

Abstract
Background and Purpose
The etiology of small fiber neuropathy (SFN) often remains unclear. Since SFN may be the only symptom of late-onset Fabry disease, it may be underdiagnosed in patients with idiopathic polyneuropathy. We aimed to uncover the etiological causes of seemingly idiopathic SFN by applying a focused investigatory procedure, to describe the clinical phenotype of true idiopathic SFN, and to elucidate the possible prevalence of late-onset Fabry disease in these patients.
Methods
Forty-seven adults younger than 60 years with seemingly idiopathic pure or predominantly small fiber sensory neuropathy underwent a standardized focused etiological and clinical investigation. The patients deemed to have true idiopathic SFN underwent genetic analysis of the alpha-galactosidase A gene (GLA) that encodes the enzyme alpha-galactosidase A (Fabry disease).
Results
The following etiologies were identified in 12 patients: impaired glucose tolerance (58.3%), diabetes mellitus (16.6%), alcohol abuse (8.3%), mitochondrial disease (8.3%), and hereditary neuropathy (8.3%). Genetic alterations of unknown clinical significance in GLA were detected in 6 of the 29 patients with true idiopathic SFN, but this rate did not differ significantly from that in healthy controls (n=203). None of the patients with genetic alterations in GLA had significant biochemical abnormalities simultaneously in blood, urine, and skin tissue.
Conclusions
A focused investigation may aid in uncovering further etiological factors in patients with seemingly idiopathic SFN, such as impaired glucose tolerance. However, idiopathic SFN in young to middle-aged Swedish patients does not seem to be due to late-onset Fabry disease.
Keywords: etiology, Fabry disease, idiopathic, impaired glucose tolerance, small fiber neuropathy
Introduction
Small fiber neuropathy (SFN) is a subgroup of peripheral neuropathy which is characterized by a disorder of the thin myelinated A-δ and unmyelinated C-fibers. Patients with SFN present with sensory symptoms and pain; the latter is often the dominant symptom, although pain is not mandatory for SFN.1,2 Autonomic dysfunction is common in SFN and may contribute to a significant level of morbidity.3 The most commonly etiology reported for SFN is diabetes mellitus.2,4 Other possible etiologies include impaired glucose tolerance (IGT),5,6 connective tissue disease,7,8 celiac disease,9 thyroid dysfunction,10,11 vitamin B12 deficiency,10 monoclonal gammopathy,12,13 HIV and hepatitis C infections,14,15 amyloidosis,16 toxicity due to alcohol or drugs,17,18 and hereditary neuropathies.19 Despite knowledge of these factors, the reported etiology of SFN has remained unclear in 23-93% of investigated patients.1,4,13,20 The clinical features and the spectrum of different types of secondary SFN due to the above-mentioned etiologies have been described previously.1,4,10,13,20,21,22 However, there have been few reports of the clinical phenotype of idiopathic SFN.23 Diagnosing SFN is difficult since the small caliber nerve fibers cannot be investigated with routine electrophysiological tests. The main methods currently used for assessing SFN are quantitative sensory testing (QST), which measures the detection thresholds for warm and cold sensations, skin biopsy for quantifying the intraepidermal nerve fiber density, and the quantitative sudomotor axon reflex test, which evaluates the postganglionic sympathetic sudomotor function.1,2
Fabry disease is an X-linked lysosomal storage disorder due to a partial or complete deficiency of the enzyme alpha-galactosidase A (α-GAL), resulting in accumulation of glycosphingolipids in different organs. Symptoms from the peripheral nervous system (PNS) involvement are common.24,25 More than 500 mutations are described in the alpha-galactosidase A gene (GLA) that encodes the enzyme α-GAL.26 It is known that heterozygous women have disease manifestations, with many of them being severely affected.25 There is also a milder variant of Fabry disease, where the onset of symptoms occurs later in life. The initial symptoms of late-onset Fabry disease are usually cardiomyopathy, renal disease, or neurological diseases such as stroke or neuropathy.27,28,29,30,31 SFN is the main manifestation of Fabry disease in the PNS, though large fiber axonal sensorimotor neuropathy may also occur.32,33 Fabry disease is suspected to be underdiagnosed in the general population and possibly also in patients with polyneuropathy of unknown cause.34,35 Very few studies have examined the possible association between late-onset Fabry disease and isolated SFN. In a recent study examining a small cohort with patients with idiopathic SFN, Tanislav et al.35 detected one patient with Fabry disease and four patients with a complex intronic haplotype in GLA and pathological biomarkers. Since the disease is treatable with enzyme replacement therapy, early diagnosis is of great importance.36,37
This study had three aims. First, we aimed to describe the clinical phenotype of SFN in patients where previous investigation at primary- or secondary-care centers had not revealed an etiology, hence leading to the diagnosis of idiopathic SFN. Second, we intended to show the spectrum of etiological factors that could be identified in this group of seemingly idiopathic SFN, if a standardized focused investigation procedure was applied. Finally, we chose to focus on one of the many uncommon causes of neuropathy, namely Fabry disease. Late-onset Fabry disease, which is known to be associated with isolated neuropathy, is suspected to be underdiagnosed. We therefore investigated whether Fabry disease could be a cause of idiopathic SFN with or without large fiber involvement in young to middle-aged patients.
Methods
Patients
Patients younger than 60 years with idiopathic SFN with or without large fiber sensorimotor axonal polyneuropathy were recruited from the Departments of Neurology and Neurophysiology at Karolinska University Hospital in Stockholm, the University Hospital in Linköping, and through collaboration with private-practice neurology and neurophysiology departments in Stockholm between September 2007 and October 2009. In addition, the diagnosis register from February 2002 to April 2009 at the Department of Neurophysiology at Karolinska University Hospital was searched for the diagnosis code "small fiber neuropathy".
Primary care referrals to the department of neurophysiology as well as medical records at the departments of neurology were studied, and patients with a possible etiology identified for SFN were excluded (Fig. 1).
Fig. 1.

Flow chart of excluded and included patients. In total, 213 patients with suspected SFN were identified originally. The numbers of patients excluded and the reasons for exclusion are noted in the figure. Ultimately, 29 patients judged to have an idiopathic SFN were included. The proportion of idiopathic SFN using the basic level of investigation/screening was 25% [33/133 (33=29 included patients plus 4 patients who declined participation in the genetic study, and 133=88 in exclusion group A plus 12 in exclusion group B plus 33)]. *Exclusion group A, †Exclusion group B. SFN: small fiber neuropathy.
The remaining patients with SFN consisted of those where a primary investigation by the family physician or at the department of neurology had not revealed an apparent etiology. These patients were asked to participate in a clinical screening visit at the outpatient clinic in an attempt to reveal any other possible etiology of SFN. All eligible patients were interviewed and underwent a standardized focused investigation (see Methods). After exclusion of other possible etiologies, the patients were invited to participate in the genetic part of the study examining for Fabry disease (Fig. 1). Written informed consent was obtained from all patients prior to participation. The study was approved by the local ethical committee at Karolinska Institutet, Stockholm.
Controls
In total, 203 age- and gender-matched healthy anonymous blood donors were used as controls for the genetic analysis, which corresponded to a patient:control ratio of 1:7.
Methods
Diagnostics
The diagnosis of SFN was based on pathological results of QST using the TSA-II NeuroSensory Analyzer (Medoc Advanced Medical Systems, Durham, NC, USA) to determine detection thresholds for warm and cold sensations, in addition to results of clinical examinations indicating small fiber dysfunction in the extremities. Pathological findings of nerve conduction studies were acceptable, but their pattern had to be axonal. A central nervous system (CNS) disorder as an alternative cause of pathological QST results was excluded through a physical examination, and in doubtful cases by MRI of the brain and spinal cord, as well as by measuring sensory evoked potentials.
Standardized focused investigation of possible etiologies of SFN
This work-up consisted of a comprehensive laboratory investigation (Table 1) and a clinical examination. All patients were examined physically by either Dr. K. Samuelsson or Dr. R. Press in the Department of Neurology, Karolinska University Hospital. The examination included determination of family history, medical history, past and present medications, exposure to environmental toxins, and alcohol drinking habits.
Table 1.
Laboratory investigation of patients with SFN
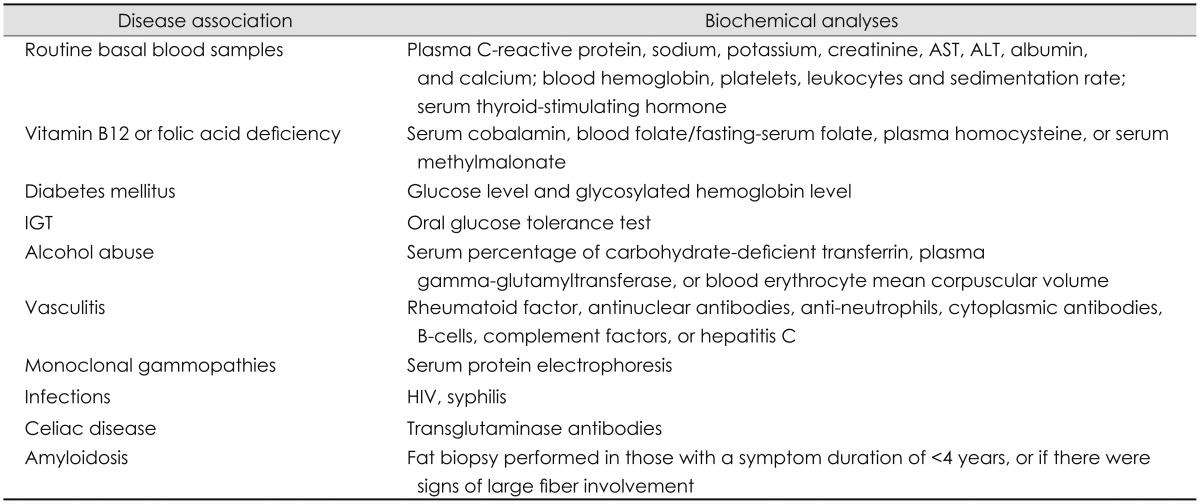
ALT: alanine aminotransferase, AST: aspartate aminotransferase, IGT: impaired glucose tolerance, SFN: small fiber neuropathy.
To be eligible for inclusion in the part of this study investigating Fabry disease, all possible etiologies of SFN had to be excluded after the above-mentioned standardized focused investigation.
DNA analysis
DNA purification was performed in the CSF laboratory of the Department of Neurology, Karolinska University Hospital using a QIAamp DNA Blood Maxi kit (Qiagen, Hilden, Germany). Sequencing analysis of GLA, including the exon-intron boundaries and parts of intron 4, was performed at the Albrecht-Kossel-Institute for Neuroregeneration, Faculty of Medicine, Rostock, Germany.35 The globotriaosylsphingosine (lyso-Gb3) level in plasma was detected in all patients according to the method described by Tanislav et al.35
The following additional biochemical analyses were performed in six patients with genetic alterations in GLA of unknown clinical relevance: the α-GAL activity in blood leucocytes38 and the globotriaosylceramide (Gb3) level in blood; and the total Gb3 and Gb3-isoform N-tetracosanoyl (Gb3_24) levels in urine were evaluated using mass spectrometry.35 A skin biopsy was carried out in the gluteal region to detect possible Gb3 accumulation. Detection of Gb3 deposits was performed with immunohistochemistry and electron microscopy.35,39 A multiplex ligation-probe assay for the exclusion of large deletions was done in specific cases.
Results
In total, 213 patients with suspected SFN were identified based on neurophysiological diagnosis codes and collaboration with other neurologists as described above. A review of referral documents and medical records led to the identification of possible etiologies in 88 patients (exclusion group A) (Fig. 1). Forty-seven patients were examined in the outpatient clinic of the Department of Neurology at Karolinska University Hospital by Dr. K. Samuelsson or Dr. R. Press, where they received a standardized focused investigation (Table 1). Eleven other patients were excluded (exclusion group B) (Fig. 1) since several possible disease-associated causes were discovered through the standardized work-up. Twenty-nine patients were finally included in the genetic part of the study (Fig. 1).
Demographics
The mean age of the 29 included patients and their matched controls was 50.2 years [19 women (66%)]. The age did not differ significantly between the included patients with idiopathic SFN and the excluded patients with secondary SFN. There were significantly more women in the inclusion group than in exclusion group A (p=0.03). The mean duration of symptoms prior to inclusion was 6.3 years (range 0.5-38 years) (Table 2).
Table 2.
Demographics of included patients with idiopathic SFN and those excluded due to identification of one or more etiological causes
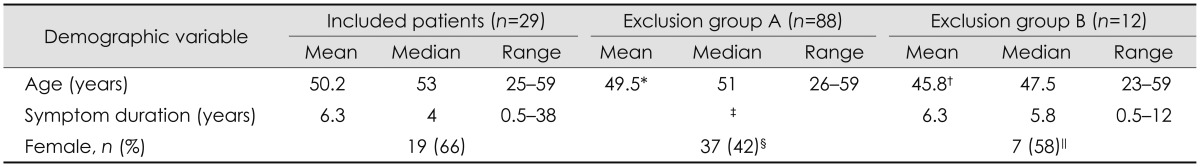
Exclusion group A=patients where etiological causes were identified by review of referral documents and medical records. Exclusion group B=patients where etiological causes were identified only after a standardized focused investigation at our tertiary center.
*No significant difference between included patients vs. exclusion group A, †No significant difference between included patients vs. exclusion group B, ‡Data missing, §Significantly more women in included patients vs. exclusion group A (p=0.03), ∥No significant difference between included patients vs. exclusion group B.
SFN: small fiber neuropathy.
Phenotype description of included patients
Sensory signs and symptoms
Twenty-four of the 29 patients (83%) had a length-dependent distribution of subjective sensory symptoms (Table 3). The remaining five patients (17%) had a non-length-dependent distribution of symptoms that was patchy in the face and trunk in addition to the extremities. Sensory symptoms included numbness, prickling, itching, tingling, coldness, and hyperesthesia. The proportions of patients with reduced pinprick, light-touch, and vibration sensations as well as those with hyperesthesia upon neurological examination are presented in Table 3. The subjective distribution of sensory symptoms usually comprised a larger area than the objective clinical findings [19 of 29 patients (66%)].
Table 3.
Symptoms and signs of the 29 included patients with idiopathic SFN
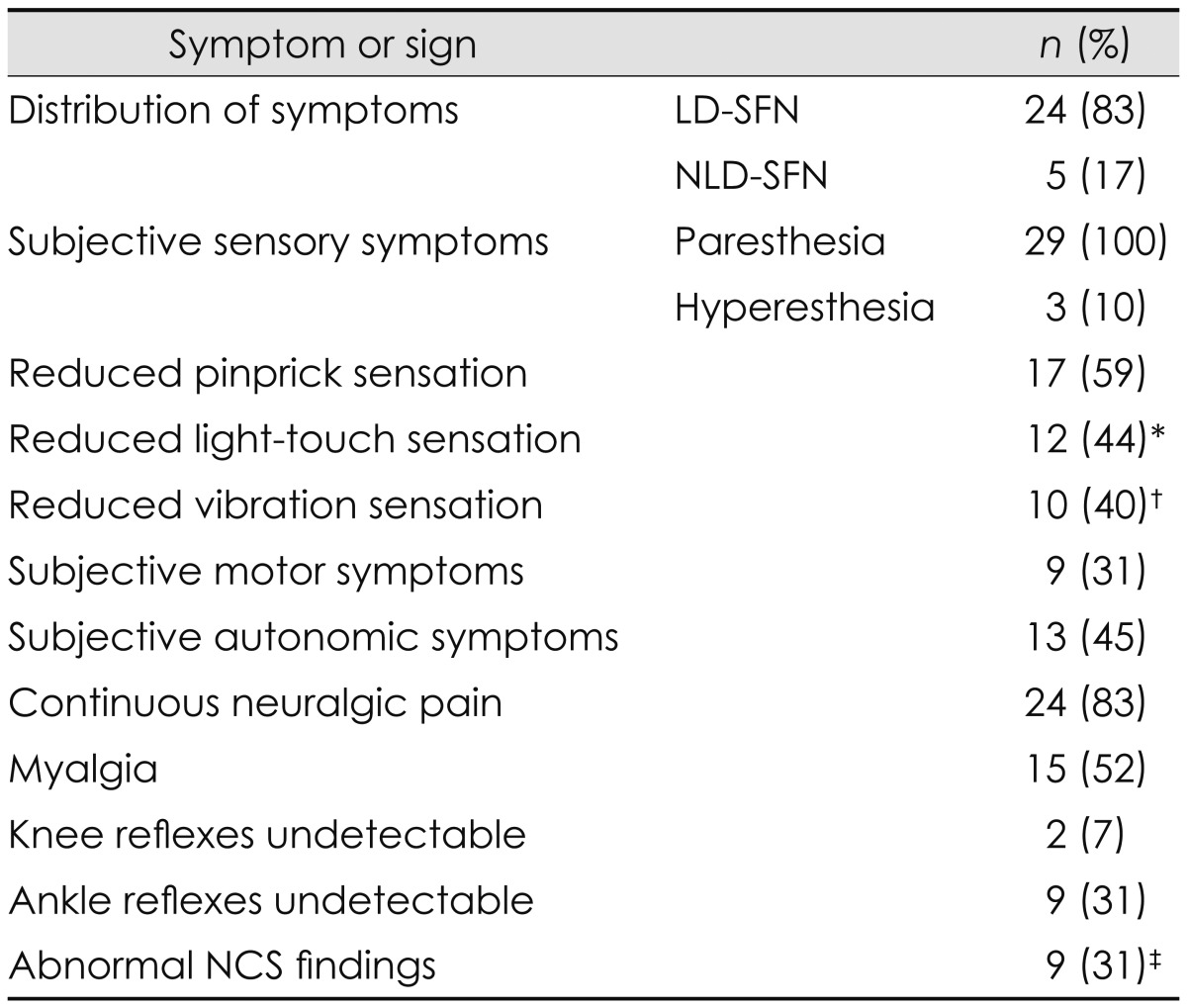
*Data missing for two patients, †Data missing for four patients, ‡The distribution of large fiber abnormalities on NCS was as follows: five patients with sensorimotor abnormality, three patients with sensory-only abnormality, and one patient with motor-only abnormality.
LD-SFN: length-dependent SFN, NCS: nerve conduction studies, NLD-SFN: non-length-dependent SFN, SFN: small fiber neuropathy.
Motor signs and symptoms
Nine of the 29 patients (31%) reported weakness in the feet, legs, or hands, while two complained of generalized weakness (Table 3). However, only two of the patients with subjective weakness in the extremities had corresponding findings on a physical examination; both of these patients had large fiber involvement.
Autonomic signs and symptoms
Autonomic symptoms consisted mainly of orthostatic lightheadedness, hyperhidrosis, erectile dysfunction, and problems related to micturition. Autonomic investigation of heart rate variation was performed in seven of the 29 patients (24%), five of whom also underwent sympathetic skin response testing. The findings of all autonomic tests were normal, regardless of the presence or absence of subjective symptoms.
Pain and concomitant medication
Twenty-six of the 29 patients (90%) had neuralgic pain, which appeared only intermittently in two of them. The pain sensation was described as burning, prickling, stabbing, aching, and icy. In addition, 15 of the 29 patients (50%) reported myalgia. Myalgia was distributed most commonly in the neck, shoulder, and back, but in two patients it was more generalized. Fourteen patients (48%) were taking regular pain medication, 10 of whom took drugs specific for neuralgic pain. All other medication used by the patients was also scrutinized in order to identify possible medications known to cause toxic neuropathy. Three patients were being treated with statins, but in neither case was there a temporal association between the initiation of medication and onset of neuropathic symptoms.
Neurophysiological findings vs. vibration loss
Nine of the 29 patients (31%) had combined SFN and large fiber axonal neuropathy according to nerve conduction studies. Notably, 41% of the patients with pure SFN had reduced vibration sensation at the ankles.
Working capacity
Sixteen of the 29 patients (55%) had reduced working capacity; in nine patients this was directly attributable to SFN.
Cardiovascular risk factors
Nine of 28 patients (32%) smoked (data were missing for one patient), and blood lipid data were available for 25 of the 29 patients. Cardiovascular risk factors were defined as follows: 1) pharmaceutical treatment of hypertension or hyperlipidemia, 2) elevated levels of fasting total cholesterol, low-density lipoprotein cholesterol, or triglycerides according to standard laboratory reference values, and 3) early cardiovascular events. Seven of the 25 patients (28%) had cardiovascular risk factors (CV+), while 18 patients did not (CV-). The mean age, gender, smoking, pain, and presence of large fiber involvement did not differ significantly between the CV+ and CV- groups.
Possible etiologies of SFN among the excluded patients
Diseases uncovered as a possible cause of SFN after a review of referral documents and medical records (exclusion group A)
Eighty-eight patients were excluded from the total of 213 patients after a review of referral documents and medical records due to identification of neuropathy-associated diseases/conditions (Fig. 1). The associated diseases are listed in Table 4; the most common was diabetes mellitus (37.5%). Sixteen patients had multiple possible etiological factors simultaneously (Table 4).
Table 4.
Exclusion group A. List of diseases identified as the probable etiology by review of referral documents and medical records of 150 patients with SFN
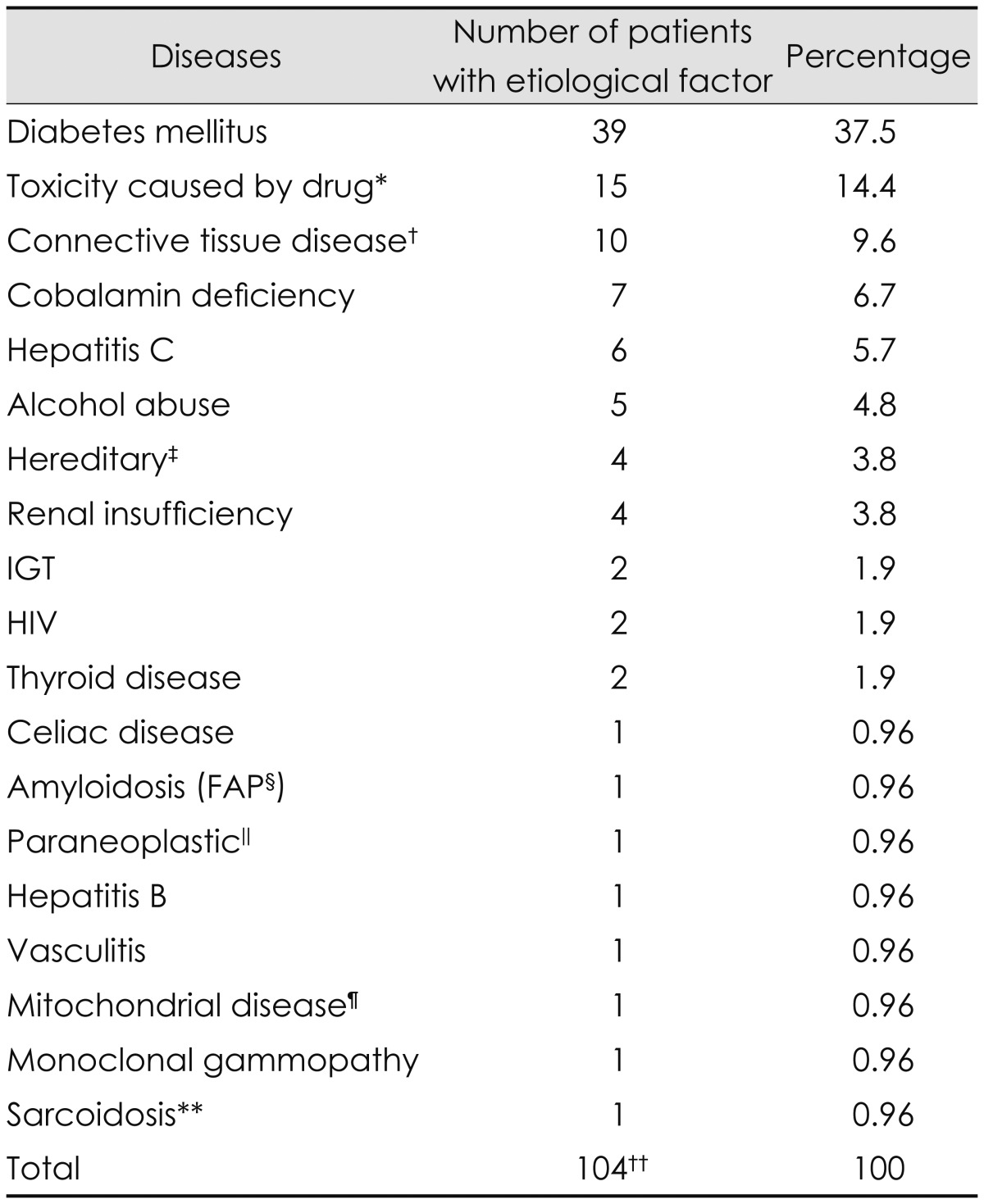
*The drugs responsible were chemotherapeutic drugs, including bortezomib, cisplatin, and thalidomide in 10 of the 15 patients; metronidazole in two of the 15 patients; and adalimumab, interferon alfa, and phenytoin in individual patients, †Systemic lupus erythematosus (n=2), psoriatic arthritis (n=2), rheumathoid arthritis (n=2), Churg-Strauss syndrome (n=1), Sjögren syndrome (n=1), unspecified connective tissue disorder (n=1), and Behçet disease (n=1), ‡Genetically verified Hereditary Sensory Neuropathy II (n=1), clinically suspected Hereditary Sensory Autonomic Neuropathy (n=1), genetically verified Spinocerebellar Ataxia 3 (n=1), and clinically suspected axonal Charcot-Marie-Tooth variant (n=1), §Familial amyloid polyneuropathy, ∥Rectal cancer, ¶Suspected based on clinical and electrophysiological data, **Suspected based on clinical data, ††Sixteen of 88 patients had multiple possible disease-associated causes simultaneously.
IGT: impaired glucose tolerance, SFN: small fiber neuropathy.
Diseases uncovered as a possible cause of SFN after a standardized focused investigation (exclusion group B)
A possible etiology was identified in 12 of the 45 patients (27%) who underwent our standardized focused investigation. The most frequent cause identified in these 12 patients was IGT (58.3%). Interestingly, the glycosylated hemoglobin levels did not differ between the subgroups with and without IGT (data not shown). Other identified causes are listed in Table 5. Two patients were excluded for other reasons (Fig. 1). The proportion of patients with idiopathic SFN who underwent our standardized focused investigation was 73% (Fig. 1).
Table 5.
Exclusion group B. List of diseases identified as an etiology by the standardized focused investigation of 45 patients with seemingly idiopathic SFN
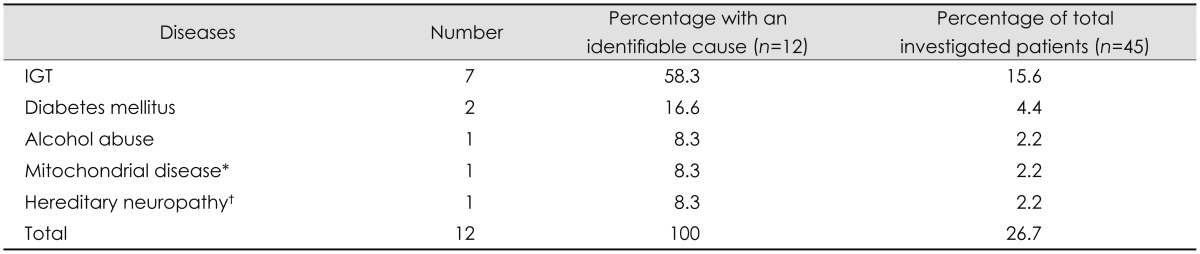
*Genetically verified novel mtDNA mutation, †Clinically suspected HSAN.
HSAN: Hereditary Sensory Autonomic Neuropathy, IGT: impaired glucose tolerance, SFN: small fiber neuropathy.
Genetic investigation of Fabry disease among the patients with idiopathic SFN
Genetic results
Six of the 29 patients had genetic alterations in GLA of unknown clinical relevance (Table 6). Two patients had the complex intronic haplotype IVS0-10C>T [rs2071225], IVS4-16A>G [rs2071397], IVS6-22C>T [rs2071228].35,40 One patient had an exon mutation, c.937G>T [D313Y], which is the so-called pseudodeficiency allele.41,42,43 Twenty-nine of the 203 controls had genetic alterations in GLA of unknown clinical relevance. One control, a 58-year-old man, had an exon mutation: c.352C>T [R118C].34,42 In all, 21% of the patients and 15% of the controls had genetic alterations of unknown clinical relevance in GLA. This difference was not significant statistically.
Table 6.
Genetic data in controls and patients with idiopathic SFN who have an alteration in GLA, as well as corresponding biochemical results
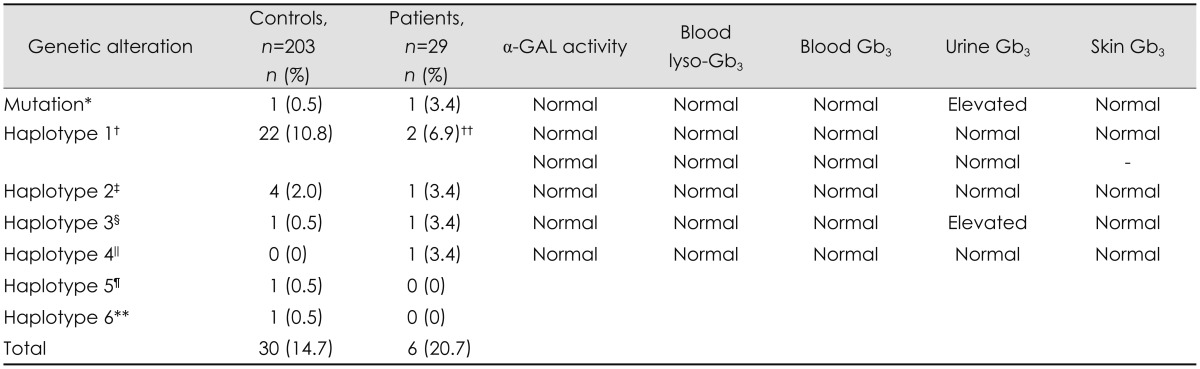
No significant differences (p<0.05) were evident between the total number of individuals with genetic alterations in GLA, nor for each respective haplotype in patients vs. controls.
*Patient c.937G>T [D313Y], control c.352C>T [R118C]. The considered pathological or unclear results of single nucleotide polymorphism combinations in the haplotypes are in italics, †Haplotype 1=IVS0-10C>T [rs2071225], IVS2-81--77delCAGCC, IVS4-16A>G [rs2071397], IVS6-22C>T [rs2071228], ‡Haplotype 2=IVS4-854--853delAG, IVS0-12A>G [rs3027585], IVS4+68A>G [rs3027589], IVS6-22C>T [rs2071228], §Haplotype 3=IVS4-854--853delAG, IVS0-12A>G [rs3027585], (IVS2-81--77delCAGCC), IVS4+68A>G [rs3027589], IVS4-16A>G [rs2071397], IVS6-22C>T [rs2071228], ∥Haplotype 4=IVS4+18 G>A, IVS4+771C>T [rs5991933], IVS6-22C>T [rs2071228], ¶Haplotype 5=IVS3-72--68delAATAA, **Haplotype 6=IVS4+739C>T, IVS0-10C>T [rs2071225], IVS6-22C>T [rs2071228], ††Two patients with haplotype 1. Skin Gb3 was not examined for one patient.
α-GAL: alpha-galactosidase A, Gb3: globotriaosylceramide, GLA: alpha-galactosidase A gene, SFN: small fiber neuropathy.
Biochemical analysis
All patients had normal levels of lyso-Gb3 in blood. Additional analyses were performed for the six patients with alterations in GLA. All Gb3 levels in blood were normal, and all skin biopsies were negative. Urine Gb3 was elevated in two patients; one had the following intronic changes: IVS4-854--853delAG, IVS0-12A>G [rs3027585], IVS2-81--77delCAGCC, IVS4+68A>G [rs3027589], IVS4-16A>G [rs2071397], IVS6-22C>T [rs2071228] (Table 6), and one had the exon mutation c.937G>T [D313Y]. The urine-Gb3_24 levels were 310.71 and 126.22 ng/mg creatinine in these two patients (reference value<35 ng/mg creatinine). The results of a multiplex ligation-probe assay were normal in both patients. Additional clinical evaluations (electrocardiography, transthoracic echocardiography, ophthalmologic investigation, and urine protein levels) were also normal in these two patients, indicating the absence of clinical signs of Fabry disease.
Clinical features
Pain and fiber size (pure small fiber or mixed small and large fiber) involvement did not differ significantly between the six patients with genetic alterations in GLA of unknown clinical relevance and the remaining 23 included patients.
Discussion
Previous studies have found the percentage of SFN patients with unknown or idiopathic etiology to vary between 23% and 93%.1,4,13,20 The proportion of idiopathic SFN among all patients with SFN depends on the quality of the evaluation for the etiology at the primary, secondary, and tertiary levels. A review of referral documents and medical records in the present study led to various conditions being identified as a possible cause of SFN in 88 of 133 screened patients (66%), with diabetes mellitus being the most common identifiable cause (37.5%). When the remainder of the patients with seemingly idiopathic SFN underwent a standardized focused investigation at our neuromuscular tertiary center, a possible etiology was uncovered in an additional 12 of 45 investigated patients (27%), with IGT being the most common associated condition (58.3% of the 12 patients).
The proportion of idiopathic SFN diagnoses based on the initial basic level of investigation was 25% (33 of 133 patients) (Fig. 1, Table 4), probably corresponding to the result of etiological investigations at the primary/secondary care centers. The proportion of patients with idiopathic SFN among the group that had undergone the standardized focused investigation was larger; that is, 73% (33 of 45 patients). This could represent the proportion of patients with idiopathic SFN in the total population of patients with SFN investigated at a tertiary center.
To our knowledge this is the first controlled study investigating if Fabry disease can be a cause of idiopathic polyneuropathy. We found genetic alterations in GLA of unknown clinical relevance in both the patients and controls, with the frequency of the genetic variants not differing significantly between these two groups.
Phenotypic description of patients with no identifiable etiology of SFN despite a standardized focused investigation
Twenty of the included patients had pure SFN and nine patients had additional axonal large fiber involvement based on the results of nerve conduction studies. The occurrence of large fiber involvement was permitted in this study since this can be present in patients with Fabry disease, especially late in the course of the disease.32,44 Also with regards to large fiber involvement, patients with idiopathic polyneuropathy are much more likely to be present in the subgroup with axonal rather than demyelinating sensorimotor neuropathy, since the latter subtype of neuropathy is more commonly due to inflammatory or hereditary causes. Length-dependent SFN (LD-SFN) is more common than non-length-dependent SFN (NLD-SFN).10 Patients with LD-SFN typically have leg-onset symmetrical sensory symptoms with a distal-to-proximal gradient, and a delayed spread of symptoms to the upper limbs. On the other hand, patients with NLD-SFN experience sensory symptoms in a rather patchy asymmetrical distribution in the face, trunk, and upper limbs, in addition to symptoms in the lower limbs. Eighty-three percent of the patients included in this study had LD-SFN. Reports on the gender distribution in SFN have been conflicting, though most studies have found a female predominance.1,4,10,13,20,21,22,23 We also found a female predominance in patients with SFN: 63% of LD-SFN patients and 80% of NLD-SFN patients were women. The opposite gender distribution of a male predominance has been reported in patients with chronic idiopathic axonal polyneuropathy.45,46,47,48 The gender difference in neuropathy patients related to the size of affected fibers may indicate gender-related etiological factors and warrants further study.
The morbidity in the group of patients with idiopathic SFN with or without large fiber involvement was high: more than 80% of the 29 patients had continuous neuralgic pain, and (notably) about 50% also reported myalgia. Myalgia is not known to be a coexisting symptom in patients with idiopathic neuropathy. The influence of polyneuropathy on daily activities such as work has been reported previously,49,50 but we found reduced work capacity as a direct consequence of idiopathic SFN in 31% of our patients.
Etiological causes of SFN among the excluded patients
The second most common etiology identified in exclusion group A was toxic neuropathy, which was mainly caused by chemotherapeutic drugs (14.4%). It is well known that neurotoxic drugs cause SFN,2,17,50,51 but the frequency was lower in previous reports1,4,10 than in our study. Sarcoidosis, another described cause of SFN,52 was found in one patient in exclusion group A. Serum angiotensin-converting enzyme (S-ACE) was not included in our initial laboratory protocol, so we lacked data regarding S-ACE from five of the 29 patients in the inclusion group. Devigili et al.1 reported no cases of sarcoidosis in their group of 67 SFN patients, though S-ACE was not included in the work-up. On the other hand, Khan and Zhou10 tested for S-ACE in their patient group (n=238), and found increased levels suggestive of sarcoidosis in three patients (1.2%), all of whom had NLD-SFN.
Mitochondrial disease was suspected in two of our patients, but this was verified by DNA analysis in only one of them. That patient was included initially (Fig. 1) and had normal electromyography (EMG) findings prior to inclusion in the study. However, follow-up EMG that was performed due to progressive muscle weakness revealed a myopathic pattern, and a muscle biopsy confirmed myopathy indicative of mitochondrial disease. Biochemical analysis of isolated mitochondria revealed severely reduced complex I activity in the respiratory chain and low adenosine-5'-triphosphate production with all substrates dependent on complex I. Sequencing the mitochondrial DNA obtained from muscle revealed a novel mutation. Polyneuropathies are prevalent in mitochondrial disorders;53,54 they often present as asymmetric, mostly axonal, sensorimotor multifocal neuropathy,53,54 although SFN has been reported as a single manifestation in the PNS in mitochondrial myopathy.55
Impaired glucose tolerance seems to be an underdiagnosed condition in patients with SFN, probably due to primary care physicians or neurologists not performing this test routinely in the work-up of most patients with SFN. Devigili et al.1 could identify a possible cause in seven of 28 patients with idiopathic SFN at a 2-year follow-up: two had IGT, four had diabetes mellitus, and one had Sjögren syndrome. We uncovered IGT in 16% of 45 patients with seemingly idiopathic SFN. However, we did not find any correlation between clinical findings in patients with idiopathic SFN and any other cardiovascular risk factors. IGT as an etiology of sensory neuropathy is still debated. However, several studies have indicated that IGT is a strong risk factor for neuropathy,4,5,6 although Hughes et al. could not confirm that IGT was the etiology for chronic idiopathic axonal polyneuropathy in a controlled study,45 and an increased prevalence of large fiber polyneuropathy was not detected among patients with impaired glycemic control.56 Hyperlipidemia has been identified as an independent risk factor for polyneuropathy.4,45,57 This could not be confirmed in our study, since 72% of our patients lacked risk factors for cardiovascular disease (including hyperlipidemia).
Genetic alterations in GLA in patients with idiopathic SFN
A recently published pilot study by Tanislav et al.35 found mutations in GLA in 5 of 24 patients (21%) with idiopathic SFN. One patient had a typical mutation for Fabry disease c.424T>C [C142R], and four had a complex intronic haplotype (hereafter referred to as haplotype 1) (Table 6).35 These four patients had pathological biomarkers of increased levels of lyso-Gb3 and urine Gb3, but no clinical manifestations of Fabry disease.35 This haplotype has also been reported in a patient with confirmed Fabry disease.40 Six of the 29 patients (21%) in the present study showed genetic alterations in GLA of unknown clinical relevance, corroborating the results obtained by Tanislav et al.35 Two of these six patients had haplotype 1, but none had altered biomarkers for Fabry disease. However, we found the same results among the controls, since 29 of the 203 controls (14%) had genetic alterations in the GLA gene of unknown clinical relevance. Haplotype 1 was found in 22 of 203 controls (11%). The proportion of individuals with genetic alterations in GLA did not differ significantly between patients and controls, which implies that Fabry disease is not a probable underlying cause of isolated SFN in Swedish patients. D313Y was found in one of our patients, who had elevated urine Gb3 but normal lyso-Gb3 and no reduction in α-GAL activity. There were no other clinical signs of Fabry disease. Whether or not D313Y is a disease-generating mutation is under debate. The classically affected individuals who have a D313Y alteration also have an additional missense mutation.41,43 Yasuda et al. found the D313Y alteration in 0.45% of subjects in a normal Caucasian population.43 However, D313Y has been found as the single genetic alteration in Fabry disease patients with highly reduced enzymatic activity and end-stage renal disease,42 in those with stroke,58 and in Fabry disease presenting with cardiovascular symptoms.59 We do not have information about any possible associated diseases in our healthy controls. Swedish regulations include Fabry disease as an exclusion criterion for being a blood donor. However, we cannot completely rule out the possibility of undiagnosed symptoms and clinical signs of Fabry disease in the male control with the exon mutation c.352C>T [R118C]. R118C was first identified by Spada et al.34 as a novel missense mutation causing late-onset disease, and was subsequently found in patients who had reduced enzyme activity and symptoms such as renal disease and stroke.42,58
The identification of decreased α-GAL activity or a genetic variation in patients with late-onset Fabry disease is not necessarily related to symptoms such as proteinuria, left ventricular hypertrophy, or pain in the hands or feet.44 Biegstraaten et al.44 propose that SFN should be considered to be due to Fabry disease only in patients with Fabry-specific SFN features; that is, a history of neuropathic pain in the hands and/or feet, with the onset of pain in either childhood or adolescence, or a course that is characterized by ongoing pain with exacerbations that are provoked by fever, exercise, or heat.
Limitations
The relatively small number of patients and the diagnostic limitation of using QST in isolation in this study weaken the conclusions drawn; however, this is the first reported controlled study regarding idiopathic SFN and Fabry disease. We chose to use QST as the main method for diagnosing SFN since its results have been found to be strongly correlated with intraepidermal nerve fiber density in detecting SFN according to the European Federation of Neurological Societies guidelines.60,61 To compensate for the shortcoming of QST sensitivity, we required that all included patients diagnosed with SFN on the basis of pathological QST results also had a clinical evaluation that excluded a CNS disorder as a cause of their sensory symptoms. Another limitation of this study is the heterogeneity of nerve fiber involvement in different patients. We therefore cannot exclude the possibility of alternative results in study groups that include only patients with isolated small fiber involvement. However, no apparent etiological or genetic differences were seen between patients with pure small fiber involvement and those with mixed small and large fiber involvement.
Though the results reported herein are based on a relatively small patient material, they indicate that idiopathic SFN predominantly affects women and is associated with high morbidity due to pain as well as reduced working capacity. A standardized focused investigation is important to uncover the etiology of SFN in patients with seemingly idiopathic neuropathy, and IGT is the most common identifiable cause. Idiopathic SFN in young to middle-aged Swedish patients seems not to be due to late-onset Fabry disease.
Acknowledgements
This study was supported by an unrestricted grant from Shire Human Genetic Therapies.
Footnotes
The authors have no financial conflicts of interest.
References
- 1.Devigili G, Tugnoli V, Penza P, Camozzi F, Lombardi R, Melli G, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain. 2008;131(Pt 7):1912–1925. doi: 10.1093/brain/awn093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lacomis D. Small-fiber neuropathy. Muscle Nerve. 2002;26:173–188. doi: 10.1002/mus.10181. [DOI] [PubMed] [Google Scholar]
- 3.Novak V, Freimer ML, Kissel JT, Sahenk Z, Periquet IM, Nash SM, et al. Autonomic impairment in painful neuropathy. Neurology. 2001;56:861–868. doi: 10.1212/wnl.56.7.861. [DOI] [PubMed] [Google Scholar]
- 4.Bednarik J, Vlckova-Moravcova E, Bursova S, Belobradkova J, Dusek L, Sommer C. Etiology of small-fiber neuropathy. J Peripher Nerv Syst. 2009;14:177–183. doi: 10.1111/j.1529-8027.2009.00229.x. [DOI] [PubMed] [Google Scholar]
- 5.Singleton JR, Smith AG, Bromberg MB. Painful sensory polyneuropathy associated with impaired glucose tolerance. Muscle Nerve. 2001;24:1225–1228. doi: 10.1002/mus.1136. [DOI] [PubMed] [Google Scholar]
- 6.Sumner CJ, Sheth S, Griffin JW, Cornblath DR, Polydefkis M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology. 2003;60:108–111. doi: 10.1212/wnl.60.1.108. [DOI] [PubMed] [Google Scholar]
- 7.Gøransson LG, Tjensvoll AB, Herigstad A, Mellgren SI, Omdal R. Small-diameter nerve fiber neuropathy in systemic lupus erythematosus. Arch Neurol. 2006;63:401–404. doi: 10.1001/archneur.63.3.401. [DOI] [PubMed] [Google Scholar]
- 8.Lopate G, Pestronk A, Al-Lozi M, Lynch T, Florence J, Miller T, et al. Peripheral neuropathy in an outpatient cohort of patients with Sjögrens' syndrome. Muscle Nerve. 2006;33:672–676. doi: 10.1002/mus.20514. [DOI] [PubMed] [Google Scholar]
- 9.Chin RL, Sander HW, Brannagan TH, Green PH, Hays AP, Alaedini A, et al. Celiac neuropathy. Neurology. 2003;60:1581–1585. doi: 10.1212/01.wnl.0000063307.84039.c7. [DOI] [PubMed] [Google Scholar]
- 10.Khan S, Zhou L. Characterization of non-length-dependent small-fiber sensory neuropathy. Muscle Nerve. 2012;45:86–91. doi: 10.1002/mus.22255. [DOI] [PubMed] [Google Scholar]
- 11.Magri F, Buonocore M, Oliviero A, Rotondi M, Gatti A, Accornero S, et al. Intraepidermal nerve fiber density reduction as a marker of preclinical asymptomatic small-fiber sensory neuropathy in hypothyroid patients. Eur J Endocrinol. 2010;163:279–284. doi: 10.1530/EJE-10-0285. [DOI] [PubMed] [Google Scholar]
- 12.Barbieri S, Sandroni P, Nobile-Orazio E, Cappellari A, Cavestro C, Baldini L, et al. Small fibre involvement in neuropathy associated with IgG, IgA and IgM monoclonal gammopathy. Electromyogr Clin Neurophysiol. 1995;35:39–44. [PubMed] [Google Scholar]
- 13.Periquet MI, Novak V, Collins MP, Nagaraja HN, Erdem S, Nash SM, et al. Painful sensory neuropathy: prospective evaluation using skin biopsy. Neurology. 1999;53:1641–1647. doi: 10.1212/wnl.53.8.1641. [DOI] [PubMed] [Google Scholar]
- 14.Polydefkis M, Yiannoutsos CT, Cohen BA, Hollander H, Schifitto G, Clifford DB, et al. Reduced intraepidermal nerve fiber density in HIV-associated sensory neuropathy. Neurology. 2002;58:115–119. doi: 10.1212/wnl.58.1.115. [DOI] [PubMed] [Google Scholar]
- 15.Yoon MS, Obermann M, Dockweiler C, Assert R, Canbay A, Haag S, et al. Sensory neuropathy in patients with cryoglobulin negative hepatitis-C infection. J Neurol. 2011;258:80–88. doi: 10.1007/s00415-010-5686-1. [DOI] [PubMed] [Google Scholar]
- 16.Adams D. Hereditary and acquired amyloid neuropathies. J Neurol. 2001;248:647–657. doi: 10.1007/s004150170109. [DOI] [PubMed] [Google Scholar]
- 17.Peltier AC, Russell JW. Advances in understanding drug-induced neuropathies. Drug Saf. 2006;29:23–30. doi: 10.2165/00002018-200629010-00002. [DOI] [PubMed] [Google Scholar]
- 18.Zambelis T, Karandreas N, Tzavellas E, Kokotis P, Liappas J. Large and small fiber neuropathy in chronic alcohol-dependent subjects. J Peripher Nerv Syst. 2005;10:375–381. doi: 10.1111/j.1085-9489.2005.00050.x. [DOI] [PubMed] [Google Scholar]
- 19.Verhoeven K, Timmerman V, Mauko B, Pieber TR, De Jonghe P, Auer-Grumbach M. Recent advances in hereditary sensory and autonomic neuropathies. Curr Opin Neurol. 2006;19:474–480. doi: 10.1097/01.wco.0000245370.82317.f6. [DOI] [PubMed] [Google Scholar]
- 20.Stewart JD, Low PA, Fealey RD. Distal small fiber neuropathy: results of tests of sweating and autonomic cardiovascular reflexes. Muscle Nerve. 1992;15:661–665. doi: 10.1002/mus.880150605. [DOI] [PubMed] [Google Scholar]
- 21.De Sousa EA, Hays AP, Chin RL, Sander HW, Brannagan TH., 3rd Characteristics of patients with sensory neuropathy diagnosed with abnormal small nerve fibres on skin biopsy. J Neurol Neurosurg Psychiatry. 2006;77:983–985. doi: 10.1136/jnnp.2006.091074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gemignani F, Giovanelli M, Vitetta F, Santilli D, Bellanova MF, Brindani F, et al. Non-length dependent small fiber neuropathy. A prospective case series. J Peripher Nerv Syst. 2010;15:57–62. doi: 10.1111/j.1529-8027.2010.00252.x. [DOI] [PubMed] [Google Scholar]
- 23.Holland NR, Crawford TO, Hauer P, Cornblath DR, Griffin JW, McArthur JC. Small-fiber sensory neuropathies: clinical course and neuropathology of idiopathic cases. Ann Neurol. 1998;44:47–59. doi: 10.1002/ana.410440111. [DOI] [PubMed] [Google Scholar]
- 24.Laaksonen SM, Röyttä M, Jääskeläinen SK, Kantola I, Penttinen M, Falck B. Neuropathic symptoms and findings in women with Fabry disease. Clin Neurophysiol. 2008;119:1365–1372. doi: 10.1016/j.clinph.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 25.MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38:769–775. doi: 10.1136/jmg.38.11.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugawara K, Ohno K, Saito S, Sakuraba H. Structural characterization of mutant alpha-galactosidases causing Fabry disease. J Hum Genet. 2008;53:812–824. doi: 10.1007/s10038-008-0316-9. [DOI] [PubMed] [Google Scholar]
- 27.Monserrat L, Gimeno-Blanes JR, Marín F, Hermida-Prieto M, García-Honrubia A, Pérez I, et al. Prevalence of fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2007;50:2399–2403. doi: 10.1016/j.jacc.2007.06.062. [DOI] [PubMed] [Google Scholar]
- 28.Nakao S, Takenaka T, Maeda M, Kodama C, Tanaka A, Tahara M, et al. An atypical variant of Fabry's disease in men with left ventricular hypertrophy. N Engl J Med. 1995;333:288–293. doi: 10.1056/NEJM199508033330504. [DOI] [PubMed] [Google Scholar]
- 29.Rolfs A, Böttcher T, Zschiesche M, Morris P, Winchester B, Bauer P, et al. Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet. 2005;366:1794–1796. doi: 10.1016/S0140-6736(05)67635-0. [DOI] [PubMed] [Google Scholar]
- 30.Sawada K, Mizoguchi K, Hishida A, Kaneko E, Koide Y, Nishimura K, et al. Point mutation in the alpha-galactosidase A gene of atypical Fabry disease with only nephropathy. Clin Nephrol. 1996;45:289–294. [PubMed] [Google Scholar]
- 31.Yang CC, Lai LW, Whitehair O, Hwu WL, Chiang SC, Lien YH. Two novel mutations in the alpha-galactosidase A gene in Chinese patients with Fabry disease. Clin Genet. 2003;63:205–209. doi: 10.1034/j.1399-0004.2003.00050.x. [DOI] [PubMed] [Google Scholar]
- 32.Dütsch M, Marthol H, Stemper B, Brys M, Haendl T, Hilz MJ. Small fiber dysfunction predominates in Fabry neuropathy. J Clin Neurophysiol. 2002;19:575–586. doi: 10.1097/00004691-200212000-00011. [DOI] [PubMed] [Google Scholar]
- 33.Luciano CA, Russell JW, Banerjee TK, Quirk JM, Scott LJ, Dambrosia JM, et al. Physiological characterization of neuropathy in Fabry's disease. Muscle Nerve. 2002;26:622–629. doi: 10.1002/mus.10236. [DOI] [PubMed] [Google Scholar]
- 34.Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, et al. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79:31–40. doi: 10.1086/504601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanislav C, Kaps M, Rolfs A, Böttcher T, Lackner K, Paschke E, et al. Frequency of Fabry disease in patients with small-fibre neuropathy of unknown aetiology: a pilot study. Eur J Neurol. 2011;18:631–636. doi: 10.1111/j.1468-1331.2010.03227.x. [DOI] [PubMed] [Google Scholar]
- 36.Hilz MJ, Brys M, Marthol H, Stemper B, Dütsch M. Enzyme replacement therapy improves function of C-, Adelta-, and Abeta-nerve fibers in Fabry neuropathy. Neurology. 2004;62:1066–1072. doi: 10.1212/01.wnl.0000118207.84514.40. [DOI] [PubMed] [Google Scholar]
- 37.Lidove O, West ML, Pintos-Morell G, Reisin R, Nicholls K, Figuera LE, et al. Effects of enzyme replacement therapy in Fabry disease--a comprehensive review of the medical literature. Genet Med. 2010;12:668–679. doi: 10.1097/GIM.0b013e3181f13b75. [DOI] [PubMed] [Google Scholar]
- 38.Desnick RJ, Allen KY, Desnick SJ, Raman MK, Bernlohr RW, Krivit W. Fabry's disease: enzymatic diagnosis of hemizygotes and heterozygotes. Alpha-galactosidase activities in plasma, serum, urine, and leukocytes. J Lab Clin Med. 1973;81:157–171. [PubMed] [Google Scholar]
- 39.Jansen T, Brokalaki E, Hillen U, Hentschke M, Grabbe S. [Manifestation of Fabry disease in a heterozygous female patient. New perspectives using enzyme replacement therapy] Dtsch Med Wochenschr. 2006;131:1590–1593. doi: 10.1055/s-2006-947801. [DOI] [PubMed] [Google Scholar]
- 40.Üçeyler N, He L, Schönfeld D, Kahn AK, Reiners K, Hilz MJ, et al. Small fibers in Fabry disease: baseline and follow-up data under enzyme replacement therapy. J Peripher Nerv Syst. 2011;16:304–314. doi: 10.1111/j.1529-8027.2011.00365.x. [DOI] [PubMed] [Google Scholar]
- 41.Froissart R, Guffon N, Vanier MT, Desnick RJ, Maire I. Fabry disease: D313Y is an alpha-galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol Genet Metab. 2003;80:307–314. doi: 10.1016/S1096-7192(03)00136-7. [DOI] [PubMed] [Google Scholar]
- 42.Gaspar P, Herrera J, Rodrigues D, Cerezo S, Delgado R, Andrade CF, et al. Frequency of Fabry disease in male and female haemodialysis patients in Spain. BMC Med Genet. 2010;11:19. doi: 10.1186/1471-2350-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yasuda M, Shabbeer J, Benson SD, Maire I, Burnett RM, Desnick RJ. Fabry disease: characterization of alpha-galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele. Hum Mutat. 2003;22:486–492. doi: 10.1002/humu.10275. [DOI] [PubMed] [Google Scholar]
- 44.Biegstraaten M, Hollak CE, Bakkers M, Faber CG, Aerts JM, van Schaik IN. Small fiber neuropathy in Fabry disease. Mol Genet Metab. 2012;106:135–141. doi: 10.1016/j.ymgme.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 45.Hughes RA, Umapathi T, Gray IA, Gregson NA, Noori M, Pannala AS, et al. A controlled investigation of the cause of chronic idiopathic axonal polyneuropathy. Brain. 2004;127(Pt 8):1723–1730. doi: 10.1093/brain/awh192. [DOI] [PubMed] [Google Scholar]
- 46.Lindh J, Tondel M, Osterberg A, Vrethem M. Cryptogenic polyneuropathy: clinical and neurophysiological findings. J Peripher Nerv Syst. 2005;10:31–37. doi: 10.1111/j.1085-9489.2005.10106.x. [DOI] [PubMed] [Google Scholar]
- 47.Notermans NC, Wokke JH, Franssen H, van der, Vermeulen M, van der Graaf Y, et al. Chronic idiopathic polyneuropathy presenting in middle or old age: a clinical and electrophysiological study of 75 patients. J Neurol Neurosurg Psychiatry. 1993;56:1066–1071. doi: 10.1136/jnnp.56.10.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolfe GI, Baker NS, Amato AA, Jackson CE, Nations SP, Saperstein DS, et al. Chronic cryptogenic sensory polyneuropathy: clinical and laboratory characteristics. Arch Neurol. 1999;56:540–547. doi: 10.1001/archneur.56.5.540. [DOI] [PubMed] [Google Scholar]
- 49.Liedberg GM, Vrethem M. Polyneuropathy, with and without neurogenic pain, and its impact on daily life activities--a descriptive study. Disabil Rehabil. 2009;31:1402–1408. doi: 10.1080/09638280802621382. [DOI] [PubMed] [Google Scholar]
- 50.Lindh J, Tondel M, Persson B, Vrethem M. Health-related quality of life in patients with cryptogenic polyneuropathy compared with the general population. Disabil Rehabil. 2011;33:617–623. doi: 10.3109/09638288.2010.505996. [DOI] [PubMed] [Google Scholar]
- 51.Hoitsma E, Reulen JP, de Baets M, Drent M, Spaans F, Faber CG. Small fiber neuropathy: a common and important clinical disorder. J Neurol Sci. 2004;227:119–130. doi: 10.1016/j.jns.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 52.Tavee J, Culver D. Sarcoidosis and small-fiber neuropathy. Curr Pain Headache Rep. 2011;15:201–206. doi: 10.1007/s11916-011-0180-8. [DOI] [PubMed] [Google Scholar]
- 53.Finsterer J. Inherited mitochondrial neuropathies. J Neurol Sci. 2011;304:9–16. doi: 10.1016/j.jns.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 54.Mancuso M, Piazza S, Volpi L, Orsucci D, Calsolaro V, Caldarazzo Ienco E, et al. Nerve and muscle involvement in mitochondrial disorders: an electrophysiological study. Neurol Sci. 2012;33:449–452. doi: 10.1007/s10072-011-0703-4. [DOI] [PubMed] [Google Scholar]
- 55.Henning F, Oey PL, Oerlemans WG, Wokke JH. Small-fiber neuropathy and the 3243A>G mutation in mitochondrial DNA. J Neurol. 2007;254:1281–1282. doi: 10.1007/s00415-006-0487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dyck PJ, Clark VM, Overland CJ, Davies JL, Pach JM, Dyck PJ, et al. Impaired glycemia and diabetic polyneuropathy: the OC IG Survey. Diabetes Care. 2012;35:584–591. doi: 10.2337/dc11-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith AG, Rose K, Singleton JR. Idiopathic neuropathy patients are at high risk for metabolic syndrome. J Neurol Sci. 2008;273:25–28. doi: 10.1016/j.jns.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baptista MV, Ferreira S, Pinho-E-Melo T, Carvalho M, Cruz VT, Carmona C, et al. Mutations of the GLA gene in young patients with stroke: the PORTYSTROKE study--screening genetic conditions in Portuguese young stroke patients. Stroke. 2010;41:431–436. doi: 10.1161/STROKEAHA.109.570499. [DOI] [PubMed] [Google Scholar]
- 59.Morita H, Larson MG, Barr SC, Vasan RS, O'Donnell CJ, Hirschhorn JN, et al. Single-gene mutations and increased left ventricular wall thickness in the community: the Framingham Heart Study. Circulation. 2006;113:2697–2705. doi: 10.1161/CIRCULATIONAHA.105.593558. [DOI] [PubMed] [Google Scholar]
- 60.Lauria G, Cornblath DR, Johansson O, McArthur JC, Mellgren SI, Nolano M, et al. EFNS guidelines on the use of skin biopsy in the diagnosis of peripheral neuropathy. Eur J Neurol. 2005;12:747–758. doi: 10.1111/j.1468-1331.2005.01260.x. [DOI] [PubMed] [Google Scholar]
- 61.Lauria G, Hsieh ST, Johansson O, Kennedy WR, Leger JM, Mellgren SI, et al. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on the use of skin biopsy in the diagnosis of small fiber neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. Eur J Neurol. 2010;17:903–912. doi: 10.1111/j.1468-1331.2010.03023.x. [DOI] [PubMed] [Google Scholar]