ray crystallography can furnish insights into the subtle features of MAPK structure and dynamics that will be important for the design of potent non-ATP competitive MAPK inhibitors.
Below we first review computational and experimental studies that have contributed to our current understanding of MAP kinase structure and dynamics. Next, progress toward the design of non-ATP competitive inhibitors is discussed for ERKs, JNKs and p38 MAPKs. Emphasis is placed on the current and future roles of advanced computational modeling methods.
THE STRUCTURE AND DYNAMICS OF MAP KINASES
We begin this section by presenting the conserved features of MAP kinases using the combination of a multiple sequence alignment and a structure of JNK1. Following this introduction, we examine the ability of modern molecular biophysics theory to optimize structural models from X-ray crystallography experiments and discuss implications for the structure-based design of MAPK inhibitors. We then discuss conformational differences between inactive and active MAP kinases based on both structural evidence and molecular dynamics simulations. Finally, non-ATP competitive MAP kinase inhibitors that rely on interactions outside the ATP binding pocket are reviewed.
The Sequence and Structure of MAP Kinases
A multiple sequence alignment of ERK1/2, JNK1/2/3 and p38 MAPKα/β/γ/δ is shown in Figure 1 and serves as a guide to both the conserved and variable regions within the binding sites discussed throughout this review [5]. Although the structure of protein kinase family members has been extensively studied, only a single structure is available for many MAP kinase isoforms. Based on the profile hidden Markov model (HMM) Pkinase/PF00069 defined in Pfam v. 25, there are 1575 protein kinase family structures in the protein data bank (PDB) as of March 2011 [6]. The MAP kinase subset includes structures of ERK1 (1), ERK2 (16), JNK1 (7), JNK2 (2), JNK3 (23), p38 MAPKα (129), p38 MAPKβ (3), p38 MAPKγ (1) and p38 MAPKδ (1). Therefore, more than twice as much structural work has been completed for p38 MAPKα than the other MAP kinases combined (129 versus 53).
Figure 1.

Shown is a multiple sequence alignment of human ERK1, ERK2, JNK1α2, JNK2α2, JNK3α2 and p38 MAPKα (CSBP2), p38 MAPKβ, p38 MAPKγ and p38 MAPKδ. The shades of blue indicate the degree of conservation for each residue and darker shades correspond to greater conservation. Note that the first 38 n-terminal residues of JNK3α2 and a variable number of c-terminal residues on each MAP kinase are not shown.
As a complement to the sequence overview, we introduce conserved structural features of MAP kinases based on the structure of JNK1 (PDB ID 3O17) shown in Figure 2A. As for all MAP kinases, it is composed of two domains. The N-terminal domain has ~135 residues and is made up mainly of β-sheets. The β1L0 and β2L0 strands of loop 0 (L0) precede a set of five conserved strands of the broader protein kinase family (β1 to β5) [7]. The final N-terminal strand β5 leads into a linker region that connects the N-terminal domain with the C-terminal domain and controls their relative orientation. The C-terminal domain is composed of ~225 residues that mainly form α-helices. The active site of MAP kinases is located between the two domains and is heavily influenced by the conformation of loop 12 (L12), which has also been referred to as the activation loop or phosphorylation lip. The β8 strand leads into three residues (DFG) whose conformation is critical for enzyme activity. Finally, we note the MAP kinase insert (helices α1L14 and α1L14) that is absent in other protein kinases, but has been shown to contribute important interactions with both MAP kinase regulators [8, 9] and substrates [10].
Figure 2.

A.) JNK1 (PDB ID: 3O17) is shown with each segment of secondary structure labeled. The subscripts for secondary structure elements that are not conserved for all protein kinases refer to loop number. For example, the first β-strand of Loop 0 is β1L0. B.) Ewald refinement of the original experimental data using prior chemical information defined by the polarizable AMOEBA force field improves the quality of the structural model via extension of energetically favorable secondary structure elements and by improving agreement with the measured diffraction data (ie. reducing Rfree from 30.6 to 27.9). The 3 β-strands and 4 α-helices that were extended are labeled. Changes near the DRS-site (Φchg) and FRS-site (αg, activation loop) may impact downstream computational methods such as virtual screening of non-ATP MAP kinase inhibitors.
Ewald X-Ray Crystallography Refinement of MAP Kinase Structures
The rationale design of potent and specific MAP kinase inhibitors benefits from precise protein-inhibitor structural models derived from X-ray crystallography diffraction data. The reliability of structure-based virtual screening for the discovery of lead compounds diminishes as the resolution of the X-ray data used to generate the structural model is reduced (~2.5 Å or better may be preferable) [11, 12]. Furthermore, once lead compounds are identified, their further optimization depends in part on understanding the water hydrogen-bonding network in the neighborhood of the MAP kinase-inhibitor binding site [13, 14]. For these reasons, we review recent advances in X-ray crystallography refinement and their impact on MAP kinase structure quality.
Over the last decade polarizable force fields [15, 16] have been developed that are more transferable across chemical environments (i.e. vacuum, liquid and crystalline phases) than fixed charged descriptions of biomolecular energetics traditionally used for X-ray crystallography refinement [17–19]. In addition, the particle-mesh Ewald (PME) method has revolutionized the efficient, rigorous evaluation of periodic, long-range electrostatic interactions [20–23]. For example, the polarizable Atomic Multipole Optimized Energetics for Biomolecular Applications (AMOEBA) force field [24, 25] evaluated using PME [26] has recently been coupled to X-ray crystallography refinement using the program Force Field X [26, 27]. This approach will simply be referred to as Ewald refinement because it represents the first application of PME electrostatics to biomolecular X-ray crystallography refinement. Ewald refinement has been shown to improve R/Rfree statistics and model geometry for high resolution peptides [28] and proteins [29], the joint refinement of X-ray plus neutron diffraction data sets [30], and low-resolution biomolecular data sets [26].
To illustrate the usefulness of these recent advances in X-ray crystallography theory for structure-based design of MAP kinase inhibitors, Ewald refinement was performed for a subset of five MAP kinase structures with an emphasis on complexes with non-ATP competitive inhibitors. The mean resolution of the data sets (shown in Table 1) was 2.5 Å and they were originally refined to mean R and Rfree values of 22.6 and 27.4 respectively. The MolProbity structure validation tool [31, 32] was applied to the deposited structural models, which quantifies van der Waals clashes, side-chain rotamer orientation and polypeptide backbone (φ,ψ) conformation. The MolProbity score is calibrated to indicate the expected experimental resolution of the structural model. For the five deposited MAP kinase structures, the mean MolProbity score (2.9) is in the 51st percentile and indicates model quality that is approximately what is expected for experimental data to 2.9 Å (ie. half of the deposited structures at this resolution have better scores and half worse). After Ewald refinement, the mean R and Rfree values improved to 20.3 and 26.0, respectively. In addition, the mean MolProbity score (1.9) was improved to the 97th percentile and indicates model quality consistent with experimental data collected to 0.6 Å higher resolution than what was actually measured. Consistent with previous findings, Ewald refinement of MAP kinases achieved more physical models that agree more closely with the experimental data.
TABLE 1.
Shown are improvements in JNK1 and JNK2 protein-inhibitor structural models using Ewald refinement. The R/Rfree statistics for the deposited PDB structures are shown as originally reported and after recalculation using the Force Field X program. After Ewald refinement, the final structural models improved the mean Rfree value relative to the recalculated Rfree by 1.4% on average. In addition, the mean MolProbity score was improved from the 51rst percentile to the 97th percentile (see the text for a description of the MolProbity structure validation tool).
| PDB / Ligand | Original Refinements
|
Ewald Refinements
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Res (Å) | R / Rfree | MolProbity
|
Recalculated R / Rfree | Final R / Rfree | MolProbity
|
RMSD (Å) | |||
| Score | % | Score | % | ||||||
| 3O17 / Apo | 3.0 | 26.3 / 30.8 | 3.9 | 17 | 25.5 / 30.6 | 22.9 / 27.9 | 2.3 | 97 | 0.38 |
| 3O2M / 46A | 2.7 | 23.3 / 26.6 | 3.3 | 30 | 22.8 / 26.4 | 19.7 / 24.9 | 2.3 | 94 | 0.25 |
| 3NEW / 3NE | 2.5 | 21.9 / 30.6 | 3.0 | 42 | 22.0 / 30.6 | 21.6 / 29.2 | 1.6 | 99 | 0.40 |
| 3E7O / 35F | 2.1 | 21.9 / 25.0 | 2.1 | 77 | 21.7 / 24.6 | 18.4 / 23.0 | 1.6 | 97 | 0.39 |
| 3NPC / B96 | 2.4 | 21.4 / 25.4 | 2.1 | 88 | 21.0 / 24.7 | 18.8 / 25.0 | 1.7 | 98 | 0.16 |
|
| |||||||||
| Mean | 2.5 | 23.0 / 27.7 | 2.9 | 51 | 22.7 / 27.4 | 20.3 / 26.0 | 1.9 | 97 | 0.31 |
For example, Ewald refinement changed the JNK1 model shown in Figure 2A by extending energetically favorable secondary structure elements to give the structure shown in Figure 2B while improving the agreement with the measured diffraction data (ie. reducing Rfree from 30.6 to 27.9). The 3 β-strands and 4 α-helices that were extended are labeled. We expect that improvements seen near the DRS (Φchg) and the FRS (αg, activation loop) will impact downstream computational methods, such as structure-based virtual screening of non-ATP competitive MAP kinase inhibitors. In a later section, we discuss refinement of the water hydrogen-bonding network around MAP kinase inhibitors.
Conformational Dynamics of MAP Kinases
The conformational dynamics of a MAP kinase is central to processes such as its activation as well as its interactions with ligands and substrates [33, 34]. Many of the dynamic processes associated with MAP kinases are on the micro- to millisecond timescale. While computational and theoretical methods for examining long time dynamics are under active development [35], large-scale conformational changes with timescales of microseconds and beyond are still difficult to capture with atomic detail. Thus, the computational studies of the intrinsic dynamics of protein kinases, including the MAP kinases have been limited. However, some protein kinases including Abl, Src, and CDK have been investigated using targeted MD, biased sampling, String and coarse-grained methods with useful insights [36–40]. For example, molecular dynamics simulations using the String Method with Swarms-of-Trajectories, allowed Roux and colleagues to propose a stepwise mechanism of conformational transitions associated with Src kinase activation where the opening of the activation loop is followed by rotation of the αC helix [39]. Below we describe a number of conformational features of the MAP kinases, which have been described experimentally, but which lack significant detailed computational analysis.
Differences between active and inactive MAP Kinase conformations
A key conformational change of MAP kinases is driven by the phosphorylation of two residues within the activation loop between the conserved DFG and APE motifs (see the multiple sequence alignment of Figure 1). Upon phosphorylation of the threonine and tyrosine residues of the T-X-Y motif, activation loop reorientation is accompanied by other alterations such as lobe closure, αC helix rotation, and C-terminal reorganization [33]. It has been suggested that the allosteric coupling of these substructures prepares the kinase for ATP and substrate binding [41]. Structures of both inactive (PDB: 1ERK, Figure 3A) and active (PDB: 2ERK, Figure 3B) forms of ERK2 have been solved, which demonstrates the conformational changes of MAP kinases upon activation [42, 43]. The active structure is dual phosphorylated on the conserved T-X-Y motif (183T-E-Y185 for ERK2) within the phosphorylation lip (L12). The negative charges on the phosphorylated residues favor refolding of the phosphorylation lip via electrostatic interactions with surface arginine-rich binding sites. The phosphorylated tyrosine residue moves ~9 Å into the domain interface and contributes to a modest closure of the two domains [42, 43]. The phosphorylated tyrosine comes to the surface via a backbone rotation and forms the proline-directed P+1 specificity site [42, 43]. The amino acid sequence of ERK1 is 90% identical to ERK2 (see Figure 1) and is also a proline-directed Ser/Thr kinase that is activated via dual phosphorylation of the threonine and tyrosine residues of 202T-E-Y204. While there are currently 16 structures of ERK2 available, only a single ERK1 data set has been deposited in the PDB; that of the mono-phosphorylated Y204 form (PDB: 2ZOQ). This structure demonstrates a novel conformation that is distinguishable from the un-phosphorylated and dual-phosphorylated forms [44]. Despite the similarities between ERK1/2, the specific inhibition of ERK2 is more promising therapeutically because studies of knockout mice have shown that ERK2 can largely compensate for the absence of ERK1 [45, 46].
Figure 3.

A.) The inactive structure of ERK2 (PDB ID: 1ERK) shows the unphosphorylated T-X-Y motif (cyan) located on the activation loop (L12) of protein kinases (183T-E-Y185 for ERK1/2). B.) The active structure of ERK2 (PDB ID: 2ERK) has been phosphorylated on both the threonine and tyrosine residues that favor conformational rearrangements stabilized by electrostatic interactions between the negatively charged phosphorylated residues and positively charged arginine residues (grey).
Conformations associated with autophosphorylation
Protein kinases can be activated via phosphorylation by upstream kinases or through auto-phosphorylation (either through an inter- or an intra-molecular mechanism) [47]. While MAPKs are typically activated by an upstream MAP kinase kinase (MKK, MEK, or MAP2K), there has been increasing evidence that certain MAPKs are capable of auto-phosphorylation. For example, TAB1-mediated p38 MAPKα auto-phosphorylation in MEF cells accounted for a small portion of the total p38 MAPKα phosphorylation that was induced by hyperosmolarity and anisomycin [48]. It was further suggested, based on crystal structures of intrinsically active mutants, that the conformational change in loop 16 plays a critical role in inducing auto-phosphorylation through an intermolecular mechanism [49]. Similarly, ERK mutants capable of auto-phosphorylation have been reported [50]. Emrick et al. discovered that the mutation of a gatekeeper residue in ERK2 led to auto-phosphorylation. In this case phosphoryl transfer was suggested to occur through an intra-molecular mechanism [51]. It has been proposed that JNK2α2 auto-phosphorylates through an intermolecular mechanism [52].
Auto-phosphorylation may be stimulated through allosteric activation upon interaction with protein binding partners, such as scaffold proteins [47]. For example, a segment of Ste5 allosterically activated auto-phosphorylation of MAPK Fus3 [53]. Recently, we performed MD simulations of JIP1 peptide binding to JNK1 [54]. The simulations clearly demonstrated that the binding of pepJIP1 has a significant effect on the inter-domain motion and structure near the active site. Removal of pepJIP1 causes an increase in domain separation. Interestingly, the activation loop in apo JNK1 is similar to the inactive form of apo ERK2, while in the JNK1•L-pepJIP1 complex it resembles the active form of apo ERK2, or the inactive form ERK2 complexed to a docking peptide derived from pepHePTP [55].
Although essential for understanding MAP kinase activities and regulation under different conditions, the auto-phosphorylation mechanism is not well understood. Due to the dynamic nature of this molecular mechanism, computational studies could potentially bring critical insights, which will in turn open up new opportunities for MAP kinase based therapeutics.
Conformations associated with the DFG motif
The conformational flexibility of the conserved Asp-Phe-Gly (DFG) motif at the beginning of the activation loop (see Figures 1 and 2) has been increasingly explored in the structure-based design of kinase inhibitors. In order to illustrate this flexibility and compare inhibitors that stabilize different DFG conformations we introduce structures of the c-jun N-terminal kinases (JNK) [56]. In 1998, the first JNK structure was solved by Su et al. of JNK3, which demonstrated that misalignment of the catalytic residues and occlusion of the active site by the phosphorylation lip are consistent with the low activity of un-phosphorylated JNK3 [57]. Of the two JNK2 structures in the PDB, the first (PDB: 3E7O) is of a complex of JNK2 with N-[3-[5-(1H-1,2,4-triazol-3-yl)-1H-indazol-3-yl]phenyl]furan-2-carboxamide (Figures 4a and 4b) with the activation loop in a ‘DFG-in’ conformation consistent with catalysis [58]. The second (PDB: 3NPC) shows the complex of JNK2 with BIRB-796 with the activation loop in a ‘DFG-out’ conformation, which does not support catalysis (Figures 4c and 4d) [59].
Figure 4.

Shown in each panel is a MAP kinase structure complexed with an inhibitor (cyan, spacefill) that targets DFG-in or DFG-out (magenta, ball & stick) and the corresponding conformation of the activation loop (magenta, backbone only). A.) JNK2 in the DFG-in conformation is shown in a complex with type-I inhibitor N-[3-[5-(1H 1,2,4-triazol-3-yl)-1H-indazol-3-yl]phenyl]furan-2-carboxamide (PDB ID 3E7O). B.) Ewald refinement of A orients the water hydrogen-bonding network around the JNK2 inhibitor-binding site. C.) JNK2 in the DFG-out conformation in a complex with type-II inhibitor BIRB-796 (PDB ID: 3NPC). D.) Ewald refinement of C orients the water hydrogen-bonding network around the JNK2 inhibitor-binding site. E.) p38 MAPKα in the DFG-out conformation in a complex with BIRB-796 (PDB ID 1KV2). Ewald refinement was not performed for E because no diffraction data was deposited.
Ewald refinement was performed for both 3E7O and 3NPC in order to orient the water hydrogen-bonding network around the JNK2 inhibitor-binding site [14, 29]. This information can be used to optimize lead compounds by chemical modifications in order to displace water molecules that, for example, do not have access to a full complement of hydrogen bonding partners [14]. For example, Ewald refinement of JNK2 complexed with the carboxamide inhibitor (3E7O) orients three water molecules that hydrogen bond directly to the inhibitor (Figure 4B). Figure 4A show that waters 1 and 2 interact with three hydrogen-bonding partners, while water 3 only forms a single canonical hydrogen bond to the inhibitor. This suggests that water 3 may be in an energetically unstable environment, such that the appropriate chemical modification of the inhibitor could promote displacement of water 3 into bulk solvent, resulting in the tighter binding of the modified inhibitor. Similarly, Ewald refinement of 3NPC orients a bridging water molecule that may be displaced by the addition of a hydrogen-bond donating functional group to nitrogen N12 of BIRB-796 (Figure 4D). During the shift from ‘DFG-in’ to ‘DFG-out’, the phenylalanine (Phe-169) side chain is able to move from its buried conformation (Figure 4a) by about ~10 Å (Figure 4c) and create a new hydrophobic pocket, but this interferes sterically with ATP binding. In addition to the diaryl urea containing BIRB-796, imatinib, sorafenib and doramapimod have been shown to stabilize the DFG-out conformation of various protein kinases [60–64]. Inhibitors that stabilize DFG-out conformations are sought after, because they are generally more selective and display slower dissociation kinetics relative to inhibitors that stabilize the DFG-in conformation [65, 66].
Elcock and colleagues showed that in some 60 nanosecond explicit-solvent molecular dynamics simulations the DFG-in configuration transformed to DFG-out spontaneously [66]. This observation is consistent with an NMR study [67] that suggests the DFG-out configuration exists intrinsically, although rarely. Nonetheless, a steered molecular-dynamics study of p38 MAPKα suggested the inhibitors complex with the DFG-in or a transition state structure first, which then transforms to DFG-out. Furthermore, Kufareva and Abagyan [68] showed that key features of ‘DFG-out’ inhibitor binding are present in the DFG-in state such that it is possible to predict the bound state structure based on the DFG-in conformation alone. More recently, based on the combination of long MD simulations and experimental studies, Shan et al. proposed a mechanism where protonation of ASP triggers the conformational switch [40]. These calculations have brought unique insights and guidance that are beyond the reach of experiments albeit further studies of long-time dynamics under various conditions are needed to obtain a comprehensive picture of the DFG conformational properties.
Both activated and inactive MAPKs are important drug targets. While they each possess specific dynamic properties, they are intrinsically linked. Thus, the conformation of an activated MAPK may be viewed as a high-energy conformation of the inactive enzyme stabilized by phosphorylation. Some MAPKs appear capable of self-activating and whether this self-activation is through an intermolecular or intramolecular mechanism of autophosphorylation is generally not clear and requires further experimental investigation. There is evidence to suggest that some ligands may stabilize conformations of the inactive MAPK that more closely resemble the activated enzyme while some ligands may stabilize intrinsically inactive conformations where the DFG motif is incapable of supporting catalysis. Understanding the dynamics associated with the various conformational states of each MAPK are expected to be invaluable in the search for inhibitors that take advantage of the conformational flexibilities embedded within MAPK polypeptides. The computational modeling of these states is likely to be an important line of research over the next few years.
NON-ATP SITE INHIBITORS
MAP kinase inhibitors are considered a very important class of therapeutic. The majority of available MAP kinase inhibitors target the ATP binding site, which is highly conserved among more than 500 protein kinases. This conservation promotes cross reactivity and the possibility of undesirable toxicities that may limit their potential as drugs [69]. In 2007, Bain et al. profiled the selectivity of 65 compounds considered to be relatively specific kinase inhibitors against a panel of 70–80 protein kinases [70]. The p38 MAPK inhibitor SB 203580 was shown to inhibit c-Raf and GSK3 in vitro and the JNK inhibitor SP600125 was shown to inhibit 13 other kinases with similar potency including PKD1, CHK2, Aurora B and C, MELK, CK1, DYRK2, DYRK3 and HIPK3 [70]. This work highlights the difficulty of identifying highly specific ATP-competitive protein kinase inhibitors. Another limitation of such inhibitors is that they must compete with ~1 mM endogenous ATP, which has quite high affinity for MAP kinases [71]. Finally, more than 10,000 patents or patent applications for protein kinase inhibitors have been filed since 2001. Most of these are competitive with ATP, making it a challenge for drug discovery researchers to find novel chemical space [72].
In the last few years, drug discovery efforts by a number of groups have demonstrated progress towards validating kinase-binding sites that are distinct from the ATP binding site. MAPK interactions with upstream kinases and downstream substrates are mediated by recruitment sites found on the surface of MAPKs that are recognized by modular docking sequences [73]. When a canonical substrate-docking sequence binds a MAP kinase recruitment site, the effective concentration of the substrate Ser/Thr-Pro motif is locally increased near the active site. This, in turn, increases the rate of substrate turnover [74]. Disruption of the docking site interaction can lead to an overall reduction in the rate of substrate phosphorylation. For example, D-site peptides from MKK3 inhibit the ability of p38 MAPK to phosphorylate the transcription factors MEF2A and ATF2 [75]. Unlike peptide-based inhibitors, which cover a substantial surface area, the challenge for the development of small molecule inhibitors is to target recruitment sites located on the protein kinase’s surface in such a manner that the docking process may be inhibited. A number of small molecule non-ATP competitive inhibitors have been described. In some cases in silico screening methods were the principal vehicle for their discovery.
In this section we describe the non-ATP competitive MAPK inhibitors that have been reported in the literature for ERK1/2, the JNKs and the p38MAPKs as shown in Figure 7. We also highlight the first model of a MAPK-substrate complex, which has proved to be useful in our laboratory for interpreting the mechanisms of non-ATP competitive inhibitors of ERK2.
Figure 7.

2D structures of non-ATP competitive MAP kinase inhibitors discussed throughout the text.
Extracellular Signal-Regulated Kinases
ERK2-substrate model
In recent work, we have studied the ERK2 recruitment sites in detail utilizing a range of biochemical approaches [4, 10, 76-79]. ERK2 is known to have two recruitment sites called the D-recruitment site (DRS) and the F-recruitment site (FRS) (Figures 2 and 6), which bind the D-site and F-site of protein substrates, respectively [79, 80]. The DRS is an extended peptide-binding groove found on the rear face of the MAPK, while the FRS is a shallow hydrophobic pocket found adjacent to the Ser/Thr-Pro binding site [4]. The docking sequence that targets the D-site is a modular motif formed of both hydrophobic and basic residues (€)2-3-X2-6-φA-X-φB, where φ is a hydrophobic residue and € is a basic residue. The F-site docking sequence ψXψP is smaller, where ψ is an aromatic residue [81, 82]. The structural basis of the interaction between the ERK DRS and its docking sequence has been studied using structures from X-ray crystallography [83]. The basic residues of D-site docking motifs bind to negatively charged surface residues such as Asp-316 and Asp-319 in the region identified as Φchg in Figure 5. The hydrophobic residues of the D-site docking motif (φA-X-φB) bind the nearby hydrophobic pocket Φhyd.
Figure 6.
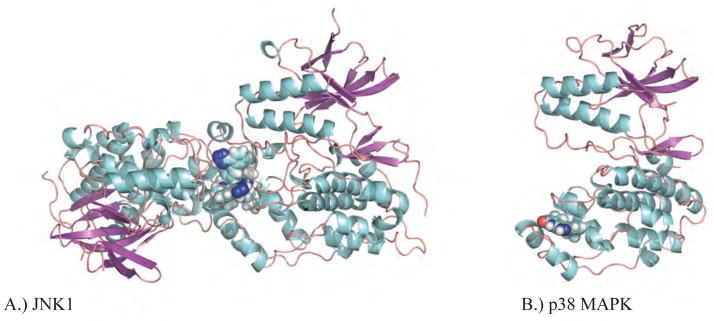
Shown in A and B are Ewald refinements of two MAP kinase complexes with non-ATP inhibitors that bind at the FRS-site. A.) The structure of the α isoform of JNK1 in complex with inhibitor 46A bound (PDB ID: 3O2M), which exhibits extensive interactions with a second copy of itself within the asymmetric unit of the crystal. B.) A promising structure of p38 MAPKα with inhibitor 3NE bound (PDB ID: 3NEW).
Figure 5.

The D-recruitment sites of A.) ERK2 with pepHePTP (PDB ID: 2GPH), B.) JNK1 with pepJIP1 (PDB IDs: 1UKH, 1UKI, 2G01, 2GMX, 2H96, 2NO3) and C.) p38 MAPKα with pepMKK3B (PDB ID: 1LEZ) / pepMEF2A (PDB ID: 1LEW) are shown. Qualitatively, to the right of the conserved C163 residue is a hydrophobic patch that contributes affinity to the interaction of MAP kinases with scaffolding proteins and to the left are interactions with negatively charged aspartate and/or glutamate residues. The superposition of JNK1 structures shows consistency for pepJIP1 residues critical to binding, but significant conformational heterogeneity otherwise.
No crystallographic structures of an active MAPK in complex with a substrate have been reported, possibly reflecting the high flexible of these complexes. As an alternative approach towards understanding MAPK-substrate interactions we modeled the ERK2•Ets-1 complex using experimental observations to guide the modeling [79]. Modeling showed that although the N-terminus of Ets-1 does not include the consensus docking sequence (€)2-3-X2-6-φA-X-φB for ERK2, it makes an important contribution to the binding of Ets-1 to ERK2 [4]. This was confirmed by NMR [79]. Lig-D (FQRKTLQRRNLKGLNLNL) is a peptide that binds the DRS of ERK2 and affects the Km, but not the kcat of Ets-1 phosphorylation by ERK2. Testing of the kinetic mechanism of inhibition by Lig-D showed a hyperbolic signature that suggests that Lig-D partially inhibits the ability of ERK2 to phosphorylate Ets-1 by displacing the N-terminus of Ets-1. In contrast, Lig-F (YAPRAPAKLAFQFPSR), which contains the F-site docking sequence (ψXψP), exhibits a mechanism of full competitive inhibition and effectively blocks the binding of Ets-1 to ERK2. Understanding the mechanisms of MAPK-substrate binding should facilitate the utilization of computational methods to identify small molecule inhibitors.
In silico approaches identify small molecules that bind the ERK2 DRS
Unlike the ATP binding site, targeting of the DRS or FRS has the potential to identify inhibitors that are specific for ERK. In 2005, Hancock et al. reported the first non-ATP competitive small molecule inhibitor of ERK identified by computer-aided drug design (CADD) [84]. By using the program SPHGEN to detect the possible binding sites on the surface of inactive ERK2 in combination with mutagenesis experiments, they recognized important residues within the Φchg (Asp-316 and Asp-319) and Φhyd regions (Thr157 and Thr158) of the DRS (Figure 5A) involved in intermolecular interactions with protein substrates. These observations served as the basis for the in-silico screening of 800,000 compounds and ultimately the selection of 80 compounds for the in vitro testing of biological activity. Compounds were assayed for their ability to inhibit the phosphorylation of p90Rsk-1 by ERK2 in HeLa cells. Compounds 1 and 2 (Figure 7) were identified as ERK2 inhibitors with an ability to inhibit p90Rsk-1 phosphorylation by more than 50% at a concentration of 100 μM. The binding of compound 1 to ERK2 was confirmed using a fluorescence-quenching assay (Kd = 5 μM). However, the direct binding of 2 has not been established. Compound 1 is predicted to contact the polar groove between Asp-316 and Asp-319 of the Φchg region and Thr-157 and Thr-158 of the Φhyd region and is suggested to have additional π-charge interactions with Arg-133.
In 2006, Chen et al. reported the characterization of another 13 compounds discovered via in silico screening of the same library against active ERK2 using the same docking site as described above for 1 [85]. Many of the compounds that were identified as targets for inactive ERK2 were also identified as targets for active ERK2, consistent with the notion that minimal conformational changes occur within the DRS of ERK2 upon activation. We note that ERK2 is a monomer in both the active and inactive forms [86]. Compounds 3 and 4 showed 70% inhibition of Elk-1 and 50% inhibition of p90Rsk-1 phosphorylation in HeLa cells at a concentration of 100 μM. Furthermore, these two compounds both showed clear selectivity for ERK2 inhibition versus p38 MAP kinase in HeLa cells. Compounds 3 and 4 were shown to directly bind ERK2 with a Kd of 13 and 16 μM, respectively.
In 2011, Boston et al. used a computational search to identify additional compounds exhibiting similar chemical features to compound 1 [87]. MAC-BITS fingerprints in conjunction with a Tanimoto Similarity index were employed to search a virtual database of more than a million commercially available compounds. Ten compounds were identified with close similarity to compound 1 and tested for their biological activity. Compounds 5 and 6 inhibited caspase-9 and p90Rsk-1 phosphorylation by ERK in HELA cells at 50 μM and inhibited HeLa cell proliferation at 100 μM. The inhibitory effect of these compounds on caspase-9 and p90Rsk-1, which are regulators of the pro-apoptotic Bad protein, caused induction of HeLa cell apoptosis.
In 2009, Li et al. synthesized a series of analogues of 1 in order to study its structure-activity relationship [88]. It was discovered that a shift of the ethoxy substitution from the 4 position to the 2 position on the phenyl ring (compound 7) improved its biological activity and specificity towards ERK over JNK and p38 MAPK. Compound 7 demonstrated an enhanced ability to inhibit U937 cell proliferation (human leukemia cells) and Elk-1 phosphorylation in U937 cells.
Based on our understanding of how MAP kinase kinases recognize MAPKs it is expected that inhibitors that target the DRS of MAP kinases have potential to inhibit both the activation of MAP kinases as well as their catalytic function. Although compounds 1–7 were able to inhibit the phosphorylation of protein substrates such as Elk-1 and/or p90Rsk-1 by ERK2, they were not found to inhibit the phosphorylation of ERK2. Further work is necessary to understand this interesting observation. The biological evaluation of compounds 1–7 confirms the usefulness of both structure-based and ligand-based computational approaches to the discovery of selective MAP kinase inhibitors that target a protein-binding site and demonstrates that in-silico drug discovery can translate into cellular activity [89].
c-Jun N-Terminal Kinases
JNK-Jip interactions
The JNK Interacting Protein (JIP) scaffolds JIP1-4 bind to JNK and serve to enhance its activation in vivo. One of the best-known classes of non-ATP competitive inhibitors of JNK is the JIP-based peptide inhibitor (see Figure 5b). JIP-based peptide inhibitors are derived from the 11-residue sequence (153RPKRPTTLNLF163) that corresponds to the single DRS docking domain of JIP1. This peptide, called pepJIP1, acts as a specific inhibitor of JNK [90] and inhibits the phosphorylation of substrates such as c-Jun, Elk, and ATF2 in in vitro cell-free assays. JIP1 exhibits selectivity for the JNKs versus the closely related MAP kinases ERK and p38 MAPK [90].
Crystal structures of the pepJIP1-JNK1 complex provide insight into the mode of JIP1 binding to the DRS of JNK (Figure 5b) [90]. Molecular dynamics simulations and alanine-scanning identified four residues (R156, P157, L160 and L162) that are crucial for the binding of pepJIP1 to JNK [54]. In one of our recent reports we engineered new JIP-based peptide inhibitors that demonstrate specificity for the JNK2-isoform with an IC50 ~90 nM. Using a flexible six carbon linker 6-aminohexanoyl (AHX) these peptides fused the JIP sequence to either the N-terminus of an inverted TAT sequence (JIP10-Δ-TATi: Ac-PKRPTTLNLF-AHX-RRRQRRKKRG-amide) or to a poly-arginine sequence (JIP10-Δ-R9: Ac-PKRPTTLNLF-AHX-RRRRRRRRR-amide) [91]. Both peptides were shown to significantly inhibit JNK activation and c-Jun phosphorylation in HEK293T cells. For example, a 5 μM concentration of either peptide is sufficient to inhibit the activation/phosphorylation of cellular JNKs by ~ 90%. This confirms the hypothesis that the targeting of the DRS of JNK represents a viable approach for the inhibition of JNK signaling in vivo.
Small molecules that bind the JNK DRS and block JNK-Jip interactions
In 2008, Stebbins et al. identified the thiadiazole BI-78D3 (8) as the first small molecule to target the JNK-JIP interaction [92]. BI-78D3 was identified as a non-ATP inhibitor of JNK that efficiently displaces biotinylated pepJIP1 from GST-JNK1 with an IC50 of 500 nM. Molecular modeling studies suggested that the benzodioxan moiety of BI-78D3 binds a small pocket in the DRS of JNK [92] characterized by an extensive hydrogen-bonding network to Arg-127 of JNK1. BI-78D3 exhibits dose-dependent inhibition of the phosphorylation of JNK substrates, both in vitro and in cells, with selectivity for JNK over p38 MAPKα, mTOR and PI3-kinase. BI-78D3 inhibits the TNF-alpha-induced phosphorylation of c-Jun in a dose-dependent manner in HeLa cells with an IC50 of 12.5 μM. Moreover, BI-78D3 abolishes ConA-induced liver damage and restores insulin sensitivity, compared to a vehicle control in a mouse model. In 2009, De et al. discovered a new series of 2-thio-benzothiazoles, which included BI-78G3 (9). It was suggested that like 8, 9 binds the DRS of JNK1 [93] with the nitrothiazole group crossing the ridge near Arg-127 and Cys-163 (Figure 5B). Compound 9 displaces pepJIP1 from JNK1 with an IC50 of 160 nM and inhibits c-Jun phosphorylation with an IC50 of 1.8 μM. While 8 and 9 were reported to more potent towards JNK compared to p38 MAPKα, their activity towards ERK2 was not reported [93]. Compound 10 is a derivative of 9 and is reported to have similar binding properties to 8 and 9 [94]. In 2010, De et al. introduced BI-90H9 (compound 11), which has similar properties to compounds 8–10 albeit with a marginally higher plasma stability [95]. Compounds 8–11 represent an interesting starting point for the development of JNK and ERK inhibitors. However, their specificities need to be evaluated further and derivatives developed that lack reactivity towards thiols.
In 2009, Chen et al. used a fluorescence polarization assay to screen for compounds that disrupt the JNK-JIP interaction [96]. Compound 12 was found to inhibit the JNK1, JNK2 and JNK3 kinase activity with IC50s of 6, 8 and 10 μM respectively. Unfortunately, while compound 12 appears to be quite selective for JNK it exhibits limited cell permeability [96]. Thus, further development of this promising class of inhibitor is required.
Chen et al. also characterized compound 13 as a dual site JNK inhibitor; it is a competitive inhibitor of ATP, which is reported to allosterically inhibit the JIP-JNK interaction. Unlike 12, however, it inhibits a number of protein kinases, consistent with the reduced selectivity expected of an inhibitor that targets the ATP binding site [96].
The thiophene-3-carboxamide derivative, 14, was reported by De et al. [97] to be a dual site ATP and JIP inhibitor through its binding to both the ATP binding site and the JIP binding site. Compound 14 inhibits JNK1 kinase activity with an IC50 of 1.3 μM (LANTHA assay) and inhibits the binding of pepJIP1 to JNK1 with an IC50 of 4.6 μM (DELFIA assay). Molecular modeling suggests that this compound binds within the ATP site and forms a hydrogen bond with the side chain of Gln-37. Binding to the DRS is favored by several hydrophobic contacts and hydrogen-bond interactions of the carboximide group with the backbone carbonyl of Asn-114 and amide nitrogen of Val-118. Compound 14 shows enhanced selectivity towards JNK (based on a panel of 26 kinases) and does not inhibit p38 MAPKα (2% inhibition at 25 μM) [97].
A novel binding site on JNK near the MAPK insert
In 2011, Compound 15 was discovered in a high throughput screen of 500,000 compounds. A unique binding site was confirmed by NMR, utilizing 13C-methyl-labeled JNK1α1 (residues 1–364, T183E and Y185E) and by crystallization of the complex of JNK1 and 15 [71]. The binding site for 15 was shown to be located in the region of the MAP insert region (Figure 6A). Interestingly, this compound binds exclusively to the inactive form of JNK1 and accordingly inhibits the phosphorylation of JNK1 by MKK7 with an IC50 of 7.7 μM.
p38 MAP Kinases
p38 MAPK is a promising target for the treatment of several inflammatory diseases due to its role in controlling the production of pro-inflammatory cytokines, such as interleukin-1 and tumor necrosis factor [98].
Small molecules targeting the DRS of p38 MAPK
In 2004, Davidson et al. showed that targeting the non-ATP binding site of a MAP kinase could produce a substrate selective inhibitor [99]. They discovered a non-ATP p38 MAPKα inhibitor (16) that inhibits the phosphorylation of MK2a with a Ki of ~330 nM. In contrast, this compound inhibits the phosphorylation of ATF2 with a Ki of more than 20 μM. Isothermal titration calorimetry analysis confirmed the binding of 16 to p38 MAPKα, but showed that this compound does not prevent the binding of MK2a. While a deuterium exchange mass spectrometry (DXMS) study suggested that 16 binds in the vicinity of the p38 MAPKα active site and disrupts the ATP binding site [99], the basis for its substrate selectivity is unknown.
A lipid-binding site on p38 MAPK
Recently, a novel lipid-binding site was noted in the C-terminal lobe of p38 MAPK formed by the MAP kinase insert [71, 100, 101]. In 2009, Perry et al. applied AutoLigand to examine both the optimal ligand size and the affinity of compounds that may bind this site [101], which represents a promising target for the design of inhibitors since it is a hydrophobic pocket furnishing hydrogen-bond acceptors. In 2011, Comess et al. reported the discovery of several inhibitors that bind this site in p38 MAPKα [71]. High-resolution NMR and crystallographic studies provided details on the binding mode for compound 17 (Figure 6B). Interestingly, the binding of 17 induces conformational changes in the region of the binding site, which have the potential to be explored in the future. Compound 17 inhibits both the ability of MKK6 to phosphorylate p38 MAPKα as well as the ability of activated p38 MAPKα to phosphorylate MK2 cascade with IC50’s of 1.2 and 0.8 μM, respectively. Surprisingly, 17 exhibits significantly less activity against p38 MAPK β, γ or δ (IC50 > 40 μM). However, unfortunately it shows marked activity against a number of other protein kinases including Pim1 (Ki = 4.5 μM), Pim3 (Ki = 1.9 μM), Map4k4 (Ki = 4.7 μM), KHS (Ki = 5.2 μM), Gsk3β (Ki = 4.9 μM) and BRSK1 (Ki = 8.5 μM).
CONCLUSIONS
An increasing number of non-ATP competitive MAPK inhibitors are being reported in the literature. Several sites found on the surfaces of MAPKs have been shown to bind small molecule inhibitors and in some cases structures of these complexes have been reported. However, given the low throughput of X-ray crystallography and the potential difficulties of determining the structures of MAP kinases complexed to surface bound ligands, in silico modeling approaches may prove to be useful, especially if prior knowledge of binding sites are available, for example through the use of high-field NMR (e.g. ERK2) [79].
The available evidence suggests that the development of small molecule non-ATP inhibitors of MAPKs represent a viable approach towards the inhibition of MAPK pathway signaling in cells. While no highly potent, i.e Ki < 10 nM, inhibitors have been developed to date further advances in computational methods should aid in the development of such inhibitors.
Over the last decade, the polarizable atomic multipole AMOEBA force field has been developed in order to provide a more accurate and transferable, albeit computationally more expensive, alternative to fixed charge descriptions of molecular electrostatics [24, 25]. In spite of their neglect of polarization and point charge approximation, fixed charged force fields have proven their usefulness for sampling the conformational heterogeneity of MAP kinases via molecular dynamics [54] and given insight into MAPK-substrate docking [4]. However, some domains of molecular modeling for the discovery of non-ATP competitive inhibitors of MAP kinases benefit from chemical accuracy (i.e. prediction of binding free energy to experimental accuracy of 1.0 kcal/mole), which has been an important goal of the AMOEBA force field.
For example, Ewald crystallography refinement of MAP kinases based on the prior chemical information in the AMOEBA force field evaluated using Ewald’s method for electrostatics improves secondary structure and orients the water hydrogen-bonding networks. Precise structural models enhance the reliability of molecular docking to predict the binding modes of new lead compounds [85, 102–106]. Understanding water structure and energetics within the relatively shallow binding sites on the surface of MAP kinases may be critical to the further optimization of the non-ATP competitive inhibitors reviewed here [91].
Once the docking pose is known and water hydrogen-bonding networks have been surveyed, the quantitative in silico prediction of relative binding affinities becomes a valuable approach for lead optimization. Progress in the sampling of molecular configurations [107], the physical model describing atomic interactions [108, 109] and more powerful computer hardware [110] all contribute to advancements in lead optimization. Our strategy has been based on coupling the AMOEBA polarizable force field to both explicit [24, 108] and implicit solvent [109, 111, 112]. For example, initial work on trypsin-benzamidine systems predicted a strong correlation (R=0.9) between binding affinity predictions and experiment. In ongoing work on MAP kinases, we are exploring enhanced sampling methods such as λ-dynamics [113], metadynamics [114] and the orthogonal space random walk (OSRW) method [115, 116] in order to compute binding free energies as efficiently as possible.
Advancements in computational technology is critically needed for us to fully understand the conformational dynamics of protein kinases, which underlie structure, function and inhibitor binding. Approaches such as Milestoning [35, 117] and a Markovian state model (MSM) based adaptive seeding method [118] are promising for achieving long-time sampling. For example, the former has been successfully applied to elucidate complex conformational dynamics such as allosteric transitions in hemoglobin [119] and the myosin stroke [35]. The later was demonstrated to recover RNA folding kinetics. In both approaches, the process of a slow conformational transition can be modeled as a series of smaller steps that can be taken in a highly parallel fashion. Coupling polarizable force fields with such efficient dynamic sampling algorithms has the potential to bring novel insight into the role of conformational dynamics in MAP kinase activities, which is essential for designing innovative strategies targeting MAP kinases.
Acknowledgments
The authors thank Johnny C. Wu and Dr. Timothy D. Fenn for helpful discussions. This research was supported in part by the grants from the Welch Foundation (F-1691) to P. Ren and (F-1390) to K.N. Dalby and from the National Institutes of Health to P. Ren (R01GM079686) and to K. N. Dalby (GM59802). Support from Texas Institute for Drug & Diagnostic Development H-F-0032 is also acknowledged by K. N. Dalby. T.S.K. acknowledges a scholarship from the Egyptian Ministry of Higher Education.
References
- 1.Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–5. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 2.Johnson LN. Protein kinase inhibitors: Contributions from structure to clinical compounds. Quarterly reviews of biophysics. 2009;42:1–40. doi: 10.1017/S0033583508004745. [DOI] [PubMed] [Google Scholar]
- 3.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocrine reviews. 2001;22:153–83. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 4.Lee S, Warthaka M, Yan C, Kaoud TS, Piserchio A, Ghose R, Ren P, Dalby KN. A model of a MAPK*substrate complex in an active conformation: A computational and experimental approach. PLoS One. 2011;6:e18594. doi: 10.1371/journal.pone.0018594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Research. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanks S, Quinn A, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 8.Robinson FL, Whitehurst AW, Raman M, Cobb MH. Identification of novel point mutations in ERK2 that selectively disrupt binding to MEK1. Journal of Biological Chemistry. 2002;277:14844–52. doi: 10.1074/jbc.M107776200. [DOI] [PubMed] [Google Scholar]
- 9.Chou F-L, Hill JM, Hsieh J-C, Pouyssegur J, Brunet A, Glading A, Überall F, Ramos JW, Werner MH, Ginsberg MH. PEA-15 binding to ERK1/2 MAPKs is required for its modulation of integrin activation. Journal of Biological Chemistry. 2003;278:52587–97. doi: 10.1074/jbc.M309322200. [DOI] [PubMed] [Google Scholar]
- 10.Callaway K, Waas WF, Rainey MA, Ren P, Dalby KN. Phosphorylation of the transcription factor ets-1 by ERK2: rapid dissociation of ADP and phospho-ets-1. Biochemistry. 2010;49:3619–30. doi: 10.1021/bi100199q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Reviews Drug Discovery. 2004;3:935–49. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 12.Hartshorn MJ, Verdonk ML, Chessari G, Brewerton SC, Mooij WTM, Mortenson PN, Murray CW. Diverse, high-quality test set for the validation of protein-ligand docking performance. Journal of Medicinal Chemistry. 2007;50:726–41. doi: 10.1021/jm061277y. [DOI] [PubMed] [Google Scholar]
- 13.Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303:1813–18. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 14.Abel R, Young T, Farid R, Berne BJ, Friesner RA. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. Journal of the American Chemical Society. 2008;130:2817–31. doi: 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ponder JW, Case DA. Advances in Protein Chemistry. Academic Press; 2003. Force fields for protein simulations; pp. 27–85. [DOI] [PubMed] [Google Scholar]
- 16.Lopes PEM, Roux B, MacKerell AD. Molecular modeling and dynamics studies with explicit inclusion of electronic polarizability: theory and applications. Theoretical Chemistry Accounts. 2009;124:11–28. doi: 10.1007/s00214-009-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunger AT, Kuriyan J, Karplus M. Crystallographic R-factor refinement by molecular dynamics. Science. 1987;235:458–60. doi: 10.1126/science.235.4787.458. [DOI] [PubMed] [Google Scholar]
- 18.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Crystallographica Section D. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 19.Brunger AT. Version 1. 2 of the crystallography and NMR system. Nature Protocols. 2007;2:2728–33. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 20.Ewald PP. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Annalen der Physik. 1921;369:253–287. [Google Scholar]
- 21.Darden T, York D, Pedersen L. Particle-mesh Ewald - An n log(n) method for Ewald sums in large systems. Journal of Chemical Physics. 1993;98:10089–92. [Google Scholar]
- 22.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle-mesh Ewald method. Journal of Chemical Physics. 1995;103:8577–93. [Google Scholar]
- 23.Sagui C, Pedersen LG, Darden TA. Towards an accurate representation of electrostatics in classical force fields: Efficient implementation of multipolar interactions in biomolecular simulations. Journal of Chemical Physics. 2004;120:73–87. doi: 10.1063/1.1630791. [DOI] [PubMed] [Google Scholar]
- 24.Ren P, Ponder JW. Polarizable atomic multipole water model for molecular mechanics simulation. Journal of Physical Chemistry B. 2003;107:5933–47. [Google Scholar]
- 25.Ponder JW, Wu C, Ren P, Pande VS, Chodera JD, Schnieders MJ, Haque I, Mobley DL, Lambrecht DS, DiStasio RA, Head-Gordon M, Clark GNI, Johnson ME, Head-Gordon T. Current status of the AMOEBA polarizable force field. Journal of Physical Chemistry B. 2010;114:2549–64. doi: 10.1021/jp910674d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schnieders MJ, Fenn TD, Pande VS. Polarizable atomic multipole X-ray refinement: Particle mesh Ewald electrostatics for macromolecular crystals. Journal of Chemical Theory and Computation. 2011;7:1141–56. doi: 10.1021/ct100506d. [DOI] [PubMed] [Google Scholar]
- 27.Fenn TD, Schnieders MJ, Brunger AT. A smooth and differentiable bulk-solvent model for macromolecular diffraction. Acta Crystallographica Section D. 2010;66:1024–31. doi: 10.1107/S0907444910031045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schnieders MJ, Fenn TD, Pande VS, Brunger AT. Polarizable atomic multipole X-ray refinement: application to peptide crystals. Acta Crystallographica Section D. 2009;65:952–65. doi: 10.1107/S0907444909022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fenn TD, Schnieders MJ, Brunger AT, Pande VS. Polarizable atomic multipole X-ray refinement: hydration geometry and application to macromolecules. Biophysical Journal. 2010;98:2984–92. doi: 10.1016/j.bpj.2010.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fenn TD, Schnieders MJ, Mustyakimov M, Wu C, Langan P, Pande VS, Brunger AT. Reintroducing electrostatics into macromolecular crystallographic refinement: application to neutron crystallography and DNA hydration. Structure. 2011;19:523–33. doi: 10.1016/j.str.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Research. 2007;35:W375–83. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D. 2009;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–82. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 34.Grant BJ, Gorfe AA, McCammon JA. Large conformational changes in proteins: signaling and other functions. Current Opinion in Structural Biology. 2010;20:142–7. doi: 10.1016/j.sbi.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Majek P, Elber R. Milestoning without a reaction coordinate. Journal of Chemical Theory and Computation. 2010;6:1805–17. doi: 10.1021/ct100114j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Banavali NK, Roux B. Anatomy of a structural pathway for activation of the catalytic domain of Src kinase Hck. Proteins. 2007;67:1096–112. doi: 10.1002/prot.21334. [DOI] [PubMed] [Google Scholar]
- 37.Ozkirimli E, Yadav SS, Miller WT, Post CB. An electrostatic network and long-range regulation of Src kinases. Protein Science. 2008;17:1871–80. doi: 10.1110/ps.037457.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang S, Roux B. Src kinase conformational activation: Thermodynamics, pathways, and mechanisms. PLoS computational biology. 2008;4:e1000047. doi: 10.1371/journal.pcbi.1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gan WX, Yang SC, Roux B. Atomistic view of the conformational activation of Src kinase using the string method with swarms-of-trajectories. Biophysical Journal. 2009;97:L8–10. doi: 10.1016/j.bpj.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shan YB, Seeliger MA, Eastwood MP, Frank F, Xu HF, Jensen MO, Dror RO, Kuriyan J, Shaw DE. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:139–144. doi: 10.1073/pnas.0811223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi Z, Resing KA, Ahn NG. Networks for the allosteric control of protein kinases. Current Opinion in Structural Biology. 2006;16:686–92. doi: 10.1016/j.sbi.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 42.Zhang F, Strand A, Robbins D, Cobb MH, Goldsmith EJ. Atomic structure of the MAP kinase ERK2 at 2. 3 Å resolution. Nature. 1994;367:704–11. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
- 43.Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–69. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 44.Kinoshita T, Yoshida I, Nakae S, Okita K, Gouda M, Matsubara M, Yokota K, Ishiguro H, Tada T. Crystal structure of human mono-phosphorylated ERK1 at Tyr204. Biochemical and Biophysical Research Communications. 2008;377:1123–7. doi: 10.1016/j.bbrc.2008.10.127. [DOI] [PubMed] [Google Scholar]
- 45.Pagès G, Guèrin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouyssègur J. Defective thymocyte maturation in p44 MAP kinase (ERK 1) knockout mice. Science. 1999;286:1374–7. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- 46.Yao Y, Li W, Wu J, Germann UA, Su MSS, Kuida K, Boucher DM. Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12759–64. doi: 10.1073/pnas.2134254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lochhead PA. Protein kinase activation loop autophosphorylation in cis: overcoming a Catch-22 situation. Science signaling. 2009;2:pe4. doi: 10.1126/scisignal.254pe4. [DOI] [PubMed] [Google Scholar]
- 48.Kang YJ, Seit-Nebi A, Davis RJ, Han J. Multiple activation mechanisms of p38alpha mitogen-activated protein kinase. Journal of Biological Chemistry. 2006;281:26225–34. doi: 10.1074/jbc.M606800200. [DOI] [PubMed] [Google Scholar]
- 49.Diskin R, Engelberg D, Livnah O. High-resolution diffracting crystals of intrinsically active p38alpha MAP kinase: a case study for low-throughput approaches. Acta Crystallographica Section D. 2007;63:260–5. doi: 10.1107/S0907444906042910. [DOI] [PubMed] [Google Scholar]
- 50.Levin-Salomon V, Kogan K, Ahn NG, Livnah O, Engelberg D. Isolation of intrinsically active (MEK-independent) variants of the ERK family of mitogen-activated protein (MAP) kinases. Journal of Biological Chemistry. 2008;283:34500–10. doi: 10.1074/jbc.M806443200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emrick MA, Lee T, Starkey PJ, Mumby MC, Resing KA, Ahn NG. The gatekeeper residue controls autoactivation of ERK2 via a pathway of intramolecular connectivity. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18101–6. doi: 10.1073/pnas.0608849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nitta RT, Chu AH, Wong AJ. Constitutive activity of JNK2α2 is dependent on a unique mechanism of MAPK activation. Journal of Biological Chemistry. 2008;283:34935–45. doi: 10.1074/jbc.M804970200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhattacharyya RP, Remenyi A, Good MC, Bashor CJ, Falick AM, Lim WA. The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science. 2006;311:822–6. doi: 10.1126/science.1120941. [DOI] [PubMed] [Google Scholar]
- 54.Yan C, Kaoud T, Lee S, Dalby KN, Ren P. Understanding the specificity of a docking interaction between JNK1 and the scaffolding protein JIP1. Journal of Physical Chemistry B. 2011;115:1491–502. doi: 10.1021/jp1073522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou T, Sun L, Humphreys J, Goldsmith EJ. Docking interactions induce exposure of activation loop in the MAP kinase ERK2. Structure. 2006;14:1011–9. doi: 10.1016/j.str.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 56.Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nature Chemical Biology. 2006;2:358–64. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 57.Xie X, Gu Y, Fox T, Coll JT, Fleming MA, Markland W, Caron PR, Wilson KP, Su MSS. Crystal structure of JNK3: a kinase implicated in neuronal apoptosis. Structure. 1998;6:983–91. doi: 10.1016/s0969-2126(98)00100-2. [DOI] [PubMed] [Google Scholar]
- 58.Shaw D, Wang SM, Villaseñor AG, Tsing S, Walter D, Browner MF, Barnett J, Kuglstatter A. The crystal structure of JNK2 reveals conformational flexibility in the MAP kinase insert and indicates its involvement in the regulation of catalytic activity. Journal of Molecular Biology. 2008;383:885–93. doi: 10.1016/j.jmb.2008.08.086. [DOI] [PubMed] [Google Scholar]
- 59.Kuglstatter A, Ghate M, Tsing S, Villaseñor AG, Shaw D, Barnett JW, Browner MF. X-ray crystal structure of JNK2 complexed with the p38 [alpha] inhibitor BIRB796: Insights into the rational design of DFG-out binding MAP kinase inhibitors. Bioorganic & Medicinal Chemistry Letters. 2010;20:5217–20. doi: 10.1016/j.bmcl.2010.06.157. [DOI] [PubMed] [Google Scholar]
- 60.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–42. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 61.Lowinger TB, Riedl B, Dumas J, Smith RA. Design and discovery of small molecules targeting raf-1 kinase. Current Pharmaceutical Design. 2002;8:2269–78. doi: 10.2174/1381612023393125. [DOI] [PubMed] [Google Scholar]
- 62.Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, Clarkson B, Kuriyan J. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571) Cancer research. 2002;62:4236–43. [PubMed] [Google Scholar]
- 63.Cowan-Jacob SW, Fendrich G, Floersheimer A, Furet P, Liebetanz J, Rummel G, Rheinberger P, Centeleghe M, Fabbro D, Manley PW. Structural biology contributions to the discovery of drugs to treat chronic myelogenous leukaemia. Acta Crystallographica Section D. 2007;63:80–93. doi: 10.1107/S0907444906047287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liao JJ. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. Journal of Medicinal Chemistry. 2007;50:409–24. doi: 10.1021/jm0608107. [DOI] [PubMed] [Google Scholar]
- 65.Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, Grob PM, Hickey ER, Moss N, Pav S, Regan J. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nature Structural Biology. 2002;9:268–72. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- 66.Frembgen-Kesner T, Elcock AH. Computational sampling of a cryptic drug binding site in a protein receptor: explicit solvent molecular dynamics and inhibitor docking to p38 MAP kinase. Journal of Molecular Biology. 2006;359:202–14. doi: 10.1016/j.jmb.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 67.Vogtherr M, Saxena K, Hoelder S, Grimme S, Betz M, Schieborr U, Pescatore B, Robin M, Delarbre L, Langer T, Wendt KU, Schwalbe H. NMR characterization of kinase p38 dynamics in free and ligand-bound forms. Angewandte Chemie. 2006;45:993–7. doi: 10.1002/anie.200502770. [DOI] [PubMed] [Google Scholar]
- 68.Kufareva I, Abagyan R. Type-II kinase inhibitor docking, screening, and profiling using modified structures of active kinase states. Journal of Medicinal Chemistry. 2008;51:7921–32. doi: 10.1021/jm8010299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fabian MA, Biggs WH, 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lelias JM, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ. A small molecule-kinase interaction map for clinical kinase inhibitors. Nature Biotechnology. 2005;23:329–36. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 70.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. The Biochemical Journal. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Comess KM, Sun C, Abad-Zapatero C, Goedken ER, Gum RJ, Borhani DW, Argiriadi M, Groebe DR, Jia Y, Clampit JE, Haasch DL, Smith HT, Wang S, Song D, Coen ML, Cloutier TE, Tang H, Cheng X, Quinn C, Liu B, Xin Z, Liu G, Fry EH, Stoll V, Ng TI, Banach D, Marcotte D, Burns DJ, Calderwood DJ, Hajduk PJ. Discovery and characterization of non-ATP site inhibitors of the mitogen activated protein (MAP) kinases. ACS Chemical Biology. 2011;6:234–44. doi: 10.1021/cb1002619. [DOI] [PubMed] [Google Scholar]
- 72.Akritopoulou-Zanze I, Hajduk PJ. Kinase-targeted libraries: The design and synthesis of novel, potent, and selective kinase inhibitors. Drug Discovery Today. 2009;14:291–7. doi: 10.1016/j.drudis.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 73.Barsyte-Lovejoy D, Galanis A, Sharrocks AD. Specificity determinants in MAPK signaling to transcription factors. Journal of Biological Chemistry. 2002;277:9896–903. doi: 10.1074/jbc.M108145200. [DOI] [PubMed] [Google Scholar]
- 74.Rainey MA, Callaway K, Barnes R, Wilson B, Dalby KN. Proximity-induced catalysis by the protein kinase ERK2. Journal of the American Chemical Society. 2005;127:10494–5. doi: 10.1021/ja052915p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bardwell AJ, Frankson E, Bardwell L. Selectivity of docking sites in MAPK kinases. Journal of Biological Chemistry. 2009;284:13165–73. doi: 10.1074/jbc.M900080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Callaway KA, Rainey MA, Riggs AF, Abramczyk O, Dalby KN. Properties and regulation of a transiently assembled ERK2. Ets-1 signaling complex. Biochemistry. 2006;45:13719–33. doi: 10.1021/bi0610451. [DOI] [PubMed] [Google Scholar]
- 77.Callaway K, Abramczyk O, Martin L, Dalby KN. The anti-apoptotic protein PEA-15 is a tight binding inhibitor of ERK1 and ERK2, which blocks docking interactions at the D-recruitment site. Biochemistry. 2007;46:9187–98. doi: 10.1021/bi700206u. [DOI] [PubMed] [Google Scholar]
- 78.Abramczyk O, Rainey MA, Barnes R, Martin L, Dalby KN. Expanding the repertoire of an ERK2 recruitment site: cysteine footprinting identifies the D-recruitment site as a mediator of Ets-1 binding. Biochemistry. 2007;46:9174–86. doi: 10.1021/bi7002058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Piserchio A, Warthaka M, Devkota AK, Kaoud TS, Lee S, Abramczyk O, Ren P, Dalby KN, Ghose R. Solution NMR insights into docking interactions involving inactive ERK2. Biochemistry. 2011 doi: 10.1021/bi2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Biondi RM, Nebreda AR. Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. The Biochemical Journal. 2003;372:1–13. doi: 10.1042/BJ20021641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Akella R, Moon TM, Goldsmith EJ. Unique MAP kinase binding sites. Biochimica et Biophysica Acta. 2008;1784:48–55. doi: 10.1016/j.bbapap.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sheridan DL, Kong Y, Parker SA, Dalby KN, Turk BE. Substrate discrimination among mitogen-activated protein kinases through distinct docking sequence motifs. Journal of Biological Chemistry. 2008;283:19511–20. doi: 10.1074/jbc.M801074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu S, Sun JP, Zhou B, Zhang ZY. Structural basis of docking interactions between ERK2 and MAP kinase phosphatase 3. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5326–31. doi: 10.1073/pnas.0510506103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hancock CN, Macias A, Lee EK, Yu SY, Mackerell AD, Jr, Shapiro P. Identification of novel extracellular signal-regulated kinase docking domain inhibitors. Journal of Medicinal Chemistry. 2005;48:4586–95. doi: 10.1021/jm0501174. [DOI] [PubMed] [Google Scholar]
- 85.Chen F, Hancock CN, Macias AT, Joh J, Still K, Zhong S, MacKerell AD, Jr, Shapiro P. Characterization of ATP-independent ERK inhibitors identified through in silico analysis of the active ERK2 structure. Bioorganic & Medicinal Chemistry Letters. 2006;16:6281–7. doi: 10.1016/j.bmcl.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kaoud TS, Devkota AK, Harris R, Rana MS, Abramczyk O, Warthaka M, Lee S, Girvin ME, Riggs AF, Dalby KN. Activated ERK2 is a monomer in vitro with or without divalent cations and when complexed to the cytoplasmic scaffold PEA15. Biochemistry. 2011 doi: 10.1021/bi200202y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Boston SR, Deshmukh R, Strome S, Priyakumar UD, MacKerell AD, Jr, Shapiro P. Characterization of ERK docking domain inhibitors that induce apoptosis by targeting Rsk-1 and caspase-9. BMC cancer. 2011;11:7. doi: 10.1186/1471-2407-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li Q, Al-Ayoubi A, Guo T, Zheng H, Sarkar A, Nguyen T, Eblen ST, Grant S, Kellogg GE, Zhang S. Structure-activity relationship (SAR) studies of 3-(2-amino-ethyl)-5-(4-ethoxy-benzylidene)-thiazolidine-2,4-dione: development of potential substrate-specific ERK1/2 inhibitors. Bioorganic & Medicinal Chemistry Letters. 2009;19:6042–6. doi: 10.1016/j.bmcl.2009.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yap JL, Worlikar S, MacKerell AD, Shapiro P, Fletcher S. Small-molecule inhibitors of the ERK signaling pathway: Towards novel anticancer therapeutics. ChemMedChem. 2011;6:38–48. doi: 10.1002/cmdc.201000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Heo YS, Kim SK, Seo CI, Kim YK, Sung BJ, Lee HS, Lee JI, Park SY, Kim JH, Hwang KY, Hyun YL, Jeon YH, Ro S, Cho JM, Lee TG, Yang CH. Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. The EMBO Journal. 2004;23:2185–95. doi: 10.1038/sj.emboj.7600212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kaoud TS, Mitra S, Lee S, Taliaferro J, Cantrell M, Linse KD, Van Den Berg CL, Dalby KN. Development of JNK2-selective peptide inhibitors that inhibit breast cancer cell migration. ACS Chemical Biology. 2011 doi: 10.1021/cb200017n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stebbins JL, De SK, Machleidt T, Becattini B, Vazquez J, Kuntzen C, Chen LH, Cellitti JF, Riel-Mehan M, Emdadi A, Solinas G, Karin M, Pellecchia M. Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16809–13. doi: 10.1073/pnas.0805677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.De SK, Chen LH, Stebbins JL, Machleidt T, Riel-Mehan M, Dahl R, Chen V, Yuan H, Barile E, Emdadi A, Murphy R, Pellecchia M. Discovery of 2-(5-nitrothiazol-2-ylthio)benzo [d]thiazoles as novel c-Jun N-terminal kinase inhibitors. Bioorganic & Medicinal Chemistry. 2009;17:2712–7. doi: 10.1016/j.bmc.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.De SK, Stebbins JL, Chen LH, Riel-Mehan M, Machleidt T, Dahl R, Yuan H, Emdadi A, Barile E, Chen V, Murphy R, Pellecchia M. Design, synthesis, and structure-activity relationship of substrate competitive, selective, and in vivo active triazole and thiadiazole inhibitors of the c-Jun N-terminal kinase. Journal of Medicinal Chemistry. 2009;52:1943–52. doi: 10.1021/jm801503n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.De SK, Chen V, Stebbins JL, Chen LH, Cellitti JF, Machleidt T, Barile E, Riel-Mehan M, Dahl R, Yang L, Emdadi A, Murphy R, Pellecchia M. Synthesis and optimization of thiadiazole derivatives as a novel class of substrate competitive c-Jun N-terminal kinase inhibitors. Bioorganic & Medicinal Chemistry. 2010;18:590–6. doi: 10.1016/j.bmc.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen T, Kablaoui N, Little J, Timofeevski S, Tschantz WR, Chen P, Feng J, Charlton M, Stanton R, Bauer P. Identification of small-molecule inhibitors of the JIP-JNK interaction. The Biochemical Journal. 2009;420:283–94. doi: 10.1042/BJ20081899. [DOI] [PubMed] [Google Scholar]
- 97.De SK, Barile E, Chen V, Stebbins JL, Cellitti JF, Machleidt T, Carlson CB, Yang L, Dahl R, Pellecchia M. Design, synthesis, and structure-activity relationship studies of thiophene-3-carboxamide derivatives as dual inhibitors of the c-Jun N-terminal kinase. Bioorganic & Medicinal Chemistry. 2011;19:2582–8. doi: 10.1016/j.bmc.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hale KK, Trollinger D, Rihanek M, Manthey CL. Differential expression and activation of p38 mitogen-activated protein kinase alpha, beta, gamma, and delta in inflammatory cell lineages. Journal of immunology. 1999;162:4246–52. [PubMed] [Google Scholar]
- 99.Davidson W, Frego L, Peet GW, Kroe RR, Labadia ME, Lukas SM, Snow RJ, Jakes S, Grygon CA, Pargellis C, Werneburg BG. Discovery and characterization of a substrate selective p38alpha inhibitor. Biochemistry. 2004;43:11658–71. doi: 10.1021/bi0495073. [DOI] [PubMed] [Google Scholar]
- 100.Diskin R, Engelberg D, Livnah O. A novel lipid binding site formed by the MAP kinase insert in p38 alpha. Journal of Molecular Biology. 2008;375:70–79. doi: 10.1016/j.jmb.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 101.Perry JJP, Harris RM, Moiani D, Olson AJ, Tainer JA. p38 alpha MAP kinase C-terminal domain binding pocket characterized by crystallographic and computational analyses. Journal of Molecular Biology. 2009;391:1–11. doi: 10.1016/j.jmb.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Abagyan R, Totrov M. Biased probability Monte-Carlo conformational searches and electrostatic calculations for peptides and proteins. Journal of Molecular Biology. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 103.Jones G, Willett P, Glen RC. Molecular recognition of receptor-sites using a genetic algorithm with a description of desolvation. Journal of Molecular Biology. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 104.Wang RX, Lu YP, Wang SM. Comparative evaluation of 11 scoring functions for molecular docking. Journal of Medicinal Chemistry. 2003;46:2287–303. doi: 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- 105.Kontoyianni M, McClellan LM, Sokol GS. Evaluation of docking performance: Comparative data on docking algorithms. Journal of Medicinal Chemistry. 2004;47:558–65. doi: 10.1021/jm0302997. [DOI] [PubMed] [Google Scholar]
- 106.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. Journal of Medicinal Chemistry. 2006;49:6177–96. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 107.Min DH, Zheng LQ, Harris W, Chen MG, Lv C, Yang W. Practically efficient QM/MM alchemical free energy simulations: The orthogonal space random walk strategy. Journal of Chemical Theory and Computation. 2010;6:2253–66. doi: 10.1021/ct100033s. [DOI] [PubMed] [Google Scholar]
- 108.Jiao D, Golubkov PA, Darden TA, Ren P. Calculation of protein-ligand binding free energy by using a polarizable potential. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6290–5. doi: 10.1073/pnas.0711686105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jiao D, Zhang JJ, Duke RE, Li GH, Schnieders MJ, Ren PY. Trypsin-ligand binding free energies from explicit and implicit solvent simulations with polarizable potential. Journal of Computational Chemistry. 2009;30:1701–11. doi: 10.1002/jcc.21268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pitera JW. Current developments in and importance of high-performance computing in drug discovery. Current Opinion in Drug Discovery & Development. 2009;12:388–96. [PubMed] [Google Scholar]
- 111.Schnieders MJ, Baker NA, Ren PY, Ponder JW. Polarizable atomic multipole solutes in a Poisson-Boltzmann continuum. Journal of Chemical Physics. 2007:126. doi: 10.1063/1.2714528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schnieders MJ, Ponder JW. Polarizable atomic multipole solutes in a generalized Kirkwood continuum. Journal of Chemical Theory and Computation. 2007;3:2083–97. doi: 10.1021/ct7001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kong X, Brooks CL., III Lambda-dynamics: A new approach to free energy calculations. Journal of Chemical Physics. 1996;105:2414–2423. [Google Scholar]
- 114.Laio A, Parrinello M. Escaping free-energy minima. Proceedings of the National Academy of Sciences. 2002;99:12562–6. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zheng L, Chen M, Yang W. Random walk in orthogonal space to achieve efficient free-energy simulation of complex systems. Proceedings of the National Academy of Sciences. 2008;105:20227–32. doi: 10.1073/pnas.0810631106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zheng L, Chen M, Yang W. Simultaneous escaping of explicit and hidden free energy barriers: Application of the orthogonal space random walk strategy in generalized ensemble based conformational sampling. Journal of Chemical Physics. 2009;130:234105. doi: 10.1063/1.3153841. [DOI] [PubMed] [Google Scholar]
- 117.Faradjian AK, Elber R. Computing time scales from reaction coordinates by milestoning. Journal of Chemical Physics. 2004;120:10880–9. doi: 10.1063/1.1738640. [DOI] [PubMed] [Google Scholar]
- 118.Huang X, Bowman GR, Bacallado S, Pande VS. Rapid equilibrium sampling initiated from nonequilibrium data. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19765–9. doi: 10.1073/pnas.0909088106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Elber R. A milestoning study of the kinetics of an allosteric transition: Atomically detailed simulations of deoxy Scapharca hemoglobin. Biophysical Journal. 2007;92:L85–7. doi: 10.1529/biophysj.106.101899. [DOI] [PMC free article] [PubMed] [Google Scholar]