

Abstract
The genetic incorporation of the 22nd proteinogenic amino acid, pyrolysine (Pyl) at amber codon is achieved by the action of pyrrolysyl-tRNA synthetase (PylRS) together with its cognate tRNAPyl. Unlike most aminoacyl-tRNA synthetases, PylRS displays high substrate side chain promiscuity, low selectivity toward its substrate α-amine, and low selectivity toward the anticodon of tRNAPyl. These unique but ordinary features of PylRS as an aminoacyl-tRNA synthetase allow the Pyl incorporation machinery to be easily engineered for the genetic incorporation of more than 100 non-canonical amino acids (NCAAs) or α-hydroxy acids into proteins at amber codon and the reassignment of other codons such as ochre UAA, opal UGA, and four-base AGGA codons to code NCAAs.
1. Introduction
With few exceptions, all biological systems code strictly 20 canonical amino acids. Given the limited functionalities of 20 amino acids, biochemists have longed for an approached for the production of proteins with unique biochemical and biophysical handles. Early efforts focused on the chemical aminoacylation of tRNAs that could be applied to undergo in vitro translation or injected into cells for the synthesis of proteins with non-canonical amino acids (NCAAs).[1] Pioneered by Schultz and others, engineered aminoacyl-tRNA synthetase (AARS)-tRNA pairs were later introduced to genetically encode NCAAs mainly at amber UAG codon in living cells, making the bulk expression of proteins with NCAAs possible.[2–4] Although artificial systems have been delicately crafted to selectively incorporate NCAAs at amber codon in living cells, nature itself has a very competitive system for reassigning amber codon to code the 22nd proteinogenic amino acid, pyrrolysine (Pyl). In this review, we will discuss the Pyl incorporation system from its discovery to its applications in the genetic incorporation of close to 100 NCAAs or α-hydroxy acids. This review intends to discuss the Pyl system alone. For more general summary of the genetic code expansion technique, please find reviews published elsewhere.
2. Pyl
2.1. Discovery
Although methanogens are phylogenetically diverse throughout the archaea domain, the methanogenic reaction in these organisms is almost universal in reducing a single substrate, carbon dioxide, to methane.[5] However, there is one order of methanogenic archaea, the Methanosarcinales, which has evolved the ability to reduce other compounds, methylamines, to methane. When sequencing clustered genes in Methanosarcina barkeri responsible for methanogenesis from monomethylamine (MMA), Krzycki et al. surprisingly discovered that the open reading frame of mtmB that codes monomethylamine methyltransferase (MMAMT) is interrupted by a single in-frame canonical amber UAG stop codon that does not appear to prevent translation of the full-length product.[6] MMAMT is a 52 kD polypeptide and is one of the most predominant proteins in MMA-grown cell extracts of M. barkeri. The truncated protein with a premature termination at the in-frame amber codon was not as abundant. Since basal levels of nonsense suppression occur at low frequencies to maintain translation integrity inside cells, this observation clearly specified that there is an endogenous but efficient amber suppression mechanism for recoding this amber stop codon in M. barkeri. The later sequencing analysis of other methylamine (trimethylamine and dimethylamine) methyltransferase genes in M. barkeri MS all showed an in-frame amber stop codon that is also well conserved in methylamine methyltransferase genes in other Methanosarcina strains.[7] This in-frame amber codon is apparently reassigned to code an amino acid that is indicatively critical for functional conservation among methylamine methyltransferases.
The crystal structure of MMAMT was later determined to a high resolution of 1.55 Å.[8] The in-frame UAG-encoded amino acid site displayed a unique electron density that was distinctive from any of the 20 canonical amino acids and selenocysteine (Sec). This electron density was refined to show a lysine (Lys) core with a modification at its side-chain amine. The identity of this modification was determined to be a (4R,5R)-4-methyl-pyrroline-5-caboxylate, with the carboxylate in the amide linkage to the Lys side chain. This novel amino acid was coined as pyrrolysine (three-letter abbreviation: Pyl; one-letter abbreviation: O). Although the installation of Pyl might result from two possible routes, one being a posttranslational modification of a pre-incorporated Lys and the other being co-translational incorporation of Pyl itself, the existence of a unique AARS and a unique tRNA in the pyl gene cluster adjacent to a methylamine methyltransferase gene cluster is most consistent with the co-translational installation route.[9] From these observations, Pyl clearly represents the 22nd naturally occurring genetically encoded amino acid to be discovered.
2.2. Incorporation and biosynthesis
Using tRANScanSE [10] to search the M. barkeri Fusaro genomic database, Krzycki et al. revealed a unique tRNA gene, pylT, near a methylamine methyltransferase gene cluster. PylT codes a special tRNA with a CUA anticodon.[9] Along with its special CUA anticodon for the recognition of amber codon, the pylT transcript, tRNAPyl, has a distinct anticodon stem of six base pairs instead of five base pairs as observed in most tRNAs, a single base between D and anticodon stems, a single base between D and acceptor stems, and a three-base small variable arm. PylT resides in the pyl gene cluster that also contains four other genes, pylS, pylB, pylC, and pylD. Sequence alignment indicated that pylS codes a class II AARS but with a low sequence identity to all AARSs in the database. Since Pyl is a Lys derivative, the recombinant pylS gene product was later tested for lysyl-tRNA synthetase (LysRS) activity. In the presence of the tRNA pool from M. barkeri, the recombinant pylS gene product succeeded in ligating [14C]Lys to tRNA. This observation led to the hypothesis that tRNAPyl is charged with Lys first and then undergoes modification at the Lys side chain to form Pyl-tRNAPyl for the co-translational incorporation of Pyl. However, following experiments with pure tRNAPyl clearly showed that the recombinant pylS gene product definitely charged tRNAPyl with Pyl instead of Lys.[11–13] Therefore, the pylS gene product is not LysRS and should be named as pyrrolysyl-tRNA synthetase (PylRS); Although it was original believed that the incorporation of Pyl requires a mRNA secondary structure similarly as observed for the Sec incorporation,[14, 15] this mRNA secondary structure was definitely not necessary for the incorporation of Pyl at an amber codon.[16–19] Therefore, the incorporation machinery of Pyl requires simply an AARS, a tRNA, a codon, and an amino acid, similar to the incorporation systems of the 20 canonical amino acids in most organisms (Figure 1).
Figure 1.

The essential four components of the Pyl incorporation machinery: pyrrolysyl-tRNA synthetase, , Pyl, and amber codon.
Kryzcki et al. also demonstrated that lateral gene transfer of the whole pyl gene cluster from M. barkeri to a naïve organism, Escherichia coli, drove the recombinant expression of functional MMAMT. The expressed MMAMT had an intact Pyl at the in-frame amber stop codon site, indicating the pyl gene cluster codes all necessary enzymes for the biosynthesis of Pyl from E. coli metabolites. The removal of each of the three genes pylB, pylC and pylD dismissed the cluster’s ability to reassign the amber codon to code Pyl. Therefore, pylB, pylC and pylD form an essential gene cassette for the biosynthesis of Pyl.[20] Following studies revealed that pylB codes a radical S-adenosyl-L-methionine enzyme.[21, 22] This enzyme catalyzes the synthesis of (3R)-3-methyl-D-ornithine from Lys whose carboxylate is then ligated to a Lys side chain amine by the action of the pylC gene product, a member of the carbamoyl phosphate synthetase family with the expense of adenosyl 5′-triphosphate (ATP). The synthesized (3R)-3-methyl-D-ornithyl-Nε-L-Lys then undergoes the oxidation at the δ-amine of its (3R)-3-methyl-D-ornithine component by the action of the pylD gene product, a NAD-dependent dehydrogenase, a simultaneous hydrolysis to from a δ-aldehyde, and then a simultaneous intramolecular condensation to form Pyl, finishing the whole biosynthetic pathway (Figure 2).[23] When D-ornithine was provided in the growth medium, the clustered genes of pylT, pylS, pylC, and pylD were able to mediate amber suppression. In this situation, the UAG-coded amino acid was desmethyl-Pyl, indicating pylC and pylD gene products are able to synthesize desmethyl-Pyl from D-ornithine and lysine and that PylRS tolerates alternative substrates.[22, 24, 25]
Figure 2.
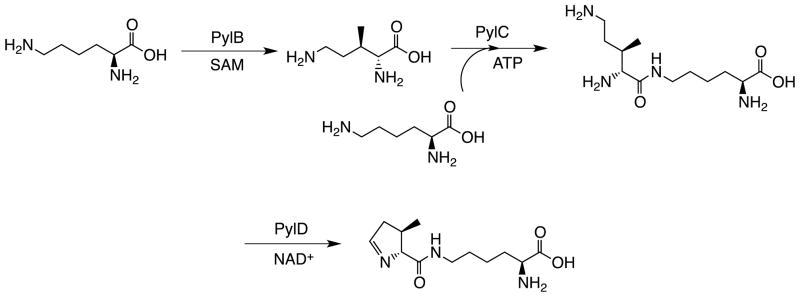
The biosynthetic pathway of Pyl.
3. PylRS
Sequence alignment indicated that full-length PylRS contains a C-terminal class II AARS catalytic core and an N-terminal domain that apparently does not share sequence homology with any structurally known protein domains. PylRS proteins from different Methanosarcina strains have variable lengths of N-terminal domains but their C-terminal catalytic cores with about 270 amino acids are highly conserved. Efforts to crystalize full-length PylRS were not successful due to the low solubility of the protein. The N-terminal domain is highly insoluble and aggregates full-length PylRS.[26, 27] Using the truncated C-terminal catalytic core of PylRS from Methanosarcina mazei, apo-PylRS and PylRS complexes with different ligands were successfully determined, attributed mostly to contributions from Yokoyama, Söll, Steitz, and their coworkers.[28–32] The three dimensional organization of the PylRS catalytic core resembles that of other synthetases from the Class II AARS family. It has a typical β-sheet core surrounded by several helices, forming a Rossmann fold for the binding of ATP. Like most other class II aminoacyl-tRNA synthetases, PylRS also forms an obligate dimer. Each subunit has an active site. Although sharing relatively low sequence identity (<30%), the PylRS catalytic core and bacterial phenylalanyl-tRNA synthetase (PheRS) are structurally similar (Figure 3).[31, 33–35] The two structures are highly superimposable and have similarly organized hydrophobic pockets for binding amino acid substrates, possibly indicating divergent evolution of the two enzymes from the same origin.
Figure 3.
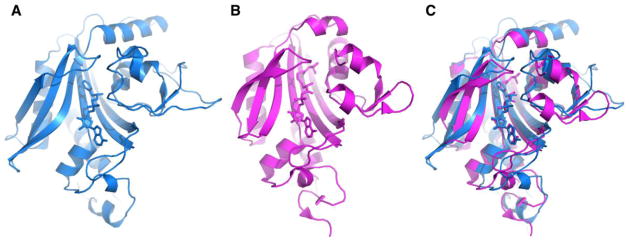
(A) The structure of Thermus thermophilus PheRS with phenyl-AMP bound at the active site (PDB entry: 1B7Y). (B) The structure of M. mazei PylRS with pyrrolysyl-AMP bound at the active site (PDB entry: 2ZIM). (C) The overlay of the two structures.
3.1. High substrate side chain promiscuity
The structure of the M. mazei PylRS catalytic core revealed a deep hydrophobic pocket for the binding of Pyl (Figure 4). A302, L305, Y306, L309, N346, C348, and W417 form a bulky cavity for the binding of the side chain (4R,5R)-4-methyl-pyrroline-5-caboxylate of Pyl.[30, 31] The Pyl side chain also forms two hydrogen bonds at the PylRS active site, with one involving the side chain amide nitrogen of N346 and the Pyl side chain amide oxygen and the other involving the pyrroline nitrogen and the phenolic oxygen of Y384. Y384 is at a flexible loop region that is random in the absence of Pyl but serves as a cap for the binding of Pyl to the active site. Although PylRS binds tightly to Pyl, with a Kd value (53 μM) relatively comparable to other AARSs, PylRS displays remarkably high tolerance toward variations of the substrate side chain, especially when a variation is at the pyrroline region.[36] As discussed above, PylRS recognizes desmethyl-Pyl and is able to direct its incorporation at amber codon when in coordination with tRNAPyl. Other NCAAs that are structurally similar to Pyl such as D-prolyl-L-Lys and Nε-cyclopentyloxycarbonyl-L-Lys can also serve as good substrates of PylRS.[36] Surprisingly, NCAAs such as Nε-(tert-butyloxycarbonyl)-L-Lys and Nε-allyloxycarbonyl-L-Lys that are structurally dramatically different from Pyl also function well as PylRS substrates.[29, 37] This high substrate side chain promiscuity of an AARS is very unique. To maintain high translation fidelity, most AARSs have evolved to specifically interact with their amino acid substrates and kinetically favor the correct tRNA aminoacylation reactions. Many AARSs also contain an editing domain that can hydrolyze misacylated tRNAs.[38–40] Therefore, in general, even a slight variation of the amino acid substrate structure will cause a typical AARS to reject it from slipping into the translation process. However, PylRS recognizes the Pyl side chain chiefly with relatively non-specific hydrophobic interactions and in addition it does not have an editing domain, explaining its high substrate side chain promiscuity. This unique but ordinary feature of PylRS as an AARS might be due to relatively relaxed evolutionary pressure in its natural hosts. Unlike the 20 canonical amino acids that are structurally and spatially similar to one another, Pyl is exceptionally large. To the best of our knowledge, non-proteinogenic amino acid metabolites with a similar structure rarely exist in cells. Therefore, there is no selection pressure for PylRS to evolve strict and exclusive recognition of Pyl. A relatively high Pyl binding affinity of PylRS alone is enough to maintain translation integrity.
Figure 4.
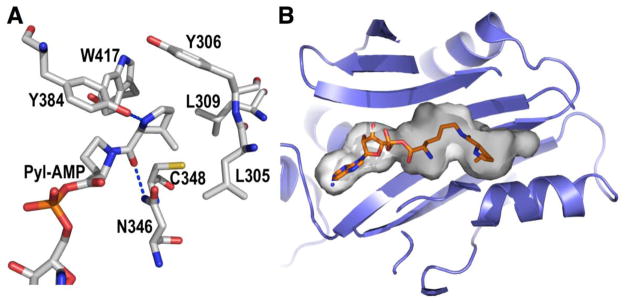
(A) The active site of Pyl. (B) The contoured deep substrate binding cavity of PylRS. Pyl-AMP is shown in sticks.
3.2. Low selectivity toward α-amine
Another striking feature of PylRS is its low selectivity toward the Pyl α-amine. In the active site, the Pyl α-amine forms two hydrogen bonds, one to the phenolic oxygen of Y384 and the other to a water molecule that has three additional hydrogen bond interactions with the side chain amide oxygen of N346 and two backbone amide nitrogens of L301 and A302 (Figure 5A). Since Y384 is in a flexible loop region that does not fold back to cap the active site until the binding of Pyl, it is possible that the hydrogen bond between the Pyl α-amine and the Y384 phenolic oxygen is flexibly formed and not essential for the catalytic activity of PylRS.[30, 31] This has been supported by the fact that several PylRS mutants with phenylalanine (Phe) or tryptophan at Y384 display higher activities toward their substrates than corresponding Y384-containing enzymes.[37, 41–43] In the PylRS catalytic core, residues that are close to the Pyl α-amine include M300 and A302. However, side chains of these two residues are too distant from the Pyl α-amine to form strong van der Waals contacts. Therefore, its hydrogen bond to the water molecule is probably the only essential interaction for the loose binding of the Pyl α-amine to the active site. Based on the hydrogen bond pattern of the water molecule, we can also conclude that the Pyl α-amine binds to the active site uncharged. Two backbone nitrogens are juxtaposed at proper positions to serve as two hydrogen bond donors for the water molecule. This leaves the water molecule with no vacancy to accept an additional hydrogen bond donor. Therefore the Pyl α-amine has to be uncharged to interact with the water molecule as a hydrogen bond acceptor. Since PylRS doesn’t involve strict recognition of the Pyl α-amine and also binds its uncharged form, presumably changing the α-amine to a α-hydroxide, a neutral amine homolog that can potentially maintain hydrogen bond interactions with both Y384 and the water molecule may not significantly change the activity of PylRS. As a matter of fact, PylRS displays high tolerance toward α-hydroxy acids. It has been demonstrated that several α-hydroxy acids serve as substrates for PylRS and its engineered mutants that, together with tRNAPyl, mediate direct incorporation of these α-hydroxy acids at amber codon in bacterial cells.[44, 45]
Figure 5.
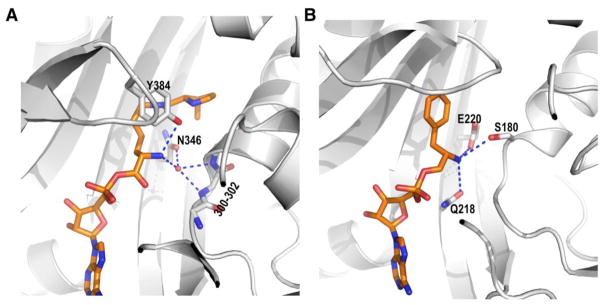
(A) The recognition of the substrate α-amine at the active site of PylRS. Side chains of residues 300–302 are not shown for a clear view of the hydrogen bond network of α-amine. (B) The recognition of the substrate α-amine at the active site of T. thermophilus PheRS.
The loose recognition of α-amine at the active site of PylRS is in striking contrast to the strict α-amine recognition feature of most AARSs. For instance, PheRS, a structural homolog of PylRS, involves unique α-amine interactions to bind Phe at its active site (Figure 5B). The α-amine forms three hydrogen bonds with side chains of S180, Q218, and E220. Since the side chain of E220 needs to be negatively charged for a strong charge-quadruple interaction with the phenyl side chain of the substrate, there is no doubt that the substrate α-amine is positively charged to generate a very strong charged hydrogen bond (salt bridge) with the E220 side chain. In addition, the positively charged substrate α-amine serves as a hydrogen bond donor in all three hydrogen bond interactions. Both the preferential binding of its charged state and three specific hydrogen bond interactions contribute to the strict recognition of Phe at the PheRS active site. Simply changing the α-substitute to a hydroxide will not only dismiss the strong charged hydrogen bond interaction but also destroy the hydrogen bond pattern in the active site since a neutral hydroxide can serve as both a hydrogen bond donor and a hydrogen bond acceptor but a charged amine can only be a hydrogen bond donor. This strict recognition of Phe can efficiently ward off the mistaken incorporation of phenyllactic acid into proteins during the translation process. Given that α-hydroxy acids that are structurally similar to canonical amino acids are regular metabolites in cells, most AARSs stay in cellular environments with high evolution pressure to maintain strict recognition of α-amine to prevent cell functions from going awry.[46–50] However, this is probably not the case for PylRS. An α-hydroxy acid with an extended side chain like the one in Pyl may not exist at all in Methanosarcinales, leaving PylRS with no pressure for evolving strict recognition of α-amine.
3.3. Low selectivity toward the tRNA anticodon
The third unique feature of PylRS is its low selectivity toward the tRNA anticodon. A recently determined crystal structure of the Deslfitobacterium hafniense PylRS complex with tRNAPyl showed that PylRS has a unique tRNA-binding domain, a special C-terminal tail, a loop region, and a bulge domain that is involved in the specific recognition of the anticodon, D, and acceptor stems of tRNAPyl (Figure 6).[32] However, D. hafniense PylRS does not directly interact with the three anticodon nucleotides. Although D. hafniense PylRS corresponds only to the C-terminal catalytic core of PylRS from Methanosarcinales, multiple observations have suggested that M. mazei PylRS does not involve specific interactions with the anticodon of tRNAPyl. Mutating the anticodon of tRNAPyl alone has not shown to abolish the aminoacylation activity of M. mazei PylRS.[18, 30] This feature is also in striking contrast to the direct synthetase-anticodon interactions that have been observed for the majority of AARSs. Anticodons in tRNAs are typically used as recognition elements of AARSs for strictly pairing amino acids with their corresponding codons to avoid misincorporation during translation.[51] Two notable exceptions are the E. coli leucyl- and seryl-tRNA synthetases.[52–56] Both leucine and serine have six corresponding codons, multiple isoacceptor tRNAs for pairing with six codons, but only a single corresponding AARS. For one synthetase to recognize multiple isoacceptor tRNAs with distinct anticodon sequences, a reasonable evolutionary route is to avoid direct interactions with the tRNA anticodon region. Therefore the evasion of direct tRNA anticodon interactions for leucyl- and seryl-tRNA synthetases might be an evolution compromise. Since Pyl has only one corresponding codon, one tRNA, and one synthetase, it is quite puzzling why PylRS has not evolved to directly interact with the anticodon of tRNAPyl for strictly pairing Pyl with an amber codon. A single mutation at the anticodon region will lead to the misincorporation of Pyl at another codon, which may be detrimental to cells. This non-selective recognition of the tRNAPyl anticodon by PylRS might be rationalized considering the pyl gene cluster is probably highly regulated and only turned on in the presence of methylamines. Its noncontinuous or even rare expression would leave PylRS with no constant evolution pressure for evolving a more optimized feature.
Figure 6.
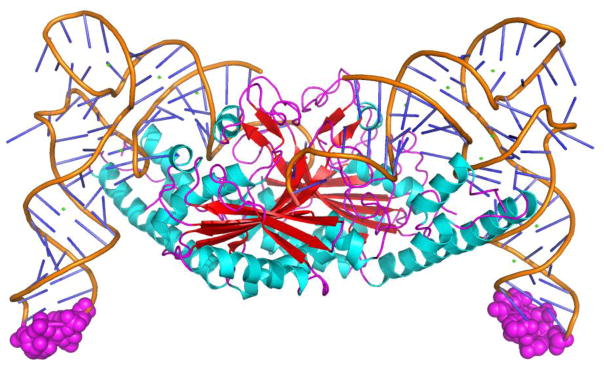
The D. hafniense PylRS complex with tRNAPyl. PylRS forms a homodimer that associates with two molecules. Three anticodon nucleotides in both molecules are shown in spheres and colored in magenta.
4. An outstanding genetic code expansion tool
Due to its unique structure, tRNAPyl has been shown to be orthogonal in commonly used bacterial and eukaryotic cells and functional to reassign amber codon when coupled with PylRS in these cell strains. Since it was first reported as a genetic code expansion tool in E. coli in 2004, the PylRS-tRNAPyl pair and its mutant forms have been used to site-specifically incorporate NCAAs into proteins at amber codon mutation sites in E. coli, Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster, and mammalian cells.[9, 29, 57–60] In E. coli, the genetic incorporation of more than 100 NCAAs and several α-hydroxy acids have been demonstrated. The PylRS-tRNAPyl pair has also shown high codon flexibility as several alternative codons for coding NCAAs have been demonstrated.[11, 61, 62] This remarkable success is largely attributed to the three unconventional features of PylRS.
4.1 The native enzyme
Shown in Figure 4A, the binding of the side chain pyrroline of Pyl to the PylRS active site involves essentially van der Waals interactions. Replacing the side chain pyrroline with a similar size chemical component with a hydrophobic nature might retain the PylRS activity to aminoacylate tRNAPyl. This potential has been explored extensively. NCAAs including Pyl and desmethyl-Pyl (1–2) which have been demonstrated to be PylRS substrates are presented in Figure 7. Söll et al. pioneered the search for alternative substrates of PylRS due to the inconvenient synthesis of Pyl and their need of a substrate to characterize PylRS.[36, 63] They showed that 3 and 4 could both serve as PylRS substrates with about 10–fold weaker binding affinities than Pyl itself. 4 was also successfully used to demonstrate that a mRNA secondary structure is not necessary for the PylRS-tRNAPyl pair to reassign amber codon to code a NCAA.[19] Two NCAAs (5–6) were discovered as PylRS substrates when Yokoyama et al. soaked PylRS crystals with different ligands for structure determination. Both 5 and 6 show strong interactions with PylRS and their electron densities can be clearly observed in the active site of PylRS.[29, 37] 5 is a lysine derivate with a Nε-Boc protecting group that can be easily removed in acidic conditions. Chin et al. have taken advantage of this chemistry to synthesize native diubiquitins and histones with a site-selectively installed Nε,Nε-dimethyllysine.[64, 65] The generated diubiquitins have been used successfully to reveal Lys29-isopeptide selectivity of an OUT deubiquitinase, TRABID.[65] 6 has a terminal alkene that can potentially undergo thio-ene photoclick, olefin metathesis, Diels-Alder, and 1,3-dipolar cycloaddition reactions. Selective labeling of proteins incorporated with 6 and another terminal alkene-containing PylRS substrate 7 using the thio-ene photoclick reaction has been demonstrated.[66] Kryzcki et al. also sought alternative PylRS substrates independently. They discovered that 8 maintained about 9% of the activity of Pyl as a PylRS substrate.[67] Encouraged by the results of 8, three NCAAs (9–11), each with a terminal alkyne, were designed and synthesized. They all worked as PylRS substrates and their incorporation into proteins allowed for site-selective modifications with azide via the Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC) reaction.[68–70] Concurrent efforts by Chin et al. revealed another two click substrates (12–13) of PylRS. Although their original goal was to select PylRS mutants that can selectively recognize 12 and 13, the evolution resulted in the native enzyme.[71] 12 and 13 contain alkyne and azide, respectively. Their incorporation into proteins allowed specific labeling with azide or alkyne dyes using the CuAAC reaction. The tetrazine-alkene Diels-Alder cycloaddition reaction is a click reaction type that has been actively preformed in the chemical biology research area due to its rapid reaction nature.[72–74] Chin et al. discovered that a norbornene-containing NCAA (14) could be efficiently recognized by PylRS and incorporated into proteins at amber codon in both E. coli and mammalian cells mediated by the PylRS-tRNAPyl pair. Proteins incorporated with 14 in mammalian cells were rapidly labeled with tetrazine dyes.[75] Two NCAAs (15–16) with a D- or L-cystyl side chain and one NCAA (17) that can be deprotected to recover a L-cystyl side chain can be weakly recognized by PylRS and genetically incorporated into proteins. Their unique abilities to undergo native chemical ligation with a peptide thioester and selective condensation with cyanobenzothiazole have been harnessed to synthesize ubiquitinated proteins and proteins selectively labeled with cyanobenzothiazole dyes.[76–80] Two NCAAs that have an additional methyl group on the side chain carbamate nitrogen also serve as PylRS substrates and have been incorporated into proteins. It has been demonstrated that both carbamate groups in the two NCAAs could be hydrolyzed in relatively mild conditions to recover Nε-methyllysine, allowing the genetic installation of a monomethylated Lys residue in proteins.[81, 82] Recently Carell et al. showed that PylRS weakly recognized three NCAAs (20–22) and mediated their incorporation into proteins at an amber codon in coordination with tRNAPyl, allowing the synthesis of proteins with site-specific lysine propionylation, butylation, and crotonylation.[83] PylRS mutants with much improved binding affinities toward 20–22 were developed concurrently in the Schultz group and in our group. They will be discussed in the next section. Another NCAA (23) that can be recognized by PylRS is a photocrosslinking amino acid.[84] Applications of this NCAA and other photocrosslinking NCAAs will also be discussed in the next section.
Figure 7.

NCAAs that serve as substrates of the native PylRS.
4.2 Engineered enzymes for lysine derivatives
Although the native PylRS displays very high substrate side chain promiscuity, its activities on certain NCAAs shown in Figure 7 such as 15–17 and 20–22 are weak. Efforts have been made in several research groups to identify PylRS mutants that have improved activities and also recognize Lys derivatives that could be targeted by the native PylRS. The distal location of the deep pyrroline binding pocket to the catalytic center of the enzyme makes the engineering process relatively straightforward without too much concern that a mutation will dramatically affect the enzymatic catalyzed aminoacylation of tRNAPyl. To date, more than 30 lysine derivatives have been genetically incorporated into proteins at amber codon in living cells using engineered PylRS-tRNAPyl pairs. Their structures are presented in Figure 8 and the sequence information of engineered PylRS mutants are shown in Table 1. For NCAAs 6–7, 14–17, and 20–22, all engineered mutants display higher activities for their genetic incorporation than the native PylRS. Other NCAAs in Figure 8 show very weak or undetectable activities toward the native PylRS. Applications of these genetically encoded NCAAs span many research areas. 6–7 and 24 have a terminal alkene that has been demonstrated to undergo thio-ene photoclick protein labeling.[66] 15–17 can be applied to undergo native chemical ligation for the synthesis of ubiquitinated proteins and a biocompatible condensation for protein labeling.[78–80] 25 and 27 has an azide that selectively undergoes the Staudinger ligation, CuAAC click, and Cu-free click reactions.[37, 85] 26 was an intermediate NCAA for the development of a 27-specific PylRS mutant. Both 28 and 31 undergo selective Cu-free click labeling with azide. They have been applied to selectively label proteins incorporated with them in living cells.[86, 87] NCAAs 14 and 28–32 react rapidly with tetrazine, allowing rapid turn-on fluorescent labeling of proteins containing these NCAAs in mammalian cells.[88–90] 33–34 have a terminal alkyne. They can be well applied for CuAAC reactions with azide and thiol-yne photoclick reactions. They also undergo biocompatible Sonogashira cross-coupling reactions with halogenated benzene derivatives in the presence of palladium catalysts. 35 undergoes a similar cross-coupling reaction.[91–93] Two groups concurrently worked out the genetic incorporation of 36. 36 has an acrylamide functionality that can selectively undergo Michael addition with a thiol, radical polymerization with an alkene, and 1,3-dipolar nitrilimine-alkene cycloaddition. We have demonstrated that a genetically incorporated 36 allowed selectively turn-on fluorescent labeling of proteins in living cells.[42, 94] Two photocrosslinking NCAAs 23 and 37 have been genetically encoded using native and engineered PylRS-tRNAPyl pairs and applied successfully to covalently crosslink associate proteins. Chen et al. has pushed the technique further in using 37 to successfully profile in vivo substrates of a major acid-protection chaperone, HdeA, in E. coli periplasm.[95–97] Two photocaged lysines 38–39 have been genetically encoded. The genetic incorporation of 39 into mammalian proteins and its following photoactivation have allowed the spatiotemporal control of p53 cellular localization, kinase activity, and gene transcription in human cells.[41, 98–100] Besides 18 and 19, two other NCAAs, 40 and 41 have been genetically encoded using mutant PylRS-tRNAPyl pairs for the indirect synthesis of proteins with site-specifically installed Nε-methyllysine. In comparison to 18 which needs to be deprotected under acidic conditions, 19 which needs ruthenium catalysts for deprotection, and 40 which can be potentially deprotected using palladium-catalyzed hydrogenation, 41, a photocaged Nε-methyllysine, can be facilely deprotected under UV irradiation. Its deprotection condition is probably the most mild. Using 41 to synthesize proteins incorporated with Nε-methyllysine has been demonstrated.[82, 101, 102] In order to undergo traceless synthesis of diubiquitins with native isopeptide bonds, Chin et al. evolved PylRS mutants that can selectively recognize 43 and 46 for their genetic incorporation into ubiquitin. 43 and 46 can selectively remove their Nε protection groups to recover δ-mercapto-Lys that undergoes native chemical ligation with ubiquitin thioester to generate diubiquitins. The δ-thiol auxiliary group can then be removed. Using this approach, several native diubiquitins were synthesized. In the process of evolving PylRS mutants for 43 and 46, PylRS mutants that recognize 42, 44, and 45 were also identified.[43] Four posttranslationally modified lysines 47 and 20–22 have been genetically encoded using PylRS mutants. PylRS mutants displayed much higher activities for the genetic incorporation of these four NCAAs than the native enzyme. 47 represents the most abundant posttranslational Lys acylation type in mammalian cells. Its genetic incorporation has been applied to study functional roles of Lys acetylation in nucleosome and p53.[103–111] Alternatively, posttranslational lysine modification mimics could be indirectly installed into proteins through the genetic incorporation of 49 followed by oxidation to form dehydroalanine and then Michael addition with N-substituted cysteamine derivatives.[112] 49 and another NCAA 48 were genetically encoded using a PylRS mutant originally evolved for 26. Another two lysine derivatives that have been incorporated into proteins using evolved PylRS mutants together with tRNAPyl are 50 and 51.[113, 114] 50 has a furan at its side chain that can be sensitized for the formation of protein-nucleotide complexes and 51 has a spin label that has been applied to measure intramolecular distance distributions of two protein residues.
Figure 8.

Lysine derivatives that have been genetically incorporated into proteins using engineering PylRS mutants in coordination with tRNAPyl.
Table 1.
Sequence information of PylRS mutants that recoganize a variety of NCAAs as substrates.
| Sequence | M276 | L301 | A302 | L305 | Y306 | L309 | N346 | C348 | M350 | Y384 | V401 | W417 | M. mazei1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Substrate | M241 | L266 | A267 | L270 | Y271 | L274 | N311 | C313 | M315 | Y349 | V367 | W383 | M. barkeri1 |
| 6, 7,14, 25, 98–101 | A | A | F | M. barkeri | |||||||||
| A | V | F | |||||||||||
| 15, 16, 17 | S | V | F | M. barkeri | |||||||||
| 17 | V | M. barkeri | |||||||||||
| 26, 27, 50 | A | F | M. mazei | ||||||||||
| 26 | A | V | M. mazei | ||||||||||
| 14, 28, 29, 30, 31 | A | F | M. mazei | ||||||||||
| 29 | G | F | M. mazei | ||||||||||
| 31, 32 | M | G | A | M. barkeri | |||||||||
| 30, 32 | A | M | A | M. barkeri | |||||||||
| 33, 34, 35, 37 | A | A | F | M. barkeri | |||||||||
| A | S | F | M. barkeri | ||||||||||
| 36, 20 | M | L | A | F | W | M. mazei | |||||||
| 23 | M | A | F | M. barkeri | |||||||||
| 38 | M | A | A | F | M. mazei | ||||||||
| I | A | A | F | M. mazei | |||||||||
| 39 | F | S | C | M | C | M. barkeri | |||||||
| 18 | F | M. barkeri | |||||||||||
| 26, 38, 40, 41, 48, 49 | M | A | T | M. mazei | |||||||||
| M | A | C | M. mazei | ||||||||||
| M | P | C | M. mazei | ||||||||||
| 41 | I | M | A | M. barkeri | |||||||||
| 42, 43 | W | M. barkeri | |||||||||||
| 44 | M | G | A | M. barkeri | |||||||||
| 45, 46 | M | G | A | W | M. barkeri | ||||||||
| 20, 36, 47, 102 | M | L | A | F | M. mazei | ||||||||
| M | L | L | S | M. mazei | |||||||||
| V | I | F | A | F | M. barkeri | ||||||||
| L | I | L | A | F | M. barkeri | ||||||||
| M | I | F | A | F | M. barkeri | ||||||||
| 51 | A | M | F | M. mazei | |||||||||
| 21, 22 | W | M. mazei | |||||||||||
| 21, 22 | A | F | F | M. barkeri | |||||||||
| 52 | A | L | M. mazei | ||||||||||
| 52 | A | K | M. mazei | ||||||||||
| 53, 54 | L | M | S | L | L | M. mazei | |||||||
| F | L | T | F | L | M. mazei | ||||||||
| M | L | S | S | M | M. mazei | ||||||||
| 55–95 | A | A | M. mazei | ||||||||||
| 56 | T | V | W | F | L | M. mazei | |||||||
| 77 | F | M | G | G | M. barkeri | ||||||||
| 96 | T | T | T | M. mazei | |||||||||
| 97 | T | G | T | I | Y | M. mazei |
Native enzyme origins.
4.3 Engineered enzymes for Phe derivatives
PylRS displays a high structural similarity to bacterial PheRS, though they share low sequence identities. This high structural similarity made us wonder whether we would be able to evolve PylRS to selectively recognize Phe. The evolution turned out to be very simple. After a PylRS library with randomization at six active site residues was selected for amber suppression ability in E. coli in LB medium, multiple clones responsive to Phe were identified.[115, 116] This result opened a new avenue of the PylRS research area which is to develop PylRS mutants for the genetic incorporation of Phe derivatives. Although a number of Phe derivatives have been genetically encoded in bacteria and eukaryotes using engineered tyrosyl-tRNA synthase-tRNAPyl pairs that were originally from Methanocaldococcus jannaschii and E. coli,[3, 117–119] the relatively relaxed substrate binding makes the engineering of PylRS more straightforward and the high orthogonally of the native Pyl system in different cell strains also allows the more convenient transfer of engineering Pyl systems between different cells. As a demonstration, we evolved PylRS mutants that can selectively incorporate 53 and 54 at amber codons in E. coli in coordination with tRNAPyl.[115] When the sequence information of PylRS clones for 52 was available, we noticed two residues N346 and C348 had convergent mutations. N346 was mutated to either alanine or serine and C348 was always mutated to a large amino acid. The structure of the PylRS complex with Pyl-AMP showed one hydrogen bond between the N346 side chain and the Pyl side chain amide. We suspected that removing the side chain of N346 dismisses this hydrogen bond and also leaves space for the binding of Phe. A large residue at C348 might provide strong van der Waals interactions with Phe for its binding to the active site. Therefore, removing the side chain of C348 could potentially leave space for adding substituents to the phenylalanine side chain. We constructed the N346A/C348A mutant of PylRS and tested this mutant on a series of p-alkoxy-Phe molecules 55–61. They all served as substrates and could be genetically incorporated.[120] Surprisingly this mutant also recognized m-substituted Phe derivatives 62–73.[121] Encouraged by these results, we synthesized Phe derivatives 74–87 to test the substrate scope of this mutant. All of these NCAAs can be readily taken and selectively incorporated at amber codons in E. coli cells expressing the N346A/C348 mutant and tRNAPyl.[122] Our most recent efforts also demonstrated that the N346A/C348 mutant recognizes o-substituted Phe derivatives 88–95 and the N346A/C348 mutant–tRNAPyl pair is able to mediate site-selective incorporation of these NCAAs into proteins in human cells.[123] NCAAs 52–95 contain reactive functional groups such as alkyne, azide, ketone, and halobenzene. As we demonstrated, their incorporation into proteins allowed the use of multiple click reaction types for selective protein modifications. Similar to the native PylRS, the N346A/C348A mutant represents a single enzyme that allows the installation of a large variety of functionalities into proteins. Efforts to identify PylRS mutants selective for Phe derivatives were also made in other groups. Chin et al. showed that a PylRS mutant together with tRNAPyl was able to genetically incorporate 77 into a tyrosine residue in STAT1 for regulating its phosphorylation.[124] Wang et al. showed that PylRS mutants selective for 56, 96, and 97 could be evolved.[125, 126]
4.4 α-Hydroxy acids
Yokoyama et al. first noticed the ability of PylRS to recognize substrates with α-substituents different from amine. In vitro aminoacylation analysis revealed that PylRS was able to charge tRNAPyl with 5 analogs that were α-hydroxy acid, non-α-amino-carboxylic acid, Nα-methylamino acid, and D-amino acid. By expressing PylRS and tRNAPyl in E. coli, the UAG amber codon was successfully reassigned to code the α-hydroxy acid analog of 5 (98).[44] In comparison to α-amine which forms a backbone amide bond and serves as a hydrogen bond donor in proteins, α-hydroxy acid forms a relatively weaker ester bond and can solely function as a hydrogen bond acceptor. A readily hydrolysable ester bond in a protein may allow for easy removal of its C-terminal tag. Yokoyama et al. showed that, after its site-specific incorporation into glutathione S-transferase, the C-terminal side of 98 could be removed with the incubation with aqueous ammonia. The weak bond nature of the protein ester bond also potentially allows appending additional chemical functionalities to protein C-terminus. Chen, Liu, and their coworkers showed that α-hydroxy acids 98–101 could be selectively incorporated into proteins using an evolved PylRS-tRNAPyl pair. A synthesized protein, HdeA with an ester bond could undergo aminolysis with hydrazine to append a C-terminal hydrazide that could further undergo alternative native chemical ligation to a peptide with a N-terminal cysteine.[45] Another potential application of the α-hydroxy acid incorporation to be demonstrated is to probe the contribution of a single hydrogen bond to protein stability. Incorporation of α-hydroxy acid to an α-helix will remove one hydrogen bond and may dissolve the α-helical folding.
4.5 Alternative codons
Previous studies have indicated that PylRS does not specifically recognize the CUA anticodon of tRNAPyl. By simply mutating the tRNAPyl anticodon, we and others have demonstrated that the system can be well applied to reassign other codons such as ochre UAA, opal UGA, and four-base AGGA codons.[61, 62, 127] The PylRS-tRNAPyl pair also displays remarkable orthogonality toward two engineered tyrosyl-tRNA synthase-tRNAPyl pairs that were originally from M. jannaschii and E. coli and used to expand the genetic code of bacteria and eukaryotes. By coupling a pair with these two pairs, two distinctive NCAAs have been genetically encoded at two separate codons namely amber UAG and ochre UAA codons in E. coli and mammalian cells.[61, 128, 129] We recently showed that p-azido-Phe which served as a substrate of an evolved M. jannaschii tyrosyl-tRNA pair and 102 which served as a substrate of an evolved pair could be site-specifically incorporated at amber and ochre mutation sites respectively into one protein that allow site-selectively dual labeling of a protein in a catalyst-free and one-pot fashion (Figure 11).[130, 131] This development provides a readily adoptable platform for generating proteins labeled with two dyes for their folding/dynamic analysis using Förster resonance energy transfer.
Figure 11.
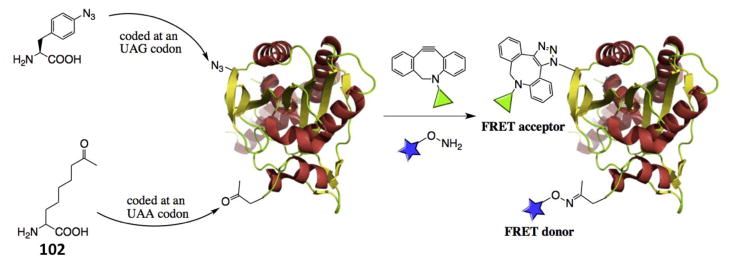
A general platform of incorporating two different NCAAs into one protein for a catalyst-free and one-pot dual labeling.
5. Conclusion remarks
In about a decade after the Pyl incorporation mechanism was discovered, the system has been transferred to bacteria, yeast, and mammalian cells for the genetic incorporation of more than 100 NCAAs or α-hydroxy acids into proteins. These NCAAs or α-hydroxy acids contain many chemically unique reactive groups that allow for the installation of a large variety of biochemical and biophysical probes for protein function investigation and biotechnological development. The system has also allowed access to proteins with multiple posttranslational modifications mainly on Lys both in vitro and in vivo. This makes it possible to tackle important issues such as how these modifications regulate protein functions, enzyme activities, and nucleosome interactions with transcription factors, addressing a lot of questions related to epigenetics. With these magnificent tools developed, we envisage the field will soon diverge to focusing on a large variety of applications from basic study to biotechnological development.
Figure 9.

Phenylalanine derivatives that have been genetically incorporated into proteins using engineering PylRS mutants in coordination with tRNAPyl.
Figure 10.
α-Hydroxy acids that have been genetically incorporated into proteins using engineering PylRS mutants in coordination with tRNAPyl.
Acknowledgments
Financial support of the PylRS engineering work in the authors’ laboratory is kindly provided from the National Institute of Health (grant 1R01CA161158), the National Science Foundation (grant CHEM-1148684), and the Welch Foundation (grant A-1715).
Abbreviations used
- Pyl
Pyrrolysine
- PylRS
pyrrolysyl-tRNA synthetase
- AARS
aminoacyl-tRNA synthetase
- NCAA
non-canonical amino acid
- MMA
monomethylamine
- MMAMT
monomethylamine methyltransferase
- LysRS
lysyl-tRNA synthetase
- Lys
lysine
- Sec
selenocysteine
- ATP
adenosine 5′-phosphate
- PheRS
phenylalanyl-tRNA synthetase
- Phe
phenylalanine
- CuAAC
Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hendrickson TL, de Crecy-Lagard V, Schimmel P. Incorporation of nonnatural amino acids into proteins. Annu Rev Biochem. 2004;73:147–176. doi: 10.1146/annurev.biochem.73.012803.092429. [DOI] [PubMed] [Google Scholar]
- 2.Furter R. Expansion of the genetic code: site-directed p-fluoro-phenylalanine incorporation in Escherichia coli. Protein Sci. 1998;7:419–426. doi: 10.1002/pro.5560070223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, Schultz PG. An expanded eukaryotic genetic code. Science. 2003;301:964–967. doi: 10.1126/science.1084772. [DOI] [PubMed] [Google Scholar]
- 4.Ngo JT, Tirrell DA. Noncanonical amino acids in the interrogation of cellular protein synthesis. Acc Chem Res. 2011;44:677–685. doi: 10.1021/ar200144y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao B, Gupta RS. Phylogenomic analysis of proteins that are distinctive of Archaea and its main subgroups and the origin of methanogenesis. Bmc Genomics. 2007;8:86. doi: 10.1186/1471-2164-8-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burke SA, Lo SL, Krzycki JA. Clustered genes encoding the methyltransferases of methanogenesis from monomethylamine. J Bacteriol. 1998;180:3432–3440. doi: 10.1128/jb.180.13.3432-3440.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paul L, Ferguson DJ, Jr, Krzycki JA. The trimethylamine methyltransferase gene and multiple dimethylamine methyltransferase genes of Methanosarcina barkeri contain in-frame and read-through amber codons. J Bacteriol. 2000;182:2520–2529. doi: 10.1128/jb.182.9.2520-2529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hao B, Gong W, Ferguson TK, James CM, Krzycki JA, Chan MK. A new UAG-encoded residue in the structure of a methanogen methyltransferase. Science. 2002;296:1462–1466. doi: 10.1126/science.1069556. [DOI] [PubMed] [Google Scholar]
- 9.Srinivasan G, James CM, Krzycki JA. Pyrrolysine encoded by UAG in Archaea: charging of a UAG-decoding specialized tRNA. Science. 2002;296:1459–1462. doi: 10.1126/science.1069588. [DOI] [PubMed] [Google Scholar]
- 10.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hao B, Zhao G, Kang PT, Soares JA, Ferguson TK, Gallucci J, Krzycki JA, Chan MK. Reactivity and chemical synthesis of L-pyrrolysine- the 22(nd) genetically encoded amino acid. Chem Biol. 2004;11:1317–1324. doi: 10.1016/j.chembiol.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 12.Blight SK, Larue RC, Mahapatra A, Longstaff DG, Chang E, Zhao G, Kang PT, Green-Church KB, Chan MK, Krzycki JA. Direct charging of tRNA(CUA) with pyrrolysine in vitro and in vivo. Nature. 2004;431:333–335. doi: 10.1038/nature02895. [DOI] [PubMed] [Google Scholar]
- 13.Polycarpo C, Ambrogelly A, Berube A, Winbush SM, McCloskey JA, Crain PF, Wood JL, Soll D. An aminoacyl-tRNA synthetase that specifically activates pyrrolysine. Proc Natl Acad Sci U S A. 2004;101:12450–12454. doi: 10.1073/pnas.0405362101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fletcher JE, Copeland PR, Driscoll DM, Krol A. The selenocysteine incorporation machinery: interactions between the SECIS RNA and the SECIS-binding protein SBP2. RNA. 2001;7:1442–1453. [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Z, Reches M, Groisman I, Engelberg-Kulka H. The nature of the minimal ‘selenocysteine insertion sequence’ (SECIS) in Escherichia coli. Nucleic Acids Res. 1998;26:896–902. doi: 10.1093/nar/26.4.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Baranov PV, Atkins JF, Gladyshev VN. Pyrrolysine and selenocysteine use dissimilar decoding strategies. J Biol Chem. 2005;280:20740–20751. doi: 10.1074/jbc.M501458200. [DOI] [PubMed] [Google Scholar]
- 17.Longstaff DG, Blight SK, Zhang L, Green-Church KB, Krzycki JA. In vivo contextual requirements for UAG translation as pyrrolysine. Mol Microbiol. 2007;63:229–241. doi: 10.1111/j.1365-2958.2006.05500.x. [DOI] [PubMed] [Google Scholar]
- 18.Ambrogelly A, Gundllapalli S, Herring S, Polycarpo C, Frauer C, Soll D. Pyrrolysine is not hardwired for cotranslational insertion at UAG codons. Proc Natl Acad Sci U S A. 2007;104:3141–3146. doi: 10.1073/pnas.0611634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Namy O, Zhou Y, Gundllapalli S, Polycarpo CR, Denise A, Rousset JP, Soll D, Ambrogelly A. Adding pyrrolysine to the Escherichia coli genetic code. FEBS Lett. 2007;581:5282–5288. doi: 10.1016/j.febslet.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 20.Longstaff DG, Larue RC, Faust JE, Mahapatra A, Zhang L, Green-Church KB, Krzycki JA. A natural genetic code expansion cassette enables transmissible biosynthesis and genetic encoding of pyrrolysine. Proc Natl Acad Sci U S A. 2007;104:1021–1026. doi: 10.1073/pnas.0610294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quitterer F, List A, Eisenreich W, Bacher A, Groll M. Crystal structure of methylornithine synthase (PylB): insights into the pyrrolysine biosynthesis. Angew Chem Int Ed Engl. 2012;51:1339–1342. doi: 10.1002/anie.201106765. [DOI] [PubMed] [Google Scholar]
- 22.Gaston MA, Zhang L, Green-Church KB, Krzycki JA. The complete biosynthesis of the genetically encoded amino acid pyrrolysine from lysine. Nature. 2011;471:647–650. doi: 10.1038/nature09918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krzycki JA. The path of lysine to pyrrolysine. Curr Opin Chem Biol. 2013;17:619–625. doi: 10.1016/j.cbpa.2013.06.023. [DOI] [PubMed] [Google Scholar]
- 24.Cellitti SE, Ou W, Chiu HP, Grunewald J, Jones DH, Hao X, Fan Q, Quinn LL, Ng K, Anfora AT, Lesley SA, Uno T, Brock A, Geierstanger BH. D-Ornithine coopts pyrrolysine biosynthesis to make and insert pyrroline-carboxy-lysine. Nat Chem Biol. 2011;7:528–530. doi: 10.1038/nchembio.586. [DOI] [PubMed] [Google Scholar]
- 25.Ou W, Uno T, Chiu HP, Grunewald J, Cellitti SE, Crossgrove T, Hao X, Fan Q, Quinn LL, Patterson P, Okach L, Jones DH, Lesley SA, Brock A, Geierstanger BH. Site-specific protein modifications through pyrroline-carboxy-lysine residues. Proc Natl Acad Sci U S A. 2011;108:10437–10442. doi: 10.1073/pnas.1105197108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang R, Krzycki JA. PylSn and the homologous N-terminal domain of pyrrolysyl-tRNA synthetase bind the tRNA that is essential for the genetic encoding of pyrrolysine. J Biol Chem. 2012;287:32738–32746. doi: 10.1074/jbc.M112.396754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yanagisawa T, Ishii R, Fukunaga R, Nureki O, Yokoyama S. Crystallization and preliminary X-ray crystallographic analysis of the catalytic domain of pyrrolysyl-tRNA synthetase from the methanogenic archaeon Methanosarcina mazei. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:1031–1033. doi: 10.1107/S1744309106036700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yanagisawa T, Sumida T, Ishii R, Yokoyama S. A novel crystal form of pyrrolysyl-tRNA synthetase reveals the pre- and post-aminoacyl-tRNA synthesis conformational states of the adenylate and aminoacyl moieties and an asparagine residue in the catalytic site. Acta Crystallogr D Biol Crystallogr. 2013;69:5–15. doi: 10.1107/S0907444912039881. [DOI] [PubMed] [Google Scholar]
- 29.Mukai T, Kobayashi T, Hino N, Yanagisawa T, Sakamoto K, Yokoyama S. Adding l-lysine derivatives to the genetic code of mammalian cells with engineered pyrrolysyl-tRNA synthetases. Biochem Biophys Res Commun. 2008;371:818–822. doi: 10.1016/j.bbrc.2008.04.164. [DOI] [PubMed] [Google Scholar]
- 30.Yanagisawa T, Ishii R, Fukunaga R, Kobayashi T, Sakamoto K, Yokoyama S. Crystallographic studies on multiple conformational states of active-site loops in pyrrolysyl-tRNA synthetase. J Mol Biol. 2008;378:634–652. doi: 10.1016/j.jmb.2008.02.045. [DOI] [PubMed] [Google Scholar]
- 31.Kavran JM, Gundllapalli S, O’Donoghue P, Englert M, Soll D, Steitz TA. Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation. Proc Natl Acad Sci U S A. 2007;104:11268–11273. doi: 10.1073/pnas.0704769104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nozawa K, O’Donoghue P, Gundllapalli S, Araiso Y, Ishitani R, Umehara T, Soll D, Nureki O. Pyrrolysyl-tRNA synthetase-tRNA(Pyl) structure reveals the molecular basis of orthogonality. Nature. 2009;457:1163–1167. doi: 10.1038/nature07611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reshetnikova L, Moor N, Lavrik O, Vassylyev DG. Crystal structures of phenylalanyl-tRNA synthetase complexed with phenylalanine and a phenylalanyl-adenylate analogue. J Mol Biol. 1999;287:555–568. doi: 10.1006/jmbi.1999.2617. [DOI] [PubMed] [Google Scholar]
- 34.Goldgur Y, Mosyak L, Reshetnikova L, Ankilova V, Lavrik O, Khodyreva S, Safro M. The crystal structure of phenylalanyl-tRNA synthetase from thermus thermophilus complexed with cognate tRNAPhe. Structure. 1997;5:59–68. doi: 10.1016/s0969-2126(97)00166-4. [DOI] [PubMed] [Google Scholar]
- 35.Mosyak L, Reshetnikova L, Goldgur Y, Delarue M, Safro MG. Structure of phenylalanyl-tRNA synthetase from Thermus thermophilus. Nat Struct Biol. 1995;2:537–547. doi: 10.1038/nsb0795-537. [DOI] [PubMed] [Google Scholar]
- 36.Polycarpo CR, Herring S, Berube A, Wood JL, Soll D, Ambrogelly A. Pyrrolysine analogues as substrates for pyrrolysyl-tRNA synthetase. FEBS Lett. 2006;580:6695–6700. doi: 10.1016/j.febslet.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yanagisawa T, Ishii R, Fukunaga R, Kobayashi T, Sakamoto K, Yokoyama S. Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode N(epsilon)-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem Biol. 2008;15:1187–1197. doi: 10.1016/j.chembiol.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 38.Delarue M. Aminoacyl-tRNA synthetases. Curr Opin Struct Biol. 1995;5:48–55. doi: 10.1016/0959-440x(95)80008-o. [DOI] [PubMed] [Google Scholar]
- 39.Ibba M, Thomann HU, Hong KW, Sherman JM, Weygand-Durasevic I, Sever S, Stange-Thomann N, Praetorius M, Soll D. Substrate selection by aminoacyl-tRNA synthetases. Nucleic Acids Symp Ser. 1995:40–42. [PubMed] [Google Scholar]
- 40.Woese CR, Olsen GJ, Ibba M, Soll D. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiology and molecular biology reviews: MMBR. 2000;64:202–236. doi: 10.1128/mmbr.64.1.202-236.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen PR, Groff D, Guo J, Ou W, Cellitti S, Geierstanger BH, Schultz PG. A facile system for encoding unnatural amino acids in mammalian cells. Angew Chem Int Ed Engl. 2009;48:4052–4055. doi: 10.1002/anie.200900683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee YJ, Wu B, Raymond JE, Zeng Y, Fang X, Wooley KL, Liu WR. A genetically encoded acrylamide functionality. ACS Chem Biol. 2013;8:1664–1670. doi: 10.1021/cb400267m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Virdee S, Kapadnis PB, Elliott T, Lang K, Madrzak J, Nguyen DP, Riechmann L, Chin JW. Traceless and site-specific ubiquitination of recombinant proteins. J Am Chem Soc. 2011;133:10708–10711. doi: 10.1021/ja202799r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi T, Yanagisawa T, Sakamoto K, Yokoyama S. Recognition of non-alpha-amino substrates by pyrrolysyl-tRNA synthetase. J Mol Biol. 2009;385:1352–1360. doi: 10.1016/j.jmb.2008.11.059. [DOI] [PubMed] [Google Scholar]
- 45.Li YM, Yang MY, Huang YC, Li YT, Chen PR, Liu L. Ligation of expressed protein alpha-hydrazides via genetic incorporation of an alpha-hydroxy acid. ACS Chem Biol. 2012;7:1015–1022. doi: 10.1021/cb300020s. [DOI] [PubMed] [Google Scholar]
- 46.Hansen RW, Hayashi JA. Glycolate metabolism in Escherichia coli. J Bacteriol. 1962;83:679–687. doi: 10.1128/jb.83.3.679-687.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grochowski LL, Xu H, White RH. Identification of lactaldehyde dehydrogenase in Methanocaldococcus jannaschii and its involvement in production of lactate for F420 biosynthesis. J Bacteriol. 2006;188:2836–2844. doi: 10.1128/JB.188.8.2836-2844.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baldoma L, Aguilar J. Involvement of lactaldehyde dehydrogenase in several metabolic pathways of Escherichia coli K12. J Biol Chem. 1987;262:13991–13996. [PubMed] [Google Scholar]
- 49.Friedrich CA, Morizot DC, Siciliano MJ, Ferrell RE. The reduction of aromatic alpha-keto acids by cytoplasmic malate dehydrogenase and lactate dehydrogenase. Biochemical genetics. 1987;25:657–669. doi: 10.1007/BF00556210. [DOI] [PubMed] [Google Scholar]
- 50.Weber WW, Zannoni VG. Reduction of aromatic alpha-keto acids by lactic dehydrogenase isozymes and aromatic alpha-keto acid reductase. Ann N Y Acad Sci. 1968;151:627–637. doi: 10.1111/j.1749-6632.1968.tb11923.x. [DOI] [PubMed] [Google Scholar]
- 51.Giege R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26:5017–5035. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Normanly J, Ollick T, Abelson J. Eight base changes are sufficient to convert a leucine-inserting tRNA into a serine-inserting tRNA. Proc Natl Acad Sci U S A. 1992;89:5680–5684. doi: 10.1073/pnas.89.12.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Breitschopf K, Achsel T, Busch K, Gross HJ. Identity elements of human tRNA(Leu): structural requirements for converting human tRNA(Ser) into a leucine acceptor in vitro. Nucleic Acids Res. 1995;23:3633–3637. doi: 10.1093/nar/23.18.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Normanly J, Ogden RC, Horvath SJ, Abelson J. Changing the identity of a transfer RNA. Nature. 1986;321:213–219. doi: 10.1038/321213a0. [DOI] [PubMed] [Google Scholar]
- 55.Asahara H, Himeno H, Tamura K, Nameki N, Hasegawa T, Shimizu M. Escherichia coli seryl-tRNA synthetase recognizes tRNA(Ser) by its characteristic tertiary structure. J Mol Biol. 1994;236:738–748. doi: 10.1006/jmbi.1994.1186. [DOI] [PubMed] [Google Scholar]
- 56.Palencia A, Crepin T, Vu MT, Lincecum TL, Jr, Martinis SA, Cusack S. Structural dynamics of the aminoacylation and proofreading functional cycle of bacterial leucyl-tRNA synthetase. Nat Struct Mol Biol. 2012;19:677–684. doi: 10.1038/nsmb.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harper AD, Bailey CB, Edwards AD, Detelich JF, Keatinge-Clay AT. Preparative, in vitro biocatalysis of triketide lactone chiral building blocks. Chembiochem. 2012;13:2200–2203. doi: 10.1002/cbic.201200378. [DOI] [PubMed] [Google Scholar]
- 58.Piasecki SK, Taylor CA, Detelich JF, Liu J, Zheng J, Komsoukaniants A, Siegel DR, Keatinge-Clay AT. Employing modular polyketide synthase ketoreductases as biocatalysts in the preparative chemoenzymatic syntheses of diketide chiral building blocks. Chem Biol. 2011;18:1331–1340. doi: 10.1016/j.chembiol.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 59.Popp BV, Ball ZT. Structure-selective modification of aromatic side chains with dirhodium metallopeptide catalysts. J Am Chem Soc. 2010;132:6660–6662. doi: 10.1021/ja101456c. [DOI] [PubMed] [Google Scholar]
- 60.Bianco A, Townsley FM, Greiss S, Lang K, Chin JW. Expanding the genetic code of Drosophila melanogaster. Nat Chem Biol. 2012;8:748–750. doi: 10.1038/nchembio.1043. [DOI] [PubMed] [Google Scholar]
- 61.Wan W, Huang Y, Wang Z, Russell WK, Pai PJ, Russell DH, Liu WR. A facile system for genetic incorporation of two different noncanonical amino acids into one protein in Escherichia coli. Angew Chem Int Ed Engl. 2010;49:3211–3214. doi: 10.1002/anie.201000465. [DOI] [PubMed] [Google Scholar]
- 62.Odoi KA, Huang Y, Rezenom YH, Liu WR. Nonsense and sense suppression abilities of original and derivative Methanosarcina mazei pyrrolysyl-tRNA synthetase-tRNA(Pyl) pairs in the Escherichia coli BL21(DE3) cell strain. PLoS One. 2013;8:e57035. doi: 10.1371/journal.pone.0057035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wong ML, Guzei IA, Kiessling LL. An asymmetric synthesis of L-pyrrolysine. Org Lett. 2012;14:1378–1381. doi: 10.1021/ol300045c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nguyen DP, Garcia Alai MM, Virdee S, Chin JW. Genetically directing varepsilon-N, N-dimethyl-L-lysine in recombinant histones. Chem Biol. 2010;17:1072–1076. doi: 10.1016/j.chembiol.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 65.Virdee S, Ye Y, Nguyen DP, Komander D, Chin JW. Engineered diubiquitin synthesis reveals Lys29-isopeptide specificity of an OTU deubiquitinase. Nat Chem Biol. 2010;6:750–757. doi: 10.1038/nchembio.426. [DOI] [PubMed] [Google Scholar]
- 66.Li YM, Yang MY, Huang YC, Song XD, Liu L, Chen PR. Genetically encoded alkenyl-pyrrolysine analogues for thiol-ene reaction mediated site-specific protein labeling. Chem Sci. 2012;3:2766–2770. [Google Scholar]
- 67.Li WT, Mahapatra A, Longstaff DG, Bechtel J, Zhao G, Kang PT, Chan MK, Krzycki JA. Specificity of pyrrolysyl-tRNA synthetase for pyrrolysine and pyrrolysine analogs. J Mol Biol. 2009;385:1156–1164. doi: 10.1016/j.jmb.2008.11.032. [DOI] [PubMed] [Google Scholar]
- 68.Fekner T, Li X, Lee MM, Chan MK. A pyrrolysine analogue for protein click chemistry. Angew Chem Int Ed Engl. 2009;48:1633–1635. doi: 10.1002/anie.200805420. [DOI] [PubMed] [Google Scholar]
- 69.Li X, Fekner T, Chan MK. N6-(2-(R)-propargylglycyl)lysine as a clickable pyrrolysine mimic. Chem Asian J. 2010;5:1765–1769. doi: 10.1002/asia.201000205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee MM, Fekner T, Tang TH, Wang L, Chan AH, Hsu PH, Au SW, Chan MK. A click-and-release pyrrolysine analogue. Chembiochem. 2013;14:805–808. doi: 10.1002/cbic.201300124. [DOI] [PubMed] [Google Scholar]
- 71.Nguyen DP, Lusic H, Neumann H, Kapadnis PB, Deiters A, Chin JW. Genetic encoding and labeling of aliphatic azides and alkynes in recombinant proteins via a pyrrolysyl-tRNA Synthetase/tRNA(CUA) pair and click chemistry. J Am Chem Soc. 2009;131:8720–8721. doi: 10.1021/ja900553w. [DOI] [PubMed] [Google Scholar]
- 72.Devaraj NK, Weissleder R. Biomedical applications of tetrazine cycloadditions. Acc Chem Res. 2011;44:816–827. doi: 10.1021/ar200037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Devaraj NK, Weissleder R, Hilderbrand SA. Tetrazine-based cycloadditions: application to pretargeted live cell imaging. Bioconjug Chem. 2008;19:2297–2299. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blackman ML, Royzen M, Fox JM. Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J Am Chem Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lang K, Davis L, Torres-Kolbus J, Chou C, Deiters A, Chin JW. Genetically encoded norbornene directs site-specific cellular protein labelling via a rapid bioorthogonal reaction. Nat Chem. 2012;4:298–304. doi: 10.1038/nchem.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Synthesis of proteins by native chemical ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 77.Muir TW, Sondhi D, Cole PA. Expressed protein ligation: a general method for protein engineering. Proc Natl Acad Sci U S A. 1998;95:6705–6710. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ren H, Xiao F, Zhan K, Kim YP, Xie H, Xia Z, Rao J. A biocompatible condensation reaction for the labeling of terminal cysteine residues on proteins. Angew Chem Int Ed Engl. 2009;48:9658–9662. doi: 10.1002/anie.200903627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li X, Fekner T, Ottesen JJ, Chan MK. A pyrrolysine analogue for site-specific protein ubiquitination. Angew Chem Int Ed Engl. 2009;48:9184–9187. doi: 10.1002/anie.200904472. [DOI] [PubMed] [Google Scholar]
- 80.Nguyen DP, Elliott T, Holt M, Muir TW, Chin JW. Genetically encoded 1,2-aminothiols facilitate rapid and site-specific protein labeling via a bio-orthogonal cyanobenzothiazole condensation. J Am Chem Soc. 2011;133:11418–11421. doi: 10.1021/ja203111c. [DOI] [PubMed] [Google Scholar]
- 81.Nguyen DP, Garcia Alai MM, Kapadnis PB, Neumann H, Chin JW. Genetically encoding N(epsilon)-methyl-L-lysine in recombinant histones. J Am Chem Soc. 2009;131:14194–14195. doi: 10.1021/ja906603s. [DOI] [PubMed] [Google Scholar]
- 82.Ai HW, Lee JW, Schultz PG. A method to site-specifically introduce methyllysine into proteins in E. coli. Chem Commun (Camb) 2010;46:5506–5508. doi: 10.1039/c0cc00108b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gattner MJ, Vrabel M, Carell T. Synthesis of epsilon-N-propionyl-, epsilon-N-butyryl-, and epsilon-N-crotonyl-lysine containing histone H3 using the pyrrolysine system. Chem Commun (Camb) 2013;49:379–381. doi: 10.1039/c2cc37836a. [DOI] [PubMed] [Google Scholar]
- 84.Chou CJ, Uprety R, Davis L, Chin JW, Deiters A. Genetically encoding an aliphatic diazirine for protein photocrosslinking. Chem Sci. 2011;2:480–483. [Google Scholar]
- 85.Hao Z, Song Y, Lin S, Yang M, Liang Y, Wang J, Chen PR. A readily synthesized cyclic pyrrolysine analogue for site-specific protein “click” labeling. Chem Commun (Camb) 2011;47:4502–4504. doi: 10.1039/c1cc00024a. [DOI] [PubMed] [Google Scholar]
- 86.Plass T, Milles S, Koehler C, Schultz C, Lemke EA. Genetically encoded copper-free click chemistry. Angew Chem Int Ed Engl. 2011;50:3878–3881. doi: 10.1002/anie.201008178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Borrmann A, Milles S, Plass T, Dommerholt J, Verkade JM, Wiessler M, Schultz C, van Hest JC, van Delft FL, Lemke EA. Genetic encoding of a bicyclo[6.1.0]nonyne-charged amino acid enables fast cellular protein imaging by metal-free ligation. Chembiochem. 2012;13:2094–2099. doi: 10.1002/cbic.201200407. [DOI] [PubMed] [Google Scholar]
- 88.Kaya E, Vrabel M, Deiml C, Prill S, Fluxa VS, Carell T. A genetically encoded norbornene amino acid for the mild and selective modification of proteins in a copper-free click reaction. Angew Chem Int Ed Engl. 2012;51:4466–4469. doi: 10.1002/anie.201109252. [DOI] [PubMed] [Google Scholar]
- 89.Plass T, Milles S, Koehler C, Szymanski J, Mueller R, Wiessler M, Schultz C, Lemke EA. Amino acids for Diels-Alder reactions in living cells. Angew Chem Int Ed Engl. 2012;51:4166–4170. doi: 10.1002/anie.201108231. [DOI] [PubMed] [Google Scholar]
- 90.Lang K, Davis L, Wallace S, Mahesh M, Cox DJ, Blackman ML, Fox JM, Chin JW. Genetic Encoding of bicyclononynes and trans-cyclooctenes for site-specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic Diels-Alder reactions. J Am Chem Soc. 2012;134:10317–10320. doi: 10.1021/ja302832g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li J, Lin S, Wang J, Jia S, Yang M, Hao Z, Zhang X, Chen PR. Ligand-free palladium-mediated site-specific protein labeling inside gram-negative bacterial pathogens. J Am Chem Soc. 2013;135:7330–7338. doi: 10.1021/ja402424j. [DOI] [PubMed] [Google Scholar]
- 92.Li N, Lim RK, Edwardraja S, Lin Q. Copper-free Sonogashira cross-coupling for functionalization of alkyne-encoded proteins in aqueous medium and in bacterial cells. J Am Chem Soc. 2011;133:15316–15319. doi: 10.1021/ja2066913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li Y, Pan M, Li Y, Huang Y, Guo Q. Thiol-yne radical reaction mediated site-specific protein labeling via genetic incorporation of an alkynyl-L-lysine analogue. Org Biomol Chem. 2013;11:2624–2629. doi: 10.1039/c3ob27116a. [DOI] [PubMed] [Google Scholar]
- 94.Li F, Zhang H, Sun Y, Pan Y, Zhou J, Wang J. Expanding the genetic code for photoclick chemistry in E. coli, mammalian cells, and A. thaliana. Angew Chem Int Ed Engl. 2013;52:9700–9704. doi: 10.1002/anie.201303477. [DOI] [PubMed] [Google Scholar]
- 95.Zhang M, Lin S, Song X, Liu J, Fu Y, Ge X, Fu X, Chang Z, Chen PR. A genetically incorporated crosslinker reveals chaperone cooperation in acid resistance. Nat Chem Biol. 2011;7:671–677. doi: 10.1038/nchembio.644. [DOI] [PubMed] [Google Scholar]
- 96.Lin S, Zhang Z, Xu H, Li L, Chen S, Li J, Hao Z, Chen PR. Site-specific incorporation of photo-cross-linker and bioorthogonal amino acids into enteric bacterial pathogens. J Am Chem Soc. 2011;133:20581–20587. doi: 10.1021/ja209008w. [DOI] [PubMed] [Google Scholar]
- 97.Ai HW, Shen W, Sagi A, Chen PR, Schultz PG. Probing protein-protein interactions with a genetically encoded photo-crosslinking amino acid. Chembiochem. 2011;12:1854–1857. doi: 10.1002/cbic.201100194. [DOI] [PubMed] [Google Scholar]
- 98.Gautier A, Nguyen DP, Lusic H, An W, Deiters A, Chin JW. Genetically encoded photocontrol of protein localization in mammalian cells. J Am Chem Soc. 2010;132:4086–4088. doi: 10.1021/ja910688s. [DOI] [PubMed] [Google Scholar]
- 99.Gautier A, Deiters A, Chin JW. Light-activated kinases enable temporal dissection of signaling networks in living cells. J Am Chem Soc. 2011;133:2124–2127. doi: 10.1021/ja1109979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hemphill J, Chou C, Chin JW, Deiters A. Genetically encoded light-activated transcription for spatiotemporal control of gene expression and gene silencing in mammalian cells. J Am Chem Soc. 2013;135:13433–13439. doi: 10.1021/ja4051026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Groff D, Chen PR, Peters FB, Schultz PG. A genetically encoded epsilon-N-methyl lysine in mammalian cells. Chembiochem. 2010;11:1066–1068. doi: 10.1002/cbic.200900690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang YS, Wu B, Wang Z, Huang Y, Wan W, Russell WK, Pai PJ, Moe YN, Russell DH, Liu WR. A genetically encoded photocaged Nepsilon-methyl-L-lysine. Mol Biosyst. 2010;6:1557–1560. doi: 10.1039/c002155e. [DOI] [PubMed] [Google Scholar]
- 103.Elsasser SJ, Huang H, Lewis PW, Chin JW, Allis CD, Patel DJ. DAXX envelops a histone H3.3-H4 dimer for H3.3-specific recognition. Nature. 2012;491:560–565. doi: 10.1038/nature11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Arbely E, Natan E, Brandt T, Allen MD, Veprintsev DB, Robinson CV, Chin JW, Joerger AC, Fersht AR. Acetylation of lysine 120 of p53 endows DNA-binding specificity at effective physiological salt concentration. Proc Natl Acad Sci U S A. 2011;108:8251–8256. doi: 10.1073/pnas.1105028108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Neumann H, Peak-Chew SY, Chin JW. Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat Chem Biol. 2008;4:232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]
- 106.Huang Y, Russell WK, Wan W, Pai PJ, Russell DH, Liu W. A convenient method for genetic incorporation of multiple noncanonical amino acids into one protein in Escherichia coli. Mol Biosyst. 2010;6:683–686. doi: 10.1039/b920120c. [DOI] [PubMed] [Google Scholar]
- 107.Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol Cell. 2009;36:153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Umehara T, Kim J, Lee S, Guo LT, Soll D, Park HS. N-Acetyl lysyl-tRNA synthetases evolved by a CcdB-based selection possess N-acetyl lysine specificity in vitro and in vivo. FEBS Lett. 2012;586:729–733. doi: 10.1016/j.febslet.2012.01.029. [DOI] [PubMed] [Google Scholar]
- 109.Oppikofer M, Kueng S, Martino F, Soeroes S, Hancock SM, Chin JW, Fischle W, Gasser SM. A dual role of H4K16 acetylation in the establishment of yeast silent chromatin. EMBO J. 2011;30:2610–2621. doi: 10.1038/emboj.2011.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu WR, Wang YS, Wan W. Synthesis of proteins with defined posttranslational modifications using the genetic noncanonical amino acid incorporation approach. Mol Biosyst. 2011;7:38–47. doi: 10.1039/c0mb00216j. [DOI] [PubMed] [Google Scholar]
- 111.Kim CH, Kang M, Kim HJ, Chatterjee A, Schultz PG. Site-specific incorporation of epsilon-N-crotonyllysine into histones. Angew Chem Int Ed Engl. 2012;51:7246–7249. doi: 10.1002/anie.201203349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang ZU, Wang YS, Pai PJ, Russell WK, Russell DH, Liu WR. A Facile Method to Synthesize Histones with Posttranslational Modification Mimics. Biochemistry. 2012;51:5232–5234. doi: 10.1021/bi300535a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schmidt MJ, Summerer D. Red-light-controlled protein-RNA crosslinking with a genetically encoded furan. Angew Chem Int Ed Engl. 2013;52:4690–4693. doi: 10.1002/anie.201300754. [DOI] [PubMed] [Google Scholar]
- 114.Schmidt MJ, Borbas J, Drescher M, Summerer D. A genetically encoded spin label for electron paramagnetic resonance distance measurements. J Am Chem Soc. 2014;136:1238–1241. doi: 10.1021/ja411535q. [DOI] [PubMed] [Google Scholar]
- 115.Wang YS, Russell WK, Wang Z, Wan W, Dodd LE, Pai PJ, Russell DH, Liu WR. The de novo engineering of pyrrolysyl-tRNA synthetase for genetic incorporation of L-phenylalanine and its derivatives. Mol Biosyst. 2011;7:714–717. doi: 10.1039/c0mb00217h. [DOI] [PubMed] [Google Scholar]
- 116.Ko JH, Wang YS, Nakamura A, Guo LT, Soll D, Umehara T. Pyrrolysyl-tRNA synthetase variants reveal ancestral aminoacylation function. FEBS Lett. 2013;587:3243–3248. doi: 10.1016/j.febslet.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang L, Brock A, Herberich B, Schultz PG. Expanding the genetic code of Escherichia coli. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 118.Liu W, Brock A, Chen S, Chen S, Schultz PG. Genetic incorporation of unnatural amino acids into proteins in mammalian cells. Nat Methods. 2007;4:239–244. doi: 10.1038/nmeth1016. [DOI] [PubMed] [Google Scholar]
- 119.Liu CC, Schultz PG. Adding new chemistries to the genetic code. Annu Rev Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 120.Wang YS, Fang X, Wallace AL, Wu B, Liu WR. A rationally designed pyrrolysyl-tRNA synthetase mutant with a broad substrate spectrum. J Am Chem Soc. 2012;134:2950–2953. doi: 10.1021/ja211972x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang YS, Fang X, Chen HY, Wu B, Wang ZU, Hilty C, Liu WR. Genetic Incorporation of Twelve meta-Substituted Phenylalanine Derivatives Using a Single Pyrrolysyl-tRNA Synthetase Mutant. ACS Chem Biol. 2013;8:405–415. doi: 10.1021/cb300512r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tuley A, Wang YS, Fang X, Kurra Y, Rezenom YH, Liu WR. The genetic incorporation of thirteen novel non-canonical amino acids. Chem Commun (Camb) 2014;50:2673–2675. doi: 10.1039/c3cc49068h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tharp JM, Wang Y-S, Lee Y-J, Yang Y, Liu WR. The genetic incorporation of severn ortho-substituted phenylalanine derivatives. ACS Chem Biol. 2014 doi: 10.1021/cb400917a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Arbely E, Torres-Kolbus J, Deiters A, Chin JW. Photocontrol of tyrosine phosphorylation in mammalian cells via genetic encoding of photocaged tyrosine. J Am Chem Soc. 2012;134:11912–11915. doi: 10.1021/ja3046958. [DOI] [PubMed] [Google Scholar]
- 125.Takimoto JK, Dellas N, Noel JP, Wang L. Stereochemical basis for engineered pyrrolysyl-tRNA synthetase and the efficient in vivo incorporation of structurally divergent non-native amino acids. ACS Chem Biol. 2011;6:733–743. doi: 10.1021/cb200057a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lacey VK, Louie GV, Noel JP, Wang L. Expanding the Library and Substrate Diversity of the Pyrrolysyl-tRNA Synthetase to Incorporate Unnatural Amino Acids Containing Conjugated Rings. Chembiochem. 2013 doi: 10.1002/cbic.201300400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Niu W, Schultz PG, Guo J. An expanded genetic code in Mammalian cells with a functional quadruplet codon. ACS Chem Biol. 2013;8:1640–1645. doi: 10.1021/cb4001662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xiao H, Chatterjee A, Choi SH, Bajjuri KM, Sinha SC, Schultz PG. Genetic incorporation of multiple unnatural amino acids into proteins in Mammalian cells. Angew Chem Int Ed Engl. 2013;52:14080–14083. doi: 10.1002/anie.201308137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chatterjee A, Sun SB, Furman JL, Xiao H, Schultz PG. A Versatile Platform for Single- and Multiple-Unnatural Amino Acid Mutagenesis in Escherichia coli. Biochemistry. 2013;52:1828–1837. doi: 10.1021/bi4000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wu B, Wang Z, Huang Y, Liu WR. Catalyst-free and site-specific one-pot dual-labeling of a protein directed by two genetically incorporated noncanonical amino acids. Chembiochem. 2012;13:1405–1408. doi: 10.1002/cbic.201200281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Huang Y, Wan W, Russell WK, Pai PJ, Wang Z, Russell DH, Liu W. Genetic incorporation of an aliphatic keto-containing amino acid into proteins for their site-specific modifications. Bioorg Med Chem Lett. 2010;20:878–880. doi: 10.1016/j.bmcl.2009.12.077. [DOI] [PubMed] [Google Scholar]