

Abstract
Recent evidence from large cohort studies suggests that there exists a higher cancer incidence in people with type 2 diabetes (DM2). However, to date, the potential reasons for this association remain unclear. Hyperglycemia, the most important feature of diabetes, may be responsible for the excess glucose supply for these glucose-hungry cells, and it contributes to apoptosis resistance, oncogenesis, and tumor cell resistance to chemotherapy. Considering associations between diabetes and malignancies, the effect of hyperglycemia on cancer progression in cancer patients with abnormal blood glucose should not be neglected. In this paper, we describe the role that hyperglycemia plays in cancer progression and treatment and illustrate that hyperglycemia may contribute to a more malignant phenotype of cancer cells and lead to drug resistance. Therefore, controlling hyperglycemia may have important therapeutic implications in cancer patients.
1. Introduction
Hyperglycemia, or high blood glucose, is a condition in which an excessive amount of glucose circulates in the blood which develops when the body has too little insulin or when the body cannot use insulin properly. A number of medical conditions can cause hyperglycemia, including diabetes mellitus (DM) [1], obesity [2], pancreatitis [3], chronic stress [4], and cancer. Interestingly, the existing epidemiological evidence indicated that all of these hyperglycemia-related conditions are likely to be associated with tumorigenesis or tumor progression [5–7]. Nowadays, researchers mainly focus on the impacts of hyperglycemia on eyes, kidneys, nerves, and heart; little attention has been paid to the roles of hyperglycemia in cancer. Given the prevalence of hyperglycemia-related conditions existing in cancer patients, the relationship between hyperglycemia and cancer should arouse enough attention.
DM is the most common medical conditions responsible for hyperglycemia. In DM patients, blood glucose levels rise either because there is an insufficient amount of insulin in the body or the body cannot use insulin well. Diabetes mellitus has a current prevalence of 347 million people worldwide, and this number will continue to increase [8]. Epidemiologic evidence in the past suggested that people with diabetes are at significantly higher risk for many types of cancer [9]. It has been recognized that diabetes plays a crucial role in the development of solid organ malignancies including liver [10, 11], pancreatic [10, 12], colorectal [10, 13], breast [14, 15], endometrial [16–18], and bladder cancers [19, 20]. Among these cancers, liver cancer and pancreatic cancer (PC) show the strongest association with DM2. A recent meta-analysis of 23 articles demonstrated a 41% increase in cancer mortality related to endometrial, breast, and colorectal cancers in patients with preexisting diabetes compared with normoglycemic individuals [21]. Thus, many studies have provided consistent evidence on the association of diabetes with an increased risk of cancer. In contrast, diabetes occurs more frequently in patients with cancer than in the general population; therefore, new-onset diabetes may be an early indicator of subclinical cancer.
Following hyperglycemia, which was first reported in cancer patients in 1885, tumor tissues were found to sustain higher rates of glucose utilization than that in normal tissues by Warburg et al. in the 1920s [22]. Patients with various types of cancer have been examined in many clinical studies for proof of abnormalities in carbohydrate metabolism. The clinical evidence indicated a positive association between neoplasia and concomitant abnormalities in glucose metabolism. Moreover, several groups have described specific cellular mechanisms associated with glucose uptake in malignant tissues [23–25]. Most malignant tissues have increased fludeoxyglucose (18F) (18F-FDG) uptake associated with an increased rate of glycolysis and glucose transportation. The increase in 18F-FDG uptake noted in malignant tissue is related in a complex manner to the proliferative activity of malignant tissue and the number of viable tumor cells [26–28].
Increasing evidence suggests a close association between diabetes and various malignancies; however, the potential biologic links between the two diseases are incompletely understood. Given that hyperglycemia is the most important biological feature of DM and cancer that are composed of glucose-hungry cells, it is not hard to imagine that hyperglycemia may play an important role during cancer progression in cancer patients with DM. Here, we review the available evidence of the relationship between hyperglycemia and different biological characteristics of cancer. It appears that hyperglycemia may contribute to a more malignant phenotype of cancer cells, including proliferation, apoptosis inhibition, metastasis, perineural invasion, chemotherapy resistance, and chemotherapy intolerance (Figure 1).
Figure 1.
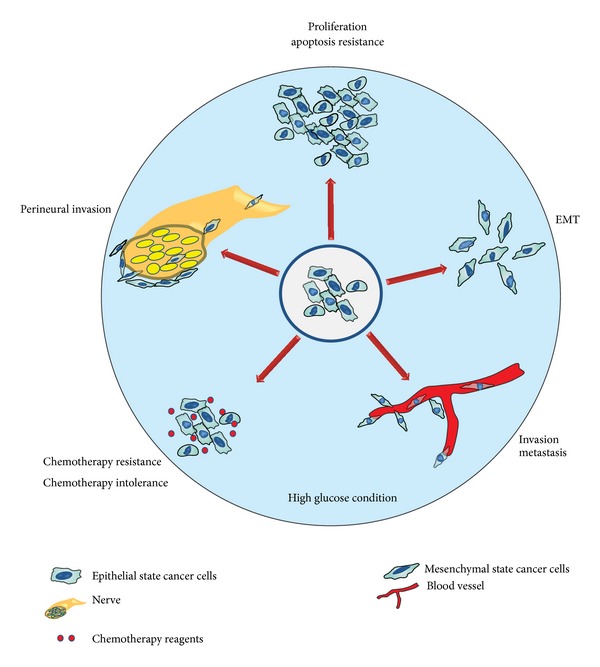
Hyperglycemia contributes to malignant cancer cell phenotypes. There is increasing evidence suggesting that there is a link between cancer and diabetes mellitus. Regardless of other shared metabolic factors, hyperglycemia, the most typical characteristic of diabetes, may be one reason to explain the prevalence of cancer incidence in patients with diabetes mellitus. Research shows that hyperglycemia may contribute to an enhanced proliferation ability, apoptosis inhibition, metastasis, perineural invasion, chemotherapy resistance, and chemotherapy intolerance.
2. Hyperglycemia and Cancer Cell Proliferation
Glucose is specifically required to meet the metabolic demands of the fast proliferation cancer cells. It has been known that glucose is a primary driving force for the growth of tumor cells for more than two decades [29]. The promoting role of hyperglycemia on cancer proliferation is not hard to understand.
Hyperglycemia is often accompanied with hyperinsulinemia in people with DM2. Proliferation assays revealed that high levels of glucose (11 mM) and insulin (100 ng/mL) promoted the proliferation of the tumor cell lines HT29 (human colon carcinoma), SW480 (human colorectal carcinoma), MCF-7 (human breast adenocarcinoma), MDA MB468 (human breast adenocarcinoma), PC3 (human prostate cancer), and T24 (human bladder carcinoma) [30]. Furthermore, the addition of oral glucose, insulin injections, or both exhibited promoting effect on mammary tumor growth in rats [31]. Recent studies showed that insulin promote cancer progression by enhancing metabolic capacities of cancer cells [32]. Since high glucose and high insulin can induce cancer cell proliferation through different mechanisms, controlling blood glucose levels and insulin levels at the appropriate level would be beneficial in cancer patients bearing DM.
Under hyperglycemic conditions, studies have found not only an increased expression of the collagen receptor but also an integrin-linked kinase and other kinases regulating many cellular processes, including growth and proliferation [33]. There is some evidence that diabetes could promote PC cell proliferation. Chu et al. examined the records of resected PC patients and found that preexisting diabetes is associated with reduced survival in patients who underwent resection for PC. Additionally, PC with new-onset diabetes may exhibit increased tumor size and decreased postresection survival [34]. When hamster H2T pancreatic carcinoma cells were implanted into the cheek pouches of Syrian hamsters, the tumor size, weight, and total DNA content were significantly greater in animals with diabetes, demonstrating that diabetes appears to promote the growth of PC cells in the hamster [35].
Increased production of reactive oxygen species (ROS) from mitochondria is the main cause of hyperglycemic complications (Figure 2). In diabetic individuals, hyperglycemia in susceptible cells results in the overproduction of superoxide by the mitochondrial electron transport chain [36]. Elevated levels of ROS can lead to cellular DNA mutations and may, therefore, play an important role in the initiation and progression of multistage carcinogenesis. More importantly, the generation of ROS was required for K-Ras-induced anchorage-independent growth through regulation of the ERK MAPK signaling pathway [37].
Figure 2.
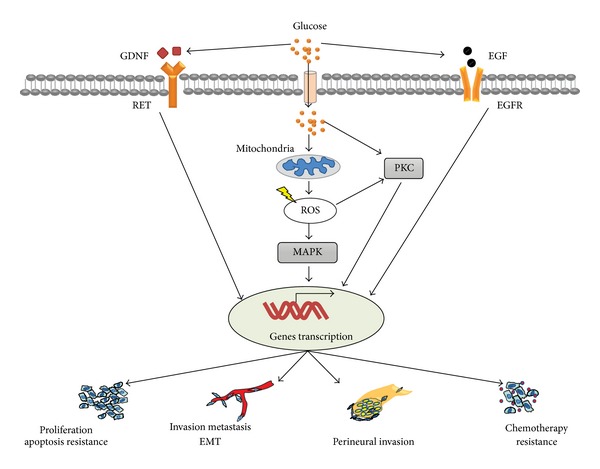
Mechanism of high glucose-induced cellular events in cancer cells. High glucose (hyperglycemia) generates cellular ROS mainly through mitochondrial metabolism; elevated ROS activate MAPK cascade, which cause cellular events by inducing related genes transcription. In addition, high glucose can induce activation of protein kinase C (PKC) through direct and indirect pathways. It is also speculated that high glucose may induce EGF transcription and EGFR transactivation, contributing to various biological behavior of cancer cells. High glucose-mediated GDNF upregulation may also involve in different cellular events through GDNF/RET cascade.
Hyperglycemia also specifically activates polyol metabolism with a subsequent decrease in Na+/K+-ATPase activity in pancreatic duct epithelial cells [38]. In addition, Tingstedt et al. found that the regenerating gene (REG) I-alpha protein was preferentially expressed in cancerous tissues and cells from PC patients with diabetes, and overexpression of this protein resulted in accelerated cell proliferation and consequently tumor growth in vitro and in vivo [39]. In addition, glucose concentration may be an important factor in breast cancer cell proliferation, and the prevalence of breast cancer is high in diabetic patients. The effects of glucose on breast cancer cell proliferation were evaluated by examining cell doubling time, DNA synthesis, the level of cell cycle related proteins, protein kinase C (PKC) isozyme expression, and the peroxisome proliferator activated receptor (PPAR) subtypes were determined following glucose exposure at normal (5.5 mM) and high (25 mM) concentrations in MCF-7 human breast cancer cells. In MCF-7 cells, high glucose stimulated cell proliferation as demonstrated by an increase in DNA synthesis and the expression of cdk2 and cyclin D1. The PKC-α, PPARγ, and PPARα protein levels were upregulated following high glucose treatment in drug-sensitive MCF-7 cells. These results suggested that hyperglycemia increases breast cancer cell proliferation through accelerated cell cycle progression with the upregulation of cdk2 and cyclin D1 [40]. Moreover, our group investigated the cell proliferation effects of glial cell line-derived neurotrophic factor (GDNF) and its tyrosine kinase receptor RET expression in BxPC-3 and MIA PaCa-2 cells with exposure to different glucose concentrations [41]. The proliferation of both BxPC-3 and MIA PaCa-2 cell lines was affected by glucose in a concentration-dependent manner. The definite expression of GDNF and RET was detected in both cell lines. The glucose concentration could alter the expression of GDNF and RET in a concentration-dependent manner correspondingly with an alteration in cell proliferation. The upregulation of GDNF and RET ligand-receptor interaction might participate in glucose-induced cancer progression (Figure 2). In addition, we demonstrated that the proliferative ability of BxPC-3 and Panc-1 cells was upregulated by high glucose in a concentration-dependent manner. Furthermore, the promotion effect of high glucose levels on EGF transcription and secretion but not its receptors in these PC cell lines was detected by using an EGF-neutralizing antibody and RT-PCR. In addition, EGFR transactivation is induced by high glucose levels in a concentration- and time-dependent manner in PC cells in the presence of an EGF-neutralizing antibody. These results suggest that high glucose levels promote PC cell proliferation via the induction of EGF expression and transactivation of EGFR [42].
3. Hyperglycemia and Cancer Cell Apoptosis
Apoptosis, a genetically regulated process that is essential to maintain individual homeostasis, is out of control in cancer. Resistance to apoptosis is one of the distinctive hallmarks of cancer cell [43]. High glucose condition is easy to induce apoptosis in normal cells [44, 45]. However, glucose metabolism protects cancer cells from cytochrome C-mediated apoptosis [46].
Recent in vitro studies suggest that an important mechanism for enhanced glucose metabolism in carcinoma cells involves the overexpression of transmembrane glucose transporters [47, 48]. Changes in glucose metabolism have also been found in many tumors, causing an increased production of lactate [49, 50]. Elevated lactate within cancer cells indicates a switch in glucose metabolism from the aerobic to anaerobic utilization of glucose, which was first described by Warburg (1956). Modern molecular biology has led to a renaissance in the Warburg effect [51, 52]. A permanent increase in anaerobic glucose utilization in primary tumors is a characteristic of more aggressive tumor cells [53]. Reduced mitochondrial respiration and an increased conversion of glucose into lactate combined with enhanced lactate secretion are associated with the acidification of a tumor and its environment [54]. This condition turns into an advantage for tumor cells with resistance to acidosis as realized by increased H+ transporter activity (e.g., Na+/H+ exchanger) [55]. However, in nonmalignant tissues, an acidic microenvironment is ordinarily toxic to mammalian cells, typically resulting in apoptosis through the activation of caspases [56].
Metformin, an oral antidiabetic drug in the biguanide class, is the first-line drug of choice for the treatment of DM2. The apoptosis-promoting effects of metformin on different cancers (e.g., ovarian cancer, breast cancer, and lung cancer) by increase of apoptotic genes have been demonstrated previously [57–59]. However, metformin-induced cancer cell apoptosis was prevented under high-glucose conditions in a carcinogen-induced rodent model of mammary tumorigenesis [60]. These data indicate that hyperglycemia can protect cancer cells from apoptosis process and thus failure to maintain glucose homeostasis may promote a more aggressive cancer phenotype.
4. Hyperglycemia and Cancer Cell Metastasis
Metastasis, which is considered a vital step in the progression of cancer, poses the largest problem for cancer treatment and is the main cause of cancer-associated deaths. Epidemiology studies showed that distant metastasis is responsible for nearly 90% of deaths from cancer [61–64].
Ever since metastasis has been investigated, models and concepts about how the metastatic disease process works have been suggested [65]. These include a “seed and soil” hypothesis in which a population of tumor cells are envisaged as seeds that require a suitable organ microenvironment, called the “soil,” to survive outside the primary tumor and grow as metastases [66, 67]. In the primary tumor site, reengineered cancer cells transform into an invasive phenotype to penetrate the tumor stroma and enter the blood circulation or lymphatic system via intravasation. In secondary lesions, a comfortable premetastatic niche must then be established for the traveling “seeds” forming macrometastases.
Recent studies revealed that hyperglycemia is associated with metastasis and might contribute to reengineer cancer cells in primary lesions. An epidemiology study demonstrated that, in cancer patients with DM2 or hyperglycemia, the proportion of tumor recurrence, metastasis, or fatal outcome is higher than in patients without metabolic disease [68]. Additionally, metformin, the most frequently used antidiabetic drug, inhibits cell migration and invasion by attenuating the cancer stem cell (CSC) function mediated by deregulating miRNAs including let-7a, let-7b, miR-26a, miR-101, miR-200b, and miR-200c, which are typically lost in PC [69]. In addition, metformin treatment also regulates the phenotype of CD44+/CD24− breast cancer stem cells by decreasing the expression of key EMT factors including the transcription factors ZEB, Twist, and Slug and the cytokine TGF-beta [70].
Vairaktaris et al. investigated the molecular basis of the association between oral cancer and diabetes (type I) in a rat model induced by a single intraperitoneal injection of streptozotocin dissolved in saline buffer [71]. This group observed that, although E26 transformation specific-1 (ets-1) expression was observed in diabetic and normal rats, its expression was higher in diabetic than normal rats in different cancer stages. It is widely recognized that ets-1 encodes a transcription factor that is involved in the transcriptional regulation of several genes implicated in tumor invasion and metastasis, such as collagenase I, stromelysin, and urokinase plasminogen activator [72–74]. Ets-1 has been implicated in human oral squamous cell carcinoma (OSCC), and ets-1 levels appear to correlate well with the grade of invasiveness and metastasis [75–77].
In recent years, the epithelial-mesenchymal transition (EMT) has received sufficient attention in metastasis. Cancer cells undergoing EMT obtain invasive properties and get into the surrounding tissue, leading to the creation of a suitable microenvironment for cancer proliferation and metastasis [78]. Accumulating data and studies have examined the relationship between EMT and hyperglycemia, mostly focusing on diabetic renal injury [79–81], diabetic vascular disease [82, 83], and peritoneal dialysis [84, 85]. Unfortunately, little attention has been focused on the role of hyperglycemia in inducing the cancer cell EMT phenotype. Our results demonstrated that high glucose could increase the production of ROS in the PC cell lines BxPC-3 and Panc-1, which further leads to cell motility and invasiveness [86]. We hypothesized that hyperglycemia facilitates PC metastasis by EMT induction and vascular destruction via oxidative stress [87].
5. Hyperglycemia and Perineural Invasion in Cancer
Perineural invasion (PNI) is defined as the presence of cancer cells within the epineural, perineural, and endoneurial spaces of the neuronal sheet and around the nerves [88, 89]. PNI is a distinct pathological entity that can be observed in the absence of lymphatic or vascular invasion, and it is associated with aggressive tumor behavior and worse clinical outcome [90]. Recent studies have demonstrated that hyperglycemia could facilitate PNI in several cancers, particularly pancreatic carcinoma [91, 92].
The mechanism of PNI in cancer is unclear. There are two prominent theories including the “path of low resistance.” There are three deficient sites around the perineurium: near the nerve ending, at the site invaded by the blood vessels present in nerves, and at the site invaded by the reticular fiber [93, 94]. Many previous studies presumed that tumor cells grow along the “path of low resistance,” and the path serves as a route for their distant migration [89]. Another possible explanation of PNI in PC is reciprocal signaling interactions. More recently, studies have demonstrated that PNI may involve reciprocal signaling interactions between tumor cells and nerves. These invading tumor cells may have acquired the ability to respond to proinvasive signals within the peripheral nerve milieu [89, 95]. The detection of increased neurotrophic factors such as nerve growth factor (NGF), glial cell line-derived neurotrophic factor (GDNF), brain-derived neurotrophic factor (BDNF), neural cell adhesion molecules (NCAMs), myelin-associated glycoprotein (MAG), and chemokines in intrapancreatic nerves and tumor cells and their receptors on tumor cells led to an increased attention to these molecules in recent years [96–98]. NGF and its receptor TrkA are the most widely observed among these factors. The receptor-ligand pair is overexpressed in PC cell lines and the perineurium of peripheral nerves. The binding of NGF to TrkA leads to the activation of the p44/42 MAPK signaling pathway, the promotion of cancer cell growth, increased invasiveness and metastasis, and eventually PNI mediation [99].
A recent study of 61 resected pancreatic tumors used histopathology to investigate many consecutive sections of tumor specimens, and the study reported an 86.9% (53/61) PNI rate in PC patients. Diabetic patients 93.75% (15/16) had a significantly higher frequency of PNI than nondiabetic patients 84.44% (38/45) [100]. A large retrospective study of 544 surgically resected pancreatic ductal adenocarcinoma patients observed similar results [101]. Diabetes or impaired glucose tolerance is often concurrently present in patients with PC and is associated with worse prognosis [34]. Nerve injury is a well-known complication of diabetes and is characterized by neuroinflammation [102]. Hyperglycemia in diabetes can cause up to a 4-fold increase in neuronal glucose levels. If persistent episodes of hyperglycemia exist, then intracellular glucose metabolism leads to neuronal damage [103, 104]. It is conceivable that, under hyperglycemic conditions, an increased level of oxidative stress and proinflammatory factors cause nerve damage and an inflammatory response [105], which simultaneously facilitates cancer cell proliferation, migration, and metastasis [106]. Li et al. revealed that nerve damage and regeneration simultaneously occur in the tumor microenvironment of PC patients with hyperglycemia; this simultaneous occurrence may aggravate the process of perineural invasion. The abnormal expression of NGF and p75 may also be involved in this process and subsequently lead to a lower rate of curative surgery [100]. In recent studies, researchers found that nerve invasion was dependent on GDNF secretion and mitogen-activated protein kinase activity. The GDNF coreceptors RET and GFRα1 were highly expressed in human pancreatic carcinomas by the same population of cells [96, 107–109]. Glucose concentrations could alter the expression of GDNF and RET in a concentration-dependent manner, and hyperglycemia could upregulate the interaction between GDNF and the RET ligand receptor [41].
In conclusion, hyperglycemia could promote PNI in several cancers, particularly pancreatic carcinoma. High glucose caused the demyelinization and axonal degeneration of nerves, which facilitate cancer cell invasion into nerves and enhanced interactions between nerve and cancer cells by increasing the expression of cytokines such as GDNF.
6. Hyperglycemia in Cancer Therapy
In addition to the effects of hyperglycemia on the biological behavior of tumor cells, the prevalence of transient hyperglycemia during induction chemotherapy has been observed, and existing evidence revealed another role of hyperglycemia in tumor treatment. There was evidence showed that hyperglycemia during chemotherapy for hematologic and solid tumors is correlated with increased toxicity [110]; thus, it appears that better glycemic control during chemotherapy could improve the toxicity and outcome of cancer patients. Moreover, hyperglycemia conferred resistance to chemotherapy for breast cancer but not nonmalignant cells, and this resistance was overcome by inhibiting fatty acid synthase (FAS) or ceramide production [111].
It has been known that DM patients were often accompanied by a disturbance in cellular innate immunity [112], and the impaired immune response may contribute to the ineffective chemotherapeutic treatment of cancer patients. For the past few years, epidemiological and laboratory evidence have shown that some antidiabetic pharmacotherapies exhibited outstanding effects for cancer prevention and treatment, such as metformin. Although some studies have revealed various molecular mechanisms for hypoglycemic agents and their anticancer effects, we should not neglect their glucose-lowering effects during cancer treatment because most malignancies constitute glucose-hungry cells. Collectively, controlling hyperglycemia may have important therapeutic implications for cancer patients. However, the role of hyperglycemia in cancer therapy and the exact mechanism remain unclear; thus, further studies are needed in this arena.
7. Conclusion and Future Directions
Increasing evidence has demonstrated a high incidence for various malignancies in patients with DM2. Although the shared associations between DM2 mellitus and cancer have been observed for a long time, the possible factors underlying cancer risk and mortality in this high-risk population remain uncertain. In this review, we discussed the effects of hyperglycemia, the key characteristic of diabetes mellitus, on various cancer biological behaviors and cancer treatment. In addition to providing rich nutrition for tumor growth directly, elevated glucose level could also induce the activation of some signaling pathways, all of which play important roles in cancer progression. Furthermore, hyperglycemiacanconfer resistance and intolerance to chemotherapy. Given the wide-ranging impact of hyperglycemia and the complexity of the microenvironment, the effect of hyperglycemia on the whole system and each component in a tumor microenvironment should not be neglected when exploring the relationship between cancer and diabetes mellitus. However, existing evidence indicates that hyperglycemia treatments may have important therapeutic implications in cancer patients.
Acknowledgment
This work was supported by Grants from the National Natural Scientific Foundation of China (no. 81172360 to Qingyong Ma and no. 81201824 to Xuqi Li).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Wanxing Duan and Xin Shen contributed equally to this work.
References
- 1.Dobbs R, Sakurai H, Sasaki H. Glucagon: role in the hyperglycemia of diabetes mellitus. Science. 1975;187(4176):544–547. doi: 10.1126/science.1089999. [DOI] [PubMed] [Google Scholar]
- 2.Martyn JAJ, Kaneki M, Yasuhara S. Obesity-induced insulin resistance and hyperglycemia: etiologic factors and molecular mechanisms. Anesthesiology. 2008;109(1):137–148. doi: 10.1097/ALN.0b013e3181799d45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fogar P, Pasquali C, Basso D, et al. Transforming growth factor β, fibrogenesis and hyperglycemia in patients with chronic pancreatitis. Journal of Medicine. 1998;29(5-6):277–287. [PubMed] [Google Scholar]
- 4.Mechanick JI. Metabolic mechanisms of stress hyperglycemia. Journal of Parenteral and Enteral Nutrition. 2006;30(2):157–163. doi: 10.1177/0148607106030002157. [DOI] [PubMed] [Google Scholar]
- 5.Kaiser J. Cancer. Cholesterol forges link between obesity and breast cancer. Science. 2013;342(6162):p. 1028. doi: 10.1126/science.342.6162.1028. [DOI] [PubMed] [Google Scholar]
- 6.Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; Aetiology, incidence, and early detection. Best Practice and Research: Clinical Gastroenterology. 2010;24(3):349–358. doi: 10.1016/j.bpg.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 7.Feng Z, Liu L, Zhang C, et al. Chronic restraint stress attenuates p53 function and promotes tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:7013–7018. doi: 10.1073/pnas.1203930109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danaei G, Finucane MM, Lu Y, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. The Lancet. 2011;378(9785):31–40. doi: 10.1016/S0140-6736(11)60679-X. [DOI] [PubMed] [Google Scholar]
- 9.Coughlin SS, Calle EE, Teras LR, Petrelli J, Thun MJ. Diabetes mellitus as a predictor of cancer mortality in a large cohort of US adults. American Journal of Epidemiology. 2004;159(12):1160–1167. doi: 10.1093/aje/kwh161. [DOI] [PubMed] [Google Scholar]
- 10.Coughlin SS, Calle EE, Teras LR, Petrelli J, Thun MJ. Diabetes mellitus as a predictor of cancer mortality in a large cohort of US adults. American Journal of Epidemiology. 2004;159(12):1160–1167. doi: 10.1093/aje/kwh161. [DOI] [PubMed] [Google Scholar]
- 11.Wideroff L, Gridley G, Mellemkjaer L, et al. Cancer incidence in a population-based cohort of patients hospitalized with diabetes mellitus in denmark. Journal of the National Cancer Institute. 1997;89(18):1360–1365. doi: 10.1093/jnci/89.18.1360. [DOI] [PubMed] [Google Scholar]
- 12.Jee SH, Ohrr H, Sull JW, Yun JE, Ji M, Samet JM. Fasting serum glucose level and cancer risk in Korean men and women. Journal of the American Medical Association. 2005;293(2):194–202. doi: 10.1001/jama.293.2.194. [DOI] [PubMed] [Google Scholar]
- 13.Yun J, Rago C, Cheong I, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325(5947):1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baron JA, Weiderpass E, Newcomb PA, et al. Metabolic disorders and breast cancer risk (United States) Cancer Causes and Control. 2001;12(10):875–880. doi: 10.1023/a:1013796112348. [DOI] [PubMed] [Google Scholar]
- 15.Lipscombe LL, Goodwin PJ, Zinman B, McLaughlin JR, Hux JE. Diabetes mellitus and breast cancer: a retrospective population-based cohort study. Breast Cancer Research and Treatment. 2006;98(3):349–356. doi: 10.1007/s10549-006-9172-5. [DOI] [PubMed] [Google Scholar]
- 16.Friberg E, Orsini N, Mantzoros CS, Wolk A. Diabetes mellitus and risk of endometrial cancer: a meta-analysis. Diabetologia. 2007;50(7):1365–1374. doi: 10.1007/s00125-007-0681-5. [DOI] [PubMed] [Google Scholar]
- 17.Anderson KE, Anderson E, Mink PJ, et al. Diabetes and endometrial cancer in the Iowa Women’s Health Study. Cancer Epidemiology Biomarkers and Prevention. 2001;10(6):611–616. [PubMed] [Google Scholar]
- 18.Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocrine-Related Cancer. 2009;16(4):1103–1123. doi: 10.1677/ERC-09-0087. [DOI] [PubMed] [Google Scholar]
- 19.Tseng C-H, Chong C-K, Tseng C-P, Chan T-T. Age-related risk of mortality from bladder cancer in diabetic patients: a 12-year follow-up of a national cohort in Taiwan. Annals of Medicine. 2009;41(5):371–379. doi: 10.1080/07853890902729778. [DOI] [PubMed] [Google Scholar]
- 20.Lewis JD, Ferrara A, Peng T, et al. Risk of bladder cancer among diabetic patients treated with pioglitazone: interim report of a longitudinal cohort study. Diabetes Care. 2011;34(4):916–922. doi: 10.2337/dc10-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barone BB, Yeh H-C, Snyder CF, et al. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: a systematic review and meta-analysis. Journal of the American Medical Association. 2008;300(23):2754–2764. doi: 10.1001/jama.2008.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. The Journal of General Physiology. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bos R, van der Hoeven JJM, van der Wall E, et al. Biologic correlates of 18fluorodeoxyglucose uptake in human breast cancer measured by positron emission tomography. Journal of Clinical Oncology. 2002;20(2):379–387. doi: 10.1200/JCO.2002.20.2.379. [DOI] [PubMed] [Google Scholar]
- 24.Maschauer S, Prante O, Hoffmann M, Deichen JT, Kuwert T. Characterization of 18F-FDG uptake in human endothelial cells in vitro . Journal of Nuclear Medicine. 2004;45(3):455–460. [PubMed] [Google Scholar]
- 25.Avril N. GLUT1 expression in tissue and 18F-FDG uptake. Journal of Nuclear Medicine. 2004;45(6):930–932. [PubMed] [Google Scholar]
- 26.Higashi K, Clavo AC, Wahl RL. Does FDG uptake measure proliferative activity of human cancer cells? In vitro comparison with DNA flow cytometry and tritiated thymidine uptake. Journal of Nuclear Medicine. 1993;34(3):414–419. [PubMed] [Google Scholar]
- 27.Haberkorn U, Strauss LG, Reisser C, et al. Glucose uptake, perfusion, and cell proliferation in head and neck tumors: relation of positron emission tomography to flow cytometry. Journal of Nuclear Medicine. 1991;32(8):1548–1555. [PubMed] [Google Scholar]
- 28.Vesselle H, Schmidt RA, Pugsley JM, et al. Lung cancer proliferation correlates with [F-18]fluorodeoxyglucose uptake by positron emission tomography. Clinical Cancer Research. 2000;6(10):3837–3844. [PubMed] [Google Scholar]
- 29.Beckner ME, Stracke ML, Liotta LA, Schiffmann E. Glycolysis as primary energy source in tumor cell chemotaxis. Journal of the National Cancer Institute. 1990;82(23):1836–1840. doi: 10.1093/jnci/82.23.1836. [DOI] [PubMed] [Google Scholar]
- 30.Masur K, Vetter C, Hinz A, et al. Diabetogenic glucose and insulin concentrations modulate transcriptom and protein levels involved in tumour cell migration, adhesion and proliferation. British Journal of Cancer. 2011;104(2):345–352. doi: 10.1038/sj.bjc.6606050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocrine-Related Cancer. 2009;16(4):1103–1123. doi: 10.1677/ERC-09-0087. [DOI] [PubMed] [Google Scholar]
- 32.Iqbal MA, Siddiqui FA, Gupta V, et al. Insulin enhances metabolic capacities of cancer cells by dual regulation of glycolytic enzyme pyruvate kinase M2. Molecular Cancer. 2013;12, article 72 doi: 10.1186/1476-4598-12-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonald PC, Fielding AB, Dedhar S. Integrin-linked kinase—essential roles in physiology and cancer biology. Journal of Cell Science. 2008;121(19):3121–3132. doi: 10.1242/jcs.017996. [DOI] [PubMed] [Google Scholar]
- 34.Chu CK, Mazo AE, Goodman M, et al. Preoperative diabetes mellitus and long-term survival after resection of pancreatic adenocarcinoma. Annals of Surgical Oncology. 2010;17(2):502–513. doi: 10.1245/s10434-009-0789-6. [DOI] [PubMed] [Google Scholar]
- 35.Fisher WE, McCullough PJ, Ray MB, Rogers DH, Bell RH., Jr. Diabetes enhances growth of pancreatic carcinoma cells. Surgery. 1988;104(2):431–436. [PubMed] [Google Scholar]
- 36.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 37.Weinberg F, Hamanaka R, Wheaton WW, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(19):8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Busik JV, Hootman SR, Greenidge CA, Henry DN. Glucose-specific regulation of aldose reductase in Capan-1 human pancreatic duct cells in vitro . Journal of Clinical Investigation. 1997;100(7):1685–1692. doi: 10.1172/JCI119693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tingstedt B, Johansson P, Andersson B, Andersson R. Predictive factors in pancreatic ductal adenocarcinoma: role of the inflammatory response. Scandinavian Journal of Gastroenterology. 2007;42(6):754–759. doi: 10.1080/00365520601058452. [DOI] [PubMed] [Google Scholar]
- 40.Okumura M, Yamamoto M, Sakuma H, et al. Leptin and high glucose stimulate cell proliferation in MCF-7 human breast cancer cells: reciprocal involvement of PKC-α and PPAR expression. Biochimica et Biophysica Acta—Molecular Cell Research. 2002;1592(2):107–116. doi: 10.1016/s0167-4889(02)00276-8. [DOI] [PubMed] [Google Scholar]
- 41.Liu H, Ma Q, Li J. High glucose promotes cell proliferation and enhances GDNF and RET expression in pancreatic cancer cells. Molecular and Cellular Biochemistry. 2011;347(1-2):95–101. doi: 10.1007/s11010-010-0617-0. [DOI] [PubMed] [Google Scholar]
- 42.Han L, Ma Q, Li J, et al. High glucose promotes pancreatic cancer cell proliferation via the induction of EGF expression and transactivation of EGFR. PLoS ONE. 2011;6(11) doi: 10.1371/journal.pone.0027074.e27074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 44.Allen DA, Harwood S, Varagunam M, Raftery MJ, Yaqoob MM. High glucose-induced oxidative stress causes apoptosis in proximal tubular epithelial cells and is mediated by multiple caspases. The FASEB Journal. 2003;17(8):908–910. doi: 10.1096/fj.02-0130fje. [DOI] [PubMed] [Google Scholar]
- 45.Ho FM, Lin WW, Chen BC, et al. High glucose-induced apoptosis in human vascular endothelial cells is mediated through NF-κB and c-Jun NH2-terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway. Cellular Signalling. 2006;18(3):391–399. doi: 10.1016/j.cellsig.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 46.Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nature Cell Biology. 2008;10(12):1477–1483. doi: 10.1038/ncb1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rudlowski C, Moser M, Becker AJ, et al. GLUT1 mRNA and protein expression in ovarian borderline tumors and cancer. Oncology. 2004;66(5):404–410. doi: 10.1159/000079489. [DOI] [PubMed] [Google Scholar]
- 48.Hauptmann S, Grünewald V, Molls D, et al. Glucose transporter GLUT1 in colorectal adenocarcinoma cell lines is inversely correlated with tumour cell proliferation. Anticancer Research. 2005;25(5):3431–3436. [PubMed] [Google Scholar]
- 49.Walenta S, Wetterling M, Lehrke M, et al. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Research. 2000;60(4):916–921. [PubMed] [Google Scholar]
- 50.Walenta S, Mueller-Klieser WF. Lactate: mirror and motor of tumor malignancy. Seminars in Radiation Oncology. 2004;14(3):267–274. doi: 10.1016/j.semradonc.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 51.Kim J-W, Dang CV. Cancer’s molecular sweet tooth and the warburg effect. Cancer Research. 2006;66(18):8927–8930. doi: 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- 52.Kondoh H. Cellular life span and the Warburg effect. Experimental Cell Research. 2008;314(9):1923–1928. doi: 10.1016/j.yexcr.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 53.Walenta S, Snyder S, Haroon ZA, et al. Tissue gradients of energy metabolites mirror oxygen tension gradients in a rat mammary carcinoma model. International Journal of Radiation Oncology Biology Physics. 2001;51(3):840–848. doi: 10.1016/s0360-3016(01)01700-x. [DOI] [PubMed] [Google Scholar]
- 54.Pelicano H, Xu R-H, Du M, et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. Journal of Cell Biology. 2006;175(6):913–923. doi: 10.1083/jcb.200512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gatenby RA, Smallbone K, Maini PK, et al. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. British Journal of Cancer. 2007;97(5):646–653. doi: 10.1038/sj.bjc.6603922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thews O, Gassner B, Kelleher DK, Schwerdt G, Gekle M. Impact of hypoxic and acidic extracellular conditions on cytotoxicity of chemotherapeutic drugs. Advances in Experimental Medicine and Biology. 2008;599:155–161. doi: 10.1007/978-0-387-71764-7_21. [DOI] [PubMed] [Google Scholar]
- 57.Yasmeen A, Beauchamp M-C, Piura E, Segal E, Pollak M, Gotlieb WH. Induction of apoptosis by metformin in epithelial ovarian cancer: involvement of the Bcl-2 family proteins. Gynecologic Oncology. 2011;121(3):492–498. doi: 10.1016/j.ygyno.2011.02.021. [DOI] [PubMed] [Google Scholar]
- 58.Malki A, Youssef A. Antidiabetic drug metformin induces apoptosis in human MCF breast cancer via targeting ERK signaling. Oncology Research. 2011;19(6):275–285. doi: 10.3727/096504011x13021877989838. [DOI] [PubMed] [Google Scholar]
- 59.Wu N, Gu C, Gu H, Hu H, Han Y, Li Q. Metformin induces apoptosis of lung cancer cells through activating JNK/p38 MAPK pathway and GADD153. Neoplasma. 2011;58(6):482–490. doi: 10.4149/neo_2011_06_482. [DOI] [PubMed] [Google Scholar]
- 60.Wahdan-Alaswad R, Fan Z, Edgerton SM, et al. Glucose promotes breast cancer aggression and reduces metformin efficacy. Cell Cycle. 2013;12:3759–3769. doi: 10.4161/cc.26641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu Y, Zhou BP. New insights of epithelial-mesenchymal transition in cancer metastasis. Acta Biochimica et Biophysica Sinica. 2008;40:643–650. doi: 10.1111/j.1745-7270.2008.00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kauppi J, Gockel I, Rantanen T, et al. Cause of death during long-term follow-up for superficial esophageal adenocarcinoma. Annals of Surgical Oncology. 2013;20(7):2428–2433. doi: 10.1245/s10434-013-2866-0. [DOI] [PubMed] [Google Scholar]
- 63.Walters S, Maringe C, Coleman MP, et al. Lung cancer survival and stage at diagnosis in Australia, Canada, Denmark, Norway, Sweden and the UK: a population-based study, 2004–2007. Thorax. 2013;68(6):551–564. doi: 10.1136/thoraxjnl-2012-202297. [DOI] [PubMed] [Google Scholar]
- 64.Walters S, Maringe C, Butler J, et al. Breast cancer survival and stage at diagnosis in Australia, Canada, Denmark, Norway, Sweden and the UK, 2000–2007: a population-based study. British Journal of Cancer. 2013;108:1195–1208. doi: 10.1038/bjc.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weiss L. Metastasis of cancer: a conceptual history from antiquity to the 1990s. Cancer and Metastasis Reviews. 2000;19(3-4):193–383. [PubMed] [Google Scholar]
- 66.Sleeman JP, Christofori G, Fodde R, et al. Concepts of metastasis in flux: the stromal progression model. Seminars in Cancer Biology. 2012;22:174–186. doi: 10.1016/j.semcancer.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 67.Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer and Metastasis Reviews. 1989;8(2):98–101. [PubMed] [Google Scholar]
- 68.Krechler T, Novotný J, Zeman M, et al. Pancreatic cancer and diabetes mellitus. Casopis Lekaru Ceskych. 2004;143(2):97–100. [PubMed] [Google Scholar]
- 69.Bao B, Wang Z, Ali S, et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prevention Research. 2012;5(3):355–364. doi: 10.1158/1940-6207.CAPR-11-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castilloand B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle. 2010;9(18):3807–3814. [PubMed] [Google Scholar]
- 71.Vairaktaris E, Goutzanis L, Kalokerinos G, et al. Diabetes increases both N-ras and Ets-1 expression during rat oral oncogenesis resulting in enhanced cell proliferation and metastatic potential. In Vivo. 2007;21(4):615–622. [PubMed] [Google Scholar]
- 72.Gutman A, Wasylyk B. The collagenase gene promoter contains a TPA and oncogene-responsive unit encompassing the PEA3 and AP-1 binding sites. The EMBO Journal. 1990;9(7):2241–2246. doi: 10.1002/j.1460-2075.1990.tb07394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wasylyk C, Gutman A, Nicholson R, Wasylyk B. The c-Ets oncoprotein activates the stromelysin promoter through the same elements as several non-nuclear oncoproteins. The EMBO Journal. 1991;10(5):1127–1134. doi: 10.1002/j.1460-2075.1991.tb08053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stacey KJ, Fowles LF, Colman MS, Ostrowski MC, Hume DA. Regulation of urokinase-type plasminogen activator gene transcription by macrophage colony-stimulating factor. Molecular and Cellular Biology. 1995;15(6):3430–3441. doi: 10.1128/mcb.15.6.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pande P, Mathur M, Shukla NK, Ralhan R. Ets-1: a plausible marker of invasive potential and lymph node metastasis in human oral squamous cell carcinomas. The Journal of Pathology. 1999;189:40–45. doi: 10.1002/(SICI)1096-9896(199909)189:1<40::AID-PATH405>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 76.Pande P, Soni S, Kaur J, et al. Prognostic factors in betel and tobacco related oral cancer. Oral Oncology. 2002;38(5):491–499. doi: 10.1016/s1368-8375(01)00090-2. [DOI] [PubMed] [Google Scholar]
- 77.Oikawa T, Yamada T. Molecular biology of the Ets family of transcription factors. Gene. 2003;303(1-2):11–34. doi: 10.1016/s0378-1119(02)01156-3. [DOI] [PubMed] [Google Scholar]
- 78.Iwatsuki M, Mimori K, Yokobori T, et al. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Science. 2010;101(2):293–299. doi: 10.1111/j.1349-7006.2009.01419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lv Z, Hu M, Zhen J, et al. Rac1/PAK1 signaling promotes epithelial-mesenchymal transition of podocytes in vitro via triggering beta-catenin transcriptional activity under high glucose conditions. The International Journal of Biochemistry & Cell Biology. 2013;45:255–264. doi: 10.1016/j.biocel.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 80.Liu R, Wang Y, Xiao Y, Shi M, Zhang G, Guo B. SnoN as a key regulator of the high glucose-induced epithelial-mesenchymal transition in cells of the proximal tubule. Kidney and Blood Pressure Research. 2012;35:517–528. doi: 10.1159/000339172. [DOI] [PubMed] [Google Scholar]
- 81.Lv Z-M, Wang Q, Wan Q, et al. The role of the p38 MAPK signaling pathway in high glucose-induced epithelial-mesenchymal transition of cultured human renal tubular epithelial cells. PLoS ONE. 2011;6(7) doi: 10.1371/journal.pone.0022806.e22806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoon JJ, Lee YJ, Kim JS, Kang DG, Lee HS. Betulinic acid inhibits high glucose-induced vascular smooth muscle cells proliferation and migration. Journal of Cellular Biochemistry. 2010;111(6):1501–1511. doi: 10.1002/jcb.22880. [DOI] [PubMed] [Google Scholar]
- 83.Urbich C, Dernbach E, Rössig L, Zeiher AM, Dimmeler S. High glucose reduces cathepsin L activity and impairs invasion of circulating progenitor cells. Journal of Molecular and Cellular Cardiology. 2008;45(3):429–436. doi: 10.1016/j.yjmcc.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 84.Zhang X, Wang J, Fan Y, Yang L, Wang L, Ma J. Zinc supplementation attenuates high glucose-induced epithelial-to-mesenchymal transition of peritoneal mesothelial cells. Biological Trace Element Research. 2012;150:229–235. doi: 10.1007/s12011-012-9451-4. [DOI] [PubMed] [Google Scholar]
- 85.Li C, Ren Y, Jia X, et al. Twist overexpression promoted epithelial-to-mesenchymal transition of human peritoneal mesothelial cells under high glucose. Nephrology Dialysis Transplantation. 2012;27:4119–4124. doi: 10.1093/ndt/gfs049. [DOI] [PubMed] [Google Scholar]
- 86.Li W, Ma Q, Li J, et al. Hyperglycemia enhances the invasive and migratory activity of pancreatic cancer cells via hydrogen peroxide. Oncology Reports. 2011;25(5):1279–1287. doi: 10.3892/or.2011.1150. [DOI] [PubMed] [Google Scholar]
- 87.Li W, Ma Q, Liu J, et al. Hyperglycemia as a mechanism of pancreatic cancer metastasis. Frontiers in Bioscience. 2012;17:1761–1774. doi: 10.2741/4017. [DOI] [PubMed] [Google Scholar]
- 88.Bapat AA, Hostetter G, Von Hoff DD, Han H. Perineural invasion and associated pain in pancreatic cancer. Nature Reviews Cancer. 2011;11(10):695–707. doi: 10.1038/nrc3131. [DOI] [PubMed] [Google Scholar]
- 89.Liebig C, Ayala G, Wilks JA, Berger DH, Albo D. Perineural invasion in cancer: a review of the literature. Cancer. 2009;115(15):3379–3391. doi: 10.1002/cncr.24396. [DOI] [PubMed] [Google Scholar]
- 90.Pour PM, Bell RH, Batra SK. Neural invasion in the staging of pancreatic cancer. Pancreas. 2003;26(4):322–325. doi: 10.1097/00006676-200305000-00002. [DOI] [PubMed] [Google Scholar]
- 91.Li J, Ma Q. Hyperglycemia promotes the perineural invasion in pancreatic cancer. Medical Hypotheses. 2008;71(3):386–389. doi: 10.1016/j.mehy.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 92.Li J, Ma Q, Liu H, et al. Relationship between neural alteration and perineural invasion in pancreatic cancer patients with hyperglycemia. PLoS ONE. 2011;6(2) doi: 10.1371/journal.pone.0017385.e17385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu B, Lu K-Y. Neural invasion in pancreatic carcinoma. Hepatobiliary and Pancreatic Diseases International. 2002;1(3):469–476. [PubMed] [Google Scholar]
- 94.Nagakawa T, Kayahara M, Ueno K, Ohta T, Konishi I, Miyazaki I. Clinicopathological study on neural invasion to the extrapancreatic nerve plexus in pancreatic cancer. Hepato-Gastroenterology. 1992;39(1):51–55. [PubMed] [Google Scholar]
- 95.Li J, Ma Q. Hyperglycemia promotes the perineural invasion in pancreatic cancer. Medical Hypotheses. 2008;71(3):386–389. doi: 10.1016/j.mehy.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 96.Ceyhan GO, Giese NA, Erkan M, et al. The neurotrophic factor artemin promotes pancreatic cancer invasion. Annals of Surgery. 2006;244(2):274–281. doi: 10.1097/01.sla.0000217642.68697.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhu Z, Friess H, DiMola FF, et al. Nerve growth factor expression correlates with perineural invasion and pain in human pancreatic cancer. Journal of Clinical Oncology. 1999;17(8):2419–2428. doi: 10.1200/JCO.1999.17.8.2419. [DOI] [PubMed] [Google Scholar]
- 98.Zhu Z-W, Friess H, Wang L, et al. Nerve growth factor exerts differential effects on the growth of human pancreatic cancer cells. Clinical Cancer Research. 2001;7(1):105–112. [PubMed] [Google Scholar]
- 99.Okada Y, Eibl G, Guha S, Duffy JP, Reber HA, Hines OJ. Nerve growth factor stimulates MMP-2 expression and activity and increases invasion by human pancreatic cancer cells. Clinical and Experimental Metastasis. 2004;21(4):285–292. doi: 10.1023/b:clin.0000046131.24625.54. [DOI] [PubMed] [Google Scholar]
- 100.Li J, Ma Q, Liu H, et al. Relationship between neural alteration and perineural invasion in pancreatic cancer patients with hyperglycemia. PLoS ONE. 2011;6(2) doi: 10.1371/journal.pone.0017385.e17385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sahin IH, Shama MA, Tanaka M, et al. Association of diabetes and perineural invasion in pancreatic cancer. Cancer Medicine. 2012;1:357–362. doi: 10.1002/cam4.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kumar A, Negi G, Sharma SS. Suppression of NF-κB and NF-κB regulated oxidative stress and neuroinflammation by BAY 11-7082 (IκB phosphorylation inhibitor) in experimental diabetic neuropathy. Biochimie. 2012;94(5):1158–1165. doi: 10.1016/j.biochi.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 103.Tomlinson DR, Gardiner NJ. Glucose neurotoxicity. Nature Reviews Neuroscience. 2008;9(1):36–45. doi: 10.1038/nrn2294. [DOI] [PubMed] [Google Scholar]
- 104.Bruckner BA, Ammini CV, Otal MP, Raizada MK, Stacpoole PW. Regulation of brain glucose transporters by glucose and oxygen deprivatiou. Metabolism: Clinical and Experimental. 1999;48(4):422–431. doi: 10.1016/s0026-0495(99)90098-7. [DOI] [PubMed] [Google Scholar]
- 105.Vincent AM, Callaghan BC, Smith AL, Feldman EL. Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nature Reviews Neurology. 2011;7(10):573–583. doi: 10.1038/nrneurol.2011.137. [DOI] [PubMed] [Google Scholar]
- 106.Bockman DE, Buchler M, Beger HG. Interaction of pancreatic ductal carcinoma with nerves leads to nerve damage. Gastroenterology. 1994;107(1):219–230. doi: 10.1016/0016-5085(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 107.Ayala GE, Dai H, Tahir SA, et al. Stromal antiapoptotic paracrine loop in perineural invasion of prostatic carcinoma. Cancer Research. 2006;66(10):5159–5164. doi: 10.1158/0008-5472.CAN-05-1847. [DOI] [PubMed] [Google Scholar]
- 108.Okada Y, Takeyama H, Sato M, et al. Experimental implication of celiac ganglionotropic invasion of pancreatic-cancer cells bearing c-ret proto-oncogene with reference to glial-cell-line-derived neurotrophic factor (GDNF) International Journal of Cancer. 1999;81:67–73. doi: 10.1002/(sici)1097-0215(19990331)81:1<67::aid-ijc13>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 109.Gil Z, Cavel O, Kelly K, et al. Paracrine regulation of pancreatic cancer cell invasion by peripheral nerves. Journal of the National Cancer Institute. 2010;102(2):107–118. doi: 10.1093/jnci/djp456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brunello A, Kapoor R, Extermann M. Hyperglycemia during chemotherapy for hematologic and solid tumors is correlated with increased toxicity. American Journal of Clinical Oncology: Cancer Clinical Trials. 2011;34(3):292–296. doi: 10.1097/COC.0b013e3181e1d0c0. [DOI] [PubMed] [Google Scholar]
- 111.Zeng L, Biernacka KM, Holly JMP, et al. Hyperglycaemia confers resistance to chemotherapy on breast cancer cells: the role of fatty acid synthase. Endocrine-Related Cancer. 2010;17(2):539–551. doi: 10.1677/ERC-09-0221. [DOI] [PubMed] [Google Scholar]
- 112.Geerlings SE, Hoepelman AIM. Immune dysfunction in patients with diabetes mellitus (DM) FEMS Immunology and Medical Microbiology. 1999;26(3-4):259–265. doi: 10.1111/j.1574-695X.1999.tb01397.x. [DOI] [PubMed] [Google Scholar]