

Abstract
Gaucher disease is the commonest lysosomal storage disease seen in India and worldwide. It should be considered in any child or adult with an unexplained splenohepatomegaly and cytopenia which are seen in the three types of Gaucher disease. Type 1 is the non-neuronopathic form and type 2 and 3 are the neuronopathic forms. Type 2 is a more severe neuronopathic form leading to mortality by 2 years of age. Definitive diagnosis is made by a blood test–the glucocerebrosidase assay. There is no role for histological examination of the bone marrow, liver or spleen for diagnosis of the disease. Molecular studies for mutations are useful for confirming diagnosis, screening family members and prognosticating the disease. A splenectomy should not be performed except for palliation or when there is no response to enzyme replacement treatment or no possibility of getting any definitive treatment. Splenectomy may worsen skeletal and lung manifestations in Gaucher disease. Enzyme replacement therapy (ERT) has completely revolutionized the prognosis and is now the standard of care for patients with this disease. Best results are seen in type 1 disease with good resolution of splenohepatomegaly, cytopenia and bone symptoms. Neurological symptoms in type 3 disease need supportive care. ERT is of no benefit in type 2 disease. Monitoring of patients on ERT involves evaluation of growth, blood counts, liver and spleen size and biomarkers such as chitotriosidase which reflect the disease burden. Therapy with ERT is very expensive and though patients in India have so far got the drug through a charitable access programme, there is a need for the government to facilitate access to treatment for this potentially curable disease. Bone marrow transplantation is an inferior option but may be considered when access to expensive ERT is not possible.
Keywords: lysosomal storage disorder, splenomegaly, glucocerebrosidase, enzyme replacement therapy, thrombocytopenia
Abbreviations: LSD, lysosomal storage disorders; GD, Gaucher disease; GD1, Gaucher disease type 1; GD2, Gaucher disease type 2; GD3, Gaucher disease type 3; GBA, acid beta-glucosidase/glucocerebrosidase; ICGC, International Collaborative Gaucher Group; TRAP, tartarate resistant acid phosphatase; ACE, angiotensin converting enzyme; USG, ultrasonography; MRI, magnetic resonance imaging; SF-36, short form 36; DEXA, dual energy X-ray absorptiometry; EEG, electroencephalography; IQ, intelligence quotient; ERT, enzyme replacement therapy; INCAP, India Charitable Access Programme
Lysosomal storage disorders (LSDs) are inherited metabolic disorders and currently more than 45 LSDs are known. Gaucher disease (GD) is the most prevalent LSD world wide.1 Gaucher (pronounced as GO-SHEY) is named after Philippe Gaucher who first described a 32-year-old woman with an enlarged spleen and described it as “primitive epithelioma of the spleen” in 1882, while he was still a medical student. GD is an autosomal recessive disorder where the metabolic defect is an inherited deficiency of glucocerebrosidase due to mutations in the GBA1 (acid-β-glucosidase) gene.1 The result is an accumulation of an abnormal lipid glucocerebroside (glucosylceramide) in the lysosomes of macrophages2 leading to a wide spectrum of phenotypic manifestations. With the availability of enzyme replacement therapy (ERT), it is also now the most treatable LSD.
Epidemiology
As around the world, Gaucher disease is also the commonest lysosomal storage disease in India.3 The risk of developing GD increases with consanguinity in the family. Its frequency differs with different populations–being most prevalent—1:450 birth incidence in individuals of Ashkenazi Jewish descent.4 Ashkenazi Jews form about 75% of the world's Jewish population. However, the overall estimated prevalence of symptomatic disease is much lower—occurring in approximately 1 in 100,000 live births.1 The International Collaborative Gaucher Group (ICGC) (http://www.gauchercare.com/healthcare/registry.aspx) launched a registry in 1991 to document clinical, laboratory, demographic, genetic and therapeutic responses in patients with GD. Most of the published data and recommendations originate from this registry.
The majority of patients have type 1 Gaucher disease (GD1), which is the non-neuronopathic form of GD. It is the main type seen in the Ashkenazi Jewish population. Type 2 Gaucher disease (GD2), is also called acute neuronopathic GD or infantile cerebral GD. It comprises about 1 percent of patients in the ICGC Registry.5 Type 3 GD (GD3) is the chronic neuronopathic form and is seen in 5% of patients overall. GD3 is mainly seen in Northern Europe, Egypt and East Asia.6 A high incidence of GD3 is found in the Swedish province of Norrbotten and is therefore also referred to as the Norrbottnian type of GD.7 In India, there are no prevalence studies but in our study of treated patients, about a third had GD3 and two thirds were GD1.8 The numbers of patients with GD1 are likely to be higher as the severe GD3 patients were not considered for treatment. Also, there are no estimates of GD2 from India.
Pathogenesis
Gaucher results from deficiency of a lysosomal enzyme glucocerebrosidase (also known as acid beta-glucosidase, GBA).2 The enzyme acts on the substrate glucocerebroside which is a component of the cell membrane. In the normal lysosome, protein saposin C presents glucocerebroside to GBA which activates the enzyme.9 This enzyme is responsible for hydrolytic breakdown of glucosylceramide to glucose and ceramide. Deficiency of the enzyme leads to accumulation of glucosylceramide and other glycolipids in the lysosomes of macrophages, primarily in the spleen, liver, bone marrow, brain, osteoclasts and less often the lungs, skin, kidneys, conjunctivae and heart. The deacylated form of glucosylceramide, glucosylsphingosine, is elevated in neuronopathic disease and correlates more with phenotype severity compared to glucosylceramide.10
Genetics
GD is an autosomal recessive disorder secondary to mutations in the glucocerebrosidase gene which is 11 exons in length and located on chromosome 1q21.11 More than 200 distinct GBA gene mutations are described (Human Gene Mutation Database www.hgmd.cf.ac.uk). Majority of these mutations are single nucleotide substitutions. The three mutations accounting for more than 90% of all mutations in Ashkenazi Jews12 are 1. The commonest mutation in the ICGC Registry5 is due to the N370S substitution in the alleles. It is a missense mutation that results in residual enzyme activity. The second and third common mutations are due to the L444P substitution and 84 GG substitution in the alleles respectively which result in affected patients who are compound heterozygotes. The mutations resulting from N370S and L444P substitutions account for approximately 70 percent of mutations in non-Ashkenazi European patients.13 In the Korean Asian population studied, N370S substitution was not found.14 To my knowledge, here are no mutation studies of GD patients from India.
Genotype–Phenotype Correlations
Mutation in alleles containing the N370S substitution is associated with the non-neurologic type 1 GD. It is commonly found in Ashkenazi Jews and non-Jewish Europeans.15 Mutations in alleles containing the L444P substitution when present in homozygous form are strongly associated with the development of neuronopathic disease.16 Therefore, the type of mutation may to some extent determine the type of Gaucher disease. However, there seem to be factors other than genotype affecting the phenotypic expression as severity may vary among siblings, even identical twins.17
Classification
Three types of GD have been described based on the clinical features, ethnicity and the natural history of the disease (Table 1 gives differences between the 3 types of GD). GD1 patients do not have neurological involvement, GD2 is the acute neuronopathic and GD3 is the chronic neuronopathic type. GD3 is further subdivided in to 3 subtypes a, b and c depending on the clinical features. Gaucher disease can also present as hydrops in the perinatal period which is often lethal. There may be congenital ichthyosis, also known as collodion baby.18 GD should therefore be considered as a differential diagnosis of any hydrops in pregnancy.
Table 1.
Differences in the Three Types of Gaucher Disease.
| Type 1 | Type 2 | Type 3 | |
|---|---|---|---|
| Disease onset | Childhood/adulthood | Infancy | Childhood/adolescence |
| Splenohepatomegaly | Present | Present | Present |
| High prevalence | Ashkenazi Jews | ? | Swedish province of Norrbotten |
| Bone involvement | Present | Absent | Present |
| Ocular signs | Absent | Present | Present |
| Neurological involvement | Absent | Present, severe | Present, mild |
| Other organ involvement | Liver cirrhosis Pulmonary hypertension | Hydrops Congenital ichthyosis | Cardiac and vascular calcifications |
| Lifespan with or without therapy | Early childhood to late adulthood | Less than 2 years | Variable—up to early adulthood |
| Response to ERT | Good | Poor, not indicated | Variable |
ERT: Enzyme replacement therapy.
Clinical presentation
The clinical presentation in GD is a broad spectrum of findings with overlap seen between the 3 types The major organs involved with their clinical manifestations have been summarized in Table 2. Gaucher disease should be considered in any child or adult who presents with a splenohepatomegaly with cytopenia which is unexplained and should not be dismissed as a malarial spleen or tropical splenomegaly.
Table 2.
Organ-wise Involvement in Gaucher Disease.
| Organ system | |
|---|---|
| General | Reduced quality of life, delayed milestones, growth retardation, pubertal status |
| Skeletal | Chronic bone pain (33%), acute bone crises (7%) Kyphosis including gibbus, scoliosis and chest deformities Bone fractures (7%) Skeletal growth retardation (36%) Bone remodeling failure (Erlenmeyer flask deformity) Osteopenia (55%) Osteonecrosis, avascular necrosis head femur Osteolysis, osteosclerosis |
| Visceral organs | Abdominal pain, early satiety, feeling of fullness, diarrhea Splenomegaly (85%), splenic infarcts Hepatomegaly (63%) (may progress to cirrhosis, portal hypertension) Cholelithiasis |
| Hematological | Anemia (34%)—Fatigue, exertional dyspnea, need for blood transfusions Thrombocytopenia (68%) spontaneous bleeding—epistaxis, bruising, menorrhagia or hemostatic problems after trauma, surgery or post-partum bleeding Leukopenia: increased risk of infection Gammopathy |
| Lungs | Dyspnea (exertional), cough, recurrent respiratory infections Pulmonary hypertension with dyspnea on exertion or at rest, syncope Hepatopulmonary syndrome—clubbing, cyanosis, orthopnea |
| CNS (Type 2/3) | Strabismus, saccade initiation failure, supranuclear gaze palsy, slow object tracking, hypertonia, rigidity, opisthotonus, bulbar palsy, seizures, ataxia, myoclonus, dementia, mental retardation |
| Skin | Yellow/brownish discoloration Bruises, petechiae |
| Heart | Valvular calcification, congestive heart failure, arrhythmias |
| Eyes | Pingueculae Corneal opacities Strabismus, saccade initiation failure (ocular motor apraxia) in type 3 disease |
| Lymphatic | Enlarged lymph nodes |
| Malignancies | Increased risk of multiple myeloma, hematological malignancy, hepatocellular carcinoma, renal cell carcinoma |
The percentages are taken from Charrow J et al of the Gaucher Registry.5 Figures for neurological involvement will differ between populations.
Type 1 GD or the non-neuronopathic type presents with symptoms which may first present in infancy to late adulthood. It is characterized by painless splenohepatomegaly, which often leads to massive abdominal distension (Figure 1) with anemia and thrombocytopenia. Fatigue, nose bleeds, and easy bruising are a manifestation of the cytopenia. Some patients are transfusion dependent. Cytopenias are secondary to hypersplenism, bone marrow infiltration by Gaucher cells and an intrinsic haemopoietic defect cells.19 The increased bleeding tendency in patients with type 1 GD is related to thrombocytopenia, coagulation abnormalities, and defective platelet function.20 The organomegaly can reach massive proportions and the splenomegaly is often the first presentation and the spleen is much more enlarged (median 15.2 times, up to 75 times) than the liver (up to 3 times).5 Palpation of the abdomen should start in the inguinal area so as to not miss the edge of the enlarged organ. Organomegaly results in early satiety, poor oral intake and delayed milestones. Splenic infarction is often the cause of acute abdominal pain. Abdominal lymphadenopathy21 also has been described in patients with this disease (Figure 2). Some individuals with this genotype remain asymptomatic throughout life. However even the “asymptomatic” ones usually have organomegaly and cytopenia.
Figure 1.

Massive splenohepatomegaly in a 18-month-old child with Type 1 Gaucher disease.
Figure 2.
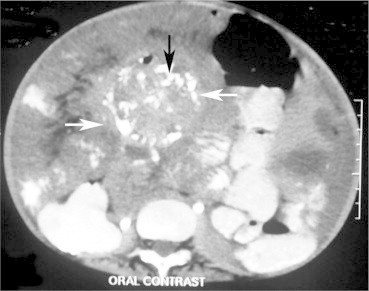
Enlarged abdominal lymph nodes with calcification (arrows) on CT scan.
Most children have poor growth and development with delayed puberty. Children with severe disease show delayed milestones and failure to thrive.5
Bone disease manifests as bone pain, painful pain crises, osteoporosis and pathologic fractures with vascular necrosis and deformities like kyphosis including gibbus (Figure 3).22–24 Bone is involved by several processes including decreased mineral density, marrow infiltration, and infarction of bone “Bone crises” typically involve the proximal tibia or distal femur with acute pain attacks and fever associated with leukocytosis and high ESR.25 Hepatic fibrosis can occur, but cirrhosis and portal hypertension are not common except in splenectomized patients.26 Hepatopulmonary syndrome with hypoxia on standing, clubbing and cyanosis may be seen in those with advanced liver disease and more so if the patient has had a splenctomy.27 Interstitial lung disease may occur when Gaucher cells infiltrate the alveoli and interstitium.28 The abnormal cells can also occlude pulmonary capillaries, perhaps contributing to pulmonary hypertension.29 By far, patients with GD1 do not have neurological involvement though peripheral polyneuropathy, has been described.30
Figure 3.
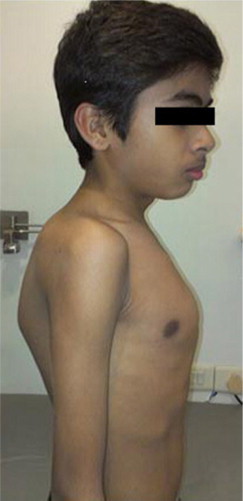
Severe kyphosis and chest deformity in a 10-year-old child with Type 1 Gaucher disease.
Type 2 GD is a neuronopathic from of disease with severe neurological disease and is usually fatal by 2 years of age.31 The neurological involvement is a more severe involvement of that seen in GD3.
Type 3 GD is the chronic neuronopathic form of GD and has a later onset than GD2. However, GD3 may have onset before age two years with slow progression. GD3 is characterized by a milder neurological involvement compared to that seen in GD2, in addition to the visceral and bone marrow involvement as in GD1. The earliest CNS involvement can be picked up on a detailed ophthalmologic examination revealing defects manifesting as horizontal saccade (fast eye movement) initiation abnormalities, strabismus (Figure 4), and bulbar palsy or paresis.32 Children with saccadic initiation abnormalities tend to move their head to shift their gaze. Neurologic progression is marked by severe hypertonia, rigidity, arching (opisthotonus), swallowing impairment, and seizures.33 Visceral disease in GD3 is often more severe than in GD1. In children, failure to thrive because of massive organomegaly can lead to delayed milestones and growth retardation and this should not be interpreted as GD3. On the other hand, subtle neurological signs may be missed initially and the patient may be initially diagnosed as having GD1 and later be diagnosed as GD3 when the neurological signs become more obvious. Three subtypes of GD3 have been described.
Figure 4.

Strabismus in a 2-year-old child with Type 3 Gaucher disease.
Type 3a is characterized by progressive dementia, ataxia, and myoclonus.34
Type 3b has extensive visceral and bone involvement with CNS involvement limited to supranuclear gaze palsy (saccade initiation failure, with compensatory head thrusting), either alone or a more slow progressive neurodegenerative syndrome.35
Type 3c (cardiovascular form) is rare and characterized by supranuclear gaze palsy, corneal opacity, and cardiovascular calcification, with little visceral disease.36 It is a unique phenotype associated with homozygosity for the D409H mutation commonly found in the Mediterranean population.37
Gaucher Disease and Parkinsonism
GD has some association with Parkinson disease. GBA mutations are associated with a spectrum of parkinsonian phenotypes ranging from Parkinson disease, mostly of the akinetic type, to a less common phenotype characteristic of Lewy body dementia.38 Literature suggests that GBA mutations may increase the risk in individuals who are otherwise prone to developing Parkinson disease39 rather than causing Parkinson's disease per se.
Liver and Gaucher Disease
Liver involvement in Gaucher disease includes hepatomegaly and ranges from mild elevation of the liver enzymes to the more severe cirrhosis with portal hypertension. In a study of 25 patients of GD who underwent liver biopsy40 three had cirrhosis and portal hypertension. Fourteen patients had infiltration of the liver with Gaucher cells mainly in the centrilobular area and five had scattered foci of gaucher cells throughout the liver. Severity of liver disease correlated with severity of other complications.
Malignancies in Gaucher Disease
Review of data from 2742 patients in the International Gaucher Registry in 2005 indicated that the overall risk of cancer, including general hematologic malignancies, is not increased compared with the expected rate of individuals of the same age and sex in the United States population except for multiple myeloma where risk was increased irrespective of ERT (RR 5.9, 95% CI 2.9–10.8).41 However, in a recent review of literature42 which included 15 cohort/cross sectional studies, and 65 case reports/series, it was concluded that GD patients have an increased risk of cancer in general [pooled relative risk of 1·70 (95% confidence interval 1·27–2·31)] and multiple myeloma and hematological malignancies in particular (estimated risk between 25·0 and 51·1 and 3·5 and 12·7, respectively). An increased risk of hepatocellular carcinoma and renal cell carcinoma has also been reported.42 Splenectomy, immune dysregulation, endoplasmic reticulum stress, genetic modifiers, altered iron metabolism and insulin resistance were thought to contribute to the development of a malignancy.
Clinical Course
The spectrum of GD is quite variable from asymptomatic disease to very severe disease. Phenotypic expression and severity may be different even in identical twins.17 Symptomatic patients may die prematurely from severe anemia, failure to thrive, sequelae of splenectomy, severe bone disease, bleeding complications, infection, liver failure, severe pulmonary disease or malignancy. The disease tends to be more rapidly progressive in children with severe disease compared to adults with milder disease where it may be progress very slowly43,44 or even spontaneously regress.45
Diagnosis
Diagnosis of Gaucher disease is made on the basis of clinical history, physical examination, laboratory test and confirmed by a blood test showing deficient glucocerebrosidase enzyme and genetic mutation studies when the diagnosis is doubtful. History of consanguinity and family history of suspected or proven GD will support the diagnosis. The diagnosis and work up of a patient with GD is summarized in Table 3.
Table 3.
Diagnosis, Work Up and Therapeutic Monitoring in Gaucher Disease (Modified from Niederau, et al55).
| Type 1—non-neuronopathic form | |
| Diagnosis and work-up |
|
| Initial monitoring |
|
| Long-term treatment |
|
| |
| |
TRAP: Tartarate resistant acid phosphatase, ACE: Angiotensin converting enzyme, USG: Ultrasonography, MRI: Magnetic resonance imaging, SF-36: Short form 36 (limited value in children), LFT: liver function test PT: prothrombin time, PTT: Partial thromboplastin time, DEXA: Dual energy X-ray absorptiometry, EEG: Electroencephalography, IQ: Intelligence quotient.
In the past, diagnosis was often made on seeing the Gaucher cells in a bone marrow aspirate or when the patient with an unexplained massive splenomegaly was subjected to a splenectomy. Gaucher cells have eccentrically placed nuclei and a cytoplasm with striations described as “wrinkled tissue paper appearance”46 (Figure 5). These can be seen on light microscopy and PAS staining. These methods of diagnosis were not accurate as many storage cells “pseudo-Gaucher cells” could be confused for Gaucher cells on the marrow examination. These are found in several hematological conditions including hemoglobinopathies47 malignancies,48–50 and mycobacterial infections.51
Figure 5.
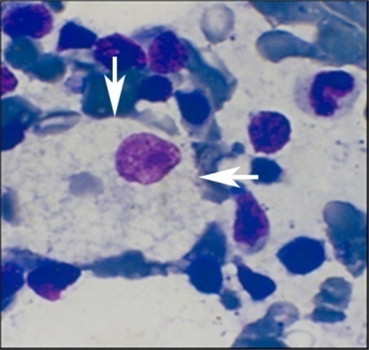
Photomicrograph of a Gaucher cell (arrows) with “crumpled tissue paper” cytoplasm and eccentric nucleus on Hematoxylin and Eosin staining.
Now, the gold standard for diagnosing Gaucher disease is measurement of glucocerebrosidase enzyme activity in leucocytes or skin fibroblasts on a skin biopsy.52 Enzyme activity in heterozygote carriers and normal individuals may shows overlap and therefore enzyme analysis by itself cannot be used to differentiate carriers from normal individuals. With the availability of the diagnostic enzyme test, there is no indication to histologically examine the bone, liver or spleen for diagnosis. Bone marrow examination may be needed if the splenomegaly does not regress on treatment or patient develops enlarged lymph nodes or B symptoms to suggest development of a lymphoma.
Laboratory Tests
Thrombocytopenia, anemia and leucopenia are seen on the blood counts. Liver enzymes may be elevated40 and coagulation factor deficiencies53 causing abnormal clotting have been described in addition to platelet function problems. Platelet adhesion defect and not aggregation defects contribute to the thrombocytopathy in GD patients and therefore predispose to an increased tendency to bleeding.20
Imaging
Organomegaly
Ultrasonography54 CT scan or ideally MRI scan of the abdomen are used to determine liver and spleen volumes (Figure 6). Focal accumulations of Gaucher cells “Gaucheromas” may be found in the liver or spleen.55 In resource poor setting like ours MRIs are rarely performed. Also, need for sedation in younger children makes it more challenging. CT scans have the disadvantage of repeated radiation and are therefore not routinely recommended.
Figure 6.

Contrast enhanced CT scan of liver reveals ill-defined minimally enhancing hypodensity in segment 7 of the liver with portal and hepatic venous branches are seen coursing through this lesion (arrows) This represent hepatic infiltration by Gaucher cells. (Gaucheroma).
Skeletal Survey
Long bone surveys are best performed by X-ray of the proximal tibia and distal femur. Loss of bone substance manifests as loss of trabecular structure, rarefaction of the cortical bone or large cystic areas.56 Fractures, bone infarcts and avascular necrosis of the femur head can also occur. Erlenmeyer flask deformity of the distal femur (a conical laboratory flask with a narrow neck and flat broad bottom) caused by abnormal modeling of the metaphysis is seen in up to half the patients of GD5 (Figure 7). However, this deformity is not specific for Gaucher disease and may be seen in Niemann–Pick disease also.
Figure 7.
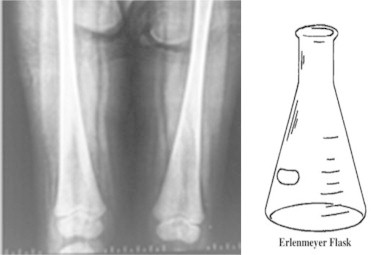
X-ray of the Erlenmeyer Flask deformity of the femora—showing lack of curve at the dia-metaphyseal junction resembling a conical flask (picture to the right) along with cortical thinning.
Bone involvement is best appreciated on MRI scans of spine (Figure 8) and femur. T1-weighted MRI is used for assessing marrow infiltration seen as “yellow bone marrow”57 T2-weighted MRI is the most sensitive method of detecting active bone infarcts.57 Dual energy X-ray absorptiometry (DEXA)58 of the lumbar spine and femora are done for determining bone density (osteopenia and osteoporosis) though values for children are not standardized.
Figure 8.

T1 and T2W1 images of the lumbar spine reveals generalized decrease in marrow signal (arrows) in the vertebral bodies representing changes of osteosclerosis.
Additional Tests
Chest X-ray and 2D echo are also desirable to rule out lung parenchymal involvement and pulmonary hypertension, respectively.55 Other additional tests may be needed in specific situations (Table 4).
Table 4.
Additional Tests Needed in Specific Situations.
| Test | Situation |
|---|---|
| Iron studies | Anemia (all Indian patients, as common), iron overload |
| Serum Vitamin B12 | Anemia (all Indian patients, as common) |
| Reticulocyte count | Anemia |
| Vitamin D3 | Bone disease (all Indian patients, as common) |
| Serum calcium, phosphorous, parathormone | Rule out other causes of bone disease |
| Platelet function studies | Thrombocytopenia resistant to treatment |
| Platelet antibodies | Thrombocytopenia resistant to treatment |
| Antibodies to imiglucerase | Poor response to ERT |
| HBsAg, anti HCV | All transfused patients |
ERT: Enzyme replacement therapy.
Biomarkers
Chitotriosidase is a prognostic biological marker which correlates with the disease burden and is useful in monitoring therapy. It is a chitinase and reflects “alternative” type macrophage activation that is overexpressed by the Gaucher cell.59 It is not a diagnostic test as it can be raised in other storage disorders as well. It may be normal in 6% of the population due to a gene mutation.60 When chitotriosidase is normal or there are no facilities for its estimation, CCl18, serum angiotensin converting enzyme (ACE) tartrate-resistant isoenzyme (TRAP), may be used as these are markers of alternatively activated macrophages. Hyperferritinemia is seen and correlates with increased liver volume and previous splenectomy.61
Molecular Diagnosis
Mutation analysis confirms the diagnosis and can prognosticate the natural course of the disease. Patients with mutations secondary to N370S substitution do not develop neurological disease15 and those with homozygosity or compound heterozygosity for L444P or D409H mutation are usually associated with neurological disease.16,62 In early childhood, the neurological signs of GD3 may not manifest and the child may be wrongly diagnosed as GD1. The neurological symptoms and signs may evolve over years. This can be obviated with a molecular diagnosis. The D409H allele is also associated with cardiovascular and corneal involvement.36,63
In an index case with a known mutation, undiagnosed affected family members and heterozygote carriers can be diagnosed with mutation analysis. It is cost effective to look for the most common mutations in the population like the N370S in the Ashkenazi Jews. However, if a mutation is not found in an otherwise clinically suspected patient of GD with low glucocerebrosidase levels, complete sequencing of the GBA gene may be needed as a second line investigation.
Prenatal Diagnosis
Prenatal diagnosis is performed by enzyme analysis of fetal cells obtained by chorionic villus sampling or amniocentesis at 16 weeks of pregnancy.64 Knowledge of the DNA mutations in the proband or in the heterozygous parents allows the use of DNA mutation analysis together with enzyme analysis for prenatal diagnosis. Mutation analysis is recommended as a confirmatory assay. Preimplantation genetic diagnosis is also possible.65
Differential Diagnosis
The differential diagnosis of GD is mainly of disease associated with splenomegaly and cytopenia. These would include hematological malignancies and storage disorders like Niemann–Pick.66 Most of these disorders have characteristic clinical, radiographic, or laboratory features that distinguish them from GD. Deficiency of saposin C also called atypical Gaucher disease or a variant of GD. It results in a severe disorder similar to GD.67
Management
The goal of therapy is to reduce the accumulation of the toxic substrate glucocerebroside and other glycolipids to prevent progressive disease with debilitating complications. This could be achieved by either by Enzyme replacement Therapy (ERT) where the deficient enzyme is administered in its modified form to metabolize the substrate or inhibition of substrate synthesis by inhibiting the enzyme–glucosylceramide synthase through substrate reduction therapy (Figure 9).
Figure 9.
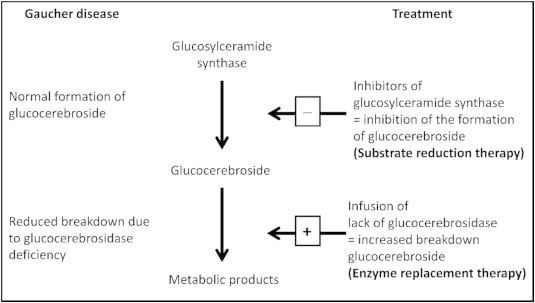
Treatment strategies in a patient with Gaucher disease (modified from Claus Niederau C52).
Enzyme replacement therapy (ERT)
ERT has revolutionized the treatment of Gaucher disease and has now become the standard of care.68 It has offered hope to patients with this disease who otherwise have a dismal prognosis. All patients do not need treatment. Only symptomatic patients of GD 1 and GD 3 merit treatment. ERT is most effective in reducing the liver and spleen size and the bone symptoms, and improving blood counts. It is partially effective in improving neurological symptoms. ERT does not cross the blood brain barrier in sufficient quantities to improve the neurological symptoms in GD2. It is also ineffective in treating lung parenchymal disease or pulmonary hypertension and lymph nodal disease. Treatment of GD2 is supportive and majority would not survive beyond second year of life.
Indications for treatment are elaborated in Table 5.69
Table 5.
Indications for Initiating Enzymes Replacement Therapy in Symptomatic Children (Modified from Kaplan, et al69).
| One or more of the following |
|
| Along with lifelong commitment to financial commitment for the drug |
Hb: Hemoglobin, MN: multiples of normal, BMD: Bone mineral density.
Before starting treatment, it may be worthwhile storing a serum sample at −70° C for future tests or in case the patient develops antibodies to ERT.
History of Enzyme Replacement Therapy
The first GD patient to have received mannose targeted ERT was in 1989. This was derived from the placenta alglucerase (Ceredase, Genzyme Corporation) and became commercially available in 1991.68 In 1994, the genetically engineered form—imiglucerase (Cerezyme—Genzyme Corporation) became available. This recombinant form of enzyme preparation is a targeted therapy towards the mannose receptors in the macrophages. The ICGC has collated data of more than 6000 patients including those who have received ERT with alglucerase/imiglucerase and several recommendations and guidelines have been published based on this data.
Administration and Dose of Enzyme Replacement Therapy
An initial test dose is given when ERT is administered for the first time. The deficient enzyme is administered in its modified form as an intravenous infusion over 2 h every 15 days.
The standard dose is 60 units/kg and this may be modified depending on need to 20–120 units/kg. Doses can be individualized as per requirements.70 This can help cut costs of this very expensive drug. On the other hand the dose may be increased if the response is not adequate. Higher doses are given to younger children with GD3 in the initial stages of the disease. Dose of 60 units/kg are recommended in those with clinical manifestations before 10 years of age and with severe bone disease.71 An interval of >2 years between diagnosis and starting ERT is associated with a risk of avascular necrosis.72
Lower doses of 15–20 units/kg may be given in those with organomegaly and cytopenia but without skeletal involvement and as maintenance doses.70
Adverse Effects of Enzyme Replacement Therapy
Reported side effects are usually mild and the drug is well tolerated. These include nausea, vomiting, abdominal pain, diarrhea, rash, fatigue, headache, fever, dizziness, chills, backache, and rapid heart rate. At the site of injection, discomfort, itching, burning swelling or uninfected abscess can occur. The commonest adverse effect of the ERT is immune mediated hypersensitivity. These can be reduced by increasing the time of infusion and pretreatment with anti-histaminics and corticosteroids. Approximately 13% of patients have developed antibodies to the enzyme though all patients tolerate treatment.73 These are IgG mediated and usually manifest as non-response of organomegaly and cytopenias to treatment.
Response to Enzyme Replacement Therapy
This is evaluated by improvement in growth, milestones and quality of life. Monitoring needed in patients on ERT is summarized in Table 3. There is regression in the liver and spleen size and blood counts improve. There is amelioration of bone symptoms except avascular necrosis which is irreversible. The therapeutic goals desired to be achieved are mentioned in Table 6.74
Table 6.
Therapeutic Goals for Children (Modified from Pastores et al74).
| Anemia |
|
| Thrombocytopenia |
|
| Hepatomegaly |
|
| Splenomegaly |
|
| Bones |
|
| Growth |
|
In a recent longterm analysis by ICGC registry of GD1 patients on ERT, 90% of patients had normal hemoglobin levels, though proportionately fewer patients had normal platelet levels after 10 years of treatment. The spleen volumes after 10 years of treatment were reduced in 97% of patients and all had reduced liver volumes on long term treatment except the ones with significant fibrosis or cirrhosis at the onset of therapy.75
Newer Enzyme Replacement Therapy
Recently, Velaglucerase alfa (VPRIV from Shire pharmaceuticals) which is produced by gene activation technology in a human cell line has been approved for treatment in GD1 in both adults and children.76,77 Taliglucerase alfa (Protalix Biotherapeutics, Carmiel, Israel) is a novel plant cell-derived recombinant human β-glucocerebrosidase for Gaucher disease where phase 3 trials support safety and efficacy of the drug.78
Substrate reduction therapy
Substrate (glucocerebroside) depletion through inhibition of glucosylceramide synthase enzyme with oral drugs like Miglustat and Eliglustat tartrate are used for milder disease where treatment with ERT is not possible79 (Figure 9). These drugs are best used as maintenance therapy after the therapeutic goals have been achieved with ERT.80
Miglustat is given in the dose of 100 g three times a day orally and has little role in neurological or bone disease, it also takes much longer than ERT to control the organomegaly or cytopenia. It is approved for use in adults only and not in children. Adverse effects include transient weight loss and GI side effects especially flatulence and diarrhea and intolerance to milk with exacerbation of pre-existing tremors.81
Eliglustat tartarate is an oral drug inhibiting production of glucosylceramide. It has been shown to be of benefit in improving organomegaly and cytopenias in patients of GD1 in phase 2 studies in adults and is being investigated for use in children. It has no major side effects described.82
Bone marrow transplantation
Bone marrow transplantation has the potential for cure of GD. Bone marrow transplant was the only treatments available prior to introduction of ERT. Since the availability of effective ERT bone marrow transplant is no longer the treatment of choice.83 as it has significant morbidity and mortality associated with it. However, when there is no access to expensive ERT which costs around Rs. 40 lakhs a year, bone marrow transplantation which costs from Rs 3 to 10 lakhs may be an option, albeit inferior to ERT. In countries like India where most patients are self paying, the immunosuppression costs are expected to be far lower than that of ERT.
Liver transplantation
Liver transplants have been anecdotally performed in GD1 patients who have cirrhosis and portal hypertension or those with the hepatopulmonary syndrome. In a report of four patients who underwent a liver transplant, two of these patients had co-infections with hepatotropic viruses, and overall good outcomes were reported.84
Supportive care
This forms an integral part of management.
Skeletal disease—Severe bone crises need analgesics and oral prednisolone.85 Patients with significant osteopenia or osteoporosis need correction of Vitamin D deficiency if present along with bisphosphonates.86 Osteomyelitis needs to be excluded.87 Avascular necrosis of the femoral head needs hip replacement surgery. Physiotherapy plays an integral role in patients with skeletal disease.
Cytopenia
Anemia—This may need blood transfusions but iron and vitamin B12 deficiency need to be ruled out. Bleeding tendency—There may be a need to transfuse platelets prior to any invasive procedures or pregnancy.
Splenectomy is rarely indicated for palliation if the patient has severe symptomatic cytopenia from a massive splenomegaly which is not responding to ERT. In the Indian setting, it is often performed as a desperate measure especially if there is no possibility of getting any definitive treatment. Surgical partial splenectomy has been described in Western literature though not done in India. It can also be performed radiologically through splenic artery embolization which carries its own risks like splenic infarction and abscess formation. Splenectomy has been associated with a higher risk of cholesterol gall stones, pulmonary complications including pulmonary hypertension, avascular necrosis of bone and iron overload states.88–91
Neuronopathic disease—This is best managed by a pediatric Neurologist and specific drugs for myoclonus or epilepsy are needed.
Malignancies–Patients of GD need to be screened for malignancy which should be suspected in case of non-response to therapy (enlarged spleen) or persistent cytopenia or development of enlarged lymph nodes. Patient can be subsequently referred to an oncologist.
Counseling
Psychological counseling is important for patients and families with GD given the likelihood of incapacitating symptoms and chronic nature of the disease. When the parents are heterozygous carriers of one of the common alleles, parental genotype can provide some guidance for prenatal counseling, each child will have a 25% chance of having the disease. The severity of the disease in the child will depend on the parental genotype and counseling can be done accordingly. Consanguineous marriages need to be discouraged.
Future therapies
Enzyme Enhancement Therapy with Chaperones
Chaperone therapy is a newly developed molecular therapeutic approach to LSDs especially those affecting the brain. Mutant enzyme proteins are unstable in the cell with inability to function. A chemical chaperone which is a substrate analog competitive inhibitor when combined with the unstable mutant enzyme protein forms a stable complex to be transported to the lysosome and restores normal activity of the mutant enzyme. This molecular interaction is a paradoxical phenomenon that an inhibitor in vitro serves as an enzyme stabilizer in situ.92 The expectorant drug ambroxol has been found to be a chaperone for glucocerebrosidase and has shown a beneficial effect in a few Gaucher patients with oculomotor dysfunction and myoclonus.93
Gene Therapy
In humans, retroviral vector transfer of the GBA gene into cultured bone marrow cells of Gaucher patients results in expression of physiologic levels of enzyme.94 In an animal model, extracorporeal transfection of stem cells achieved production of human glucocerebroside in mouse macrophages.95 Although gene therapy is an attractive option, the progress in this field has slowed because of issues of gene delivery expression, especially in stem cells derived from bone marrow and non-reproducibility of the beneficial effects in the clinical setting.
Gaucher disease in India
Gaucher disease has been described to be the commonest LSD in India.3,96 In our Indian multicentre study which included 5 centres from across the country, there was history of consanguinity in 50% of families. The first 25 patients who were treated with ERT were analyzed and found to have significant improvement in the liver and spleen sizes and blood counts.8 Diagnosis was first made on bone marrow examination in 36% and after a splenectomy in 24% of patients, though confirmed in all by the enzyme assay.
There has been a spurt of GD patients being diagnosed in India in the last decade with the availability of the diagnostic blood test–glucocerebrosidase assay. This increase in diagnosis is also related to the availability of ERT. ERT is profoundly expensive and virtually beyond the means of any patient. So far, over 65 patients from the country have been treated with ERT (imiglucerase) through the INCAP programme (personal communication). The INCAP (India Charitable Access Programme) of Genzyme Corporation, USA has made it possible for patients of Gaucher disease to get ERT free of cost. The candidacy of patients to receive ERT is evaluated by an advisory board that includes 6 physicians from centers across India, international experts on the disease and representatives of Genzyme Corporation. Similar support for a smaller number of patients is being provided for velaglucerase from Shire pharmaceuticals. However, with increasing number of patients being diagnosed, it is becoming difficult to support all patients with the disease. Few children are being financially supported by the parents employers especially those in the public sector companies. A patient advocacy group–LSDSS (lysosomal storage disorders support society www.lsdss.org) is working at various levels to get the government and employers to share the expenses of this otherwise very expensive therapy which is unaffordable on an individual basis.
Conflicts of interest
The author is a member of the Indian Medical Advisory Board (IMAB) of the Genzyme Corporation.
References
- 1.Beutler E., Grabowski G.A. Gaucher disease. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. p. 3635. [Google Scholar]
- 2.Messner M.C., Cabot M.C. Glucosylceramide in humans. Adv Exp Med Biol. 2010;688:156–164. doi: 10.1007/978-1-4419-6741-1_11. [DOI] [PubMed] [Google Scholar]
- 3.Sheth J., Mistri M., Sheth F. Burden of lysosomal storage disorders in India: experience of 387 affected children from a single diagnostic facility. JIMD Rep. 2013 Jul 13;12:51–63. doi: 10.1007/8904_2013_244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zimran A., Gelbart T., Westwood B., Grabowski G.A., Beutler E. High frequency of the Gaucher disease mutation at nucleotide 1226 among Ashkenazi Jews. Am J Hum Genet. 1991;49:855–859. [PMC free article] [PubMed] [Google Scholar]
- 5.Charrow J., Andersson H.C., Kaplan P. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000;160:2835–2843. doi: 10.1001/archinte.160.18.2835. [DOI] [PubMed] [Google Scholar]
- 6.Tylki-Szymańska A., Vellodi A., El-Beshlawy A., Cole J.A., Kolodny E. Neuronopathic Gaucher disease: demographic and clinical features of 131 patients enrolled in the International Collaborative Gaucher Group Neurological Outcomes Subregistry. J Inherit Metab Dis. 2010;33:339–346. doi: 10.1007/s10545-009-9009-6. [DOI] [PubMed] [Google Scholar]
- 7.Dreborg S., Erikson A., Hagberg B. Gaucher disease–Norrbottnian type. I. General clinical description. Eur J Pediatr. 1980;133:107–118. doi: 10.1007/BF00441578. [DOI] [PubMed] [Google Scholar]
- 8.Nagral A., Mewawalla P., Jagadeesh S. Recombinant macrophage targeted enzyme replacement therapy for Gaucher disease in India. Indian Pediatr. 2011;48:779–784. doi: 10.1007/s13312-011-0128-4. [DOI] [PubMed] [Google Scholar]
- 9.Locatelli Hoops S., Kolter T., Sandhoff K. Saposin C and other sphingolipid activator proteins. In: Futerman A.H., Zimran A., editors. Gaucher Disease. CRC Press; Boca Raton: 2006. p. 67. [Google Scholar]
- 10.Orvisky E., Park J.K., LaMarca M.E. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: correlation with phenotype and genotype. Mol Genet Metab. 2002;76:262–270. doi: 10.1016/s1096-7192(02)00117-8. [DOI] [PubMed] [Google Scholar]
- 11.Cormand B., Montfort M., Chabás A., Vilagelieu L., Grinberg D. Genetic fine localization of the beta-glucocerebrosidase (GBA) and prosaposin (PSAP) genes: implications for Gaucher disease. Hum Genet. 1997;100:75–79. doi: 10.1007/s004390050468. [DOI] [PubMed] [Google Scholar]
- 12.Beutler E., Nguyen N.J., Henneberger M.W., Giralt M., Giraldo P., Pocoví M. Gaucher disease: gene frequencies in the Ashkenazi Jewish population. Am J Hum Genet. 1993;52:85–88. [PMC free article] [PubMed] [Google Scholar]
- 13.Alfonso P., Cenarro A., Pérez-Calvo J.I. Mutation prevalence among 51 unrelated Spanish patients with Gaucher disease: identification of 11 novel mutations. Blood Cells Mol Dis. 2001;27:88–91. doi: 10.1006/bcmd.2001.0461. [DOI] [PubMed] [Google Scholar]
- 14.Jeong S.Y., Park S.J., Kim H.J. Clinical and genetic characteristics of Korean patients with Gaucher disease. Blood Cells Mol Dis. 2011;46:11–14. doi: 10.1016/j.bcmd.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 15.Koprivica V., Stone D.L., Park J.K. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am J Hum Genet. 2000;66:1777–1786. doi: 10.1086/302925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amaral O., Lacerda L., Santos R. Type 1 Gaucher disease: molecular, biochemical, and clinical characterization of patients from northern Portugal. Biochem Med Metab Biol. 1993;49:97–107. doi: 10.1006/bmmb.1993.1011. [DOI] [PubMed] [Google Scholar]
- 17.Lachmann R.H., Grant I.R., Halsall D., Cox T.M. Twin pairs showing discordance of phenotype in adult Gaucher's disease. QJM. 2004;97:199–204. doi: 10.1093/qjmed/hch036. [DOI] [PubMed] [Google Scholar]
- 18.Stone D.L., Carey W.F., Christodoulou J. Type 2 Gaucher disease: the collodion baby phenotype revisited. Arch Dis Child Fetal Neonatal Ed. 2000;82(2):F163–F166. doi: 10.1136/fn.82.2.F163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berger J., Lecourt S., Vanneaux V. Glucocerebrosidase deficiency dramatically impairs human bone marrow haematopoiesis in an in vitro model of Gaucher disease. Br J Haematol. 2010;150:93–101. doi: 10.1111/j.1365-2141.2010.08214.x. [DOI] [PubMed] [Google Scholar]
- 20.Spectre G., Roth B., Ronen G. Platelet adhesion defect in type I Gaucher Disease is associated with a risk of mucosal bleeding. Br J Haematol. 2011;153:372–378. doi: 10.1111/j.1365-2141.2011.08613.x. [DOI] [PubMed] [Google Scholar]
- 21.Burrow T.A., Cohen M.B., Bokulic R. Gaucher disease: progressive mesenteric and mediastinal lymphadenopathy despite enzyme therapy. J Pediatr. 2007;150:202–206. doi: 10.1016/j.jpeds.2006.10.062. [DOI] [PubMed] [Google Scholar]
- 22.Bembi B., Ciana G., Mengel E., Terk M.R., Martini C., Wenstrup R.J. Bone complications in children with Gaucher disease. Br J Radiol. 2002;75(suppl 1):A37. doi: 10.1259/bjr.75.suppl_1.750037. [DOI] [PubMed] [Google Scholar]
- 23.Deegan P.B., Pavlova E., Tindall J. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Med Baltim. 2011;90:52–60. doi: 10.1097/MD.0b013e3182057be4. [DOI] [PubMed] [Google Scholar]
- 24.Rodrigue S.W., Rosenthal D.I., Barton N.W., Zurakowski D., Mankin H.J. Risk factors for osteonecrosis in patients with type 1 Gaucher's disease. Clin Orthop Relat Res. 1997;362:201–207. [PubMed] [Google Scholar]
- 25.Cohen I.J. Bone crises in Gaucher disease. Isr Med Assoc J. 2003;5:838–839. [PubMed] [Google Scholar]
- 26.Lachmann R.H., Wight D.G., Lomas D.J. Massive hepatic fibrosis in Gaucher's disease: clinico-pathological and radiological features. QJM. 2000;93:237–244. doi: 10.1093/qjmed/93.4.237. [DOI] [PubMed] [Google Scholar]
- 27.Kim J.H., Park C.H., Pai M.S., Hahn M.H., Kim H.J. Hepatopulmonary syndrome in Gaucher disease with right-to-left shunt: evaluation and measurement using Tc-99m MAA. Clin Nucl Med. 1999;24(3):164–166. doi: 10.1097/00003072-199903000-00005. [DOI] [PubMed] [Google Scholar]
- 28.Miller A., Brown L.K., Pastores G.M., Desnick R.J. Pulmonary involvement in type 1 Gaucher disease: functional and exercise findings in patients with and without clinical interstitial lung disease. Clin Genet. 2003;63:368–376. doi: 10.1034/j.1399-0004.2003.00060.x. [DOI] [PubMed] [Google Scholar]
- 29.Ross D.J., Spira S., Buchbinder N.A. Gaucher cells in pulmonary-capillary blood in association with pulmonary hypertension. N Engl J Med. 1997;336:379–381. doi: 10.1056/NEJM199701303360516. [DOI] [PubMed] [Google Scholar]
- 30.Biegstraaten M., Mengel E., Maródi L. Peripheral neuropathy in adult type 1 Gaucher disease: a 2-year prospective observational study. Brain. 2010;133:2909–2919. doi: 10.1093/brain/awq198. [DOI] [PubMed] [Google Scholar]
- 31.Mignot C., Doummar D., Maire I. Type 2 Gaucher disease: 15 new cases and review of the literature. Brain Dev. 2006;28:39–48. doi: 10.1016/j.braindev.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 32.Harris C.M., Taylor D.S., Vellodi A. Ocular motor abnormalities in Gaucher disease. Neuropediatrics. 1999;30:289–293. doi: 10.1055/s-2007-973507. [DOI] [PubMed] [Google Scholar]
- 33.Blom S., Erikson A. Gaucher disease – Norrbottnian type. Neurodevelopmental, neurological, and neurophysiological aspects. Eur J Pediatr. 1983;140:316. doi: 10.1007/BF00442672. [DOI] [PubMed] [Google Scholar]
- 34.Tüzün E., Baykan B., Gürses C., Gökyigit A. Longterm follow-up of electroencephalographic and clinical findings of a case with Gaucher's disease type 3a. Seizure. 2000 Oct;9(7):469–472. doi: 10.1053/seiz.2000.0426. [DOI] [PubMed] [Google Scholar]
- 35.Patterson M.C., Horowitz M., Abel R.B. Isolated horizontal supranuclear gaze palsy as a marker of severe systemic involvement in Gaucher's disease. Neurology. 1993;43:1993. doi: 10.1212/wnl.43.10.1993. [DOI] [PubMed] [Google Scholar]
- 36.Abrahamov A., Elstein D., Gross-Tsur V. Gaucher's disease variant characterised by progressive calcification of heart valves and unique genotype. Lancet. 1995;346:1000. doi: 10.1016/s0140-6736(95)91688-1. [DOI] [PubMed] [Google Scholar]
- 37.Chabás A., Gort L., Montfort M. Recurrence of the D409H mutation in Spanish Gaucher disease patients: description of a new homozygous patient and haplotype analysis. J Med Genet. 1998;35:775. doi: 10.1136/jmg.35.9.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goker-Alpan O., Lopez G., Vithayathil J., Davis J., Hallett M., Sidransky E. The spectrum of parkinsonian manifestations associated with glucocerebrosidase mutations. Arch Neurol. 2008;65:1353–1357. doi: 10.1001/archneur.65.10.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sidransky E. Gaucher disease and Parkinsonism. Mol Genet Metab. 2005;84:302. doi: 10.1016/j.ymgme.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 40.James S.P., Stromeyer F.W., Chang C., Barranger J.A. Liver abnormalities in patients with Gaucher's disease. Gastroenterology. 1981;80:126–133. [PubMed] [Google Scholar]
- 41.Rosenbloom B.E., Weinreb N.J., Zimran A., Kacena K.A., Charrow J., Ward E. Gaucher disease and cancer incidence: a study from the Gaucher Registry. Blood. 2005;105:4569–4572. doi: 10.1182/blood-2004-12-4672. [DOI] [PubMed] [Google Scholar]
- 42.Arends M., van Dussen L., Biegstraaten M., Hollak C.E. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of literature. Br J Haematol. 2013;161:832–842. doi: 10.1111/bjh.12335. [DOI] [PubMed] [Google Scholar]
- 43.Maaswinkel-Mooij P., Hollak C., van Eysden-Plaisier M., Prins M., Aerts H., Pöll R. The natural course of Gaucher disease in The Netherlands: implications for monitoring of disease manifestations. J Inherit Metab Dis. 2000;23:77–82. doi: 10.1023/a:1005655031239. [DOI] [PubMed] [Google Scholar]
- 44.Piran S., Roberts A., Patterson M.A., Amato D. The clinical course of untreated Gaucher disease in 22 patients over 10 years: hematological and skeletal manifestations. Blood Cells Mol Dis. 2009;43:289–293. doi: 10.1016/j.bcmd.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 45.Boomsma J.M., van Dussen L., Wiersma M.G. Spontaneous regression of disease manifestations can occur in type 1 Gaucher disease; results of a retrospective cohort study. Blood Cells Mol Dis. 2010;44:181–187. doi: 10.1016/j.bcmd.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 46.Niederau C., Häussinger D. Gaucher's disease: a review for the internist and hepatologist. Hepatogastroenterology. 2000;47:984–997. [PubMed] [Google Scholar]
- 47.Sharma P., Khurana N., Singh T. Pseudo-Gaucher cells in Hb E disease and thalassemia intermedia. Hematology. 2007;12:457–459. doi: 10.1080/10245330701393675. [DOI] [PubMed] [Google Scholar]
- 48.Costello R., O'Callaghan T., Sébahoun G. Gaucher disease and multiple myeloma. Leuk Lymphoma. 2006;47:1365–1368. doi: 10.1080/10428190600565453. [DOI] [PubMed] [Google Scholar]
- 49.Stewart A.J., Jones R.D. Pseudo-Gaucher cells in myelodysplasia. J Clin Pathol. 1999;52:917–918. doi: 10.1136/jcp.52.12.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Knox-Macaulay H., Bhusnurmath S., Alwaily A. Pseudo-Gaucher's cells in association with common acute lymphoblastic leukemia. South Med J. 1997;90:69–71. doi: 10.1097/00007611-199701000-00016. [DOI] [PubMed] [Google Scholar]
- 51.Busarla S.V., Sadruddin F.A., Sohani A.R. Pseudo-Gaucher cells in disseminated mycobacterial infection. Am J Hematol. 2013;88:155. doi: 10.1002/ajh.22269. [DOI] [PubMed] [Google Scholar]
- 52.Ho M.W., Seck J., Schmidt D. Adult Gaucher's disease: kindred studies and demonstration of a deficiency of acid beta-glucosidase in cultured fibroblasts. Am J Hum Genet. 1972;24:37–45. [PMC free article] [PubMed] [Google Scholar]
- 53.Deghady A., Marzouk I., El-Shayeb A., Wali Y. Coagulation abnormalities in type 1 Gaucher disease in children. Pediatr Hematol Oncol. 2006;23:411–417. doi: 10.1080/08880010600623232. [DOI] [PubMed] [Google Scholar]
- 54.Patlas M., Hadas-Halpern I., Abrahamov A. Spectrum of abdominal sonographic findings in 103 pediatric patients with Gaucher disease. Eur Radiol. 2002;12:397–400. doi: 10.1007/s003300101031. [DOI] [PubMed] [Google Scholar]
- 55.Niederau C. Gaucher disease: pathophysiology, clinical features and management. In: vom Dal S., Wendel U., Strohmeyer G., editors. Genetic Metabolic Disorders: Management, Costs and Socio-medical Aspects. Deutscher Arzte-Verlag Koln; 2007. pp. 19–33. [Google Scholar]
- 56.Maas M., van Kuijk C., Stoker J. Quantification of bone involvement in Gaucher disease: MR imaging bone marrow burden score as an alternative to Dixon quantitative chemical shift MR imaging–initial experience. Radiology. 2003;229:554–561. doi: 10.1148/radiol.2292020296. [DOI] [PubMed] [Google Scholar]
- 57.Poll L.W., Willers R., Häussinger D. MRI bone marrow findings in 63 patients with type I Gaucher disease. Rofo. 2010;182:979–985. doi: 10.1055/s-0029-1245410. [DOI] [PubMed] [Google Scholar]
- 58.Crabtree N.J., Kibirige M.S., Fordham J.N. The relationship between lean body mass and bone mineral content in paediatric health and disease. Bone. 2004;35:965–972. doi: 10.1016/j.bone.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 59.Giraldo P., Cenarro A., Alfonso P. Chitotriosidase genotype and plasma activity in patients type 1 Gaucher's disease and their relatives (carriers and non carriers) Hematologica. 2001;86:977–984. [PubMed] [Google Scholar]
- 60.Chang K.L., Hwu W.L., Yeh H.Y., Lee N.C., Chien Y.H. CCL18 as an alternative marker in Gaucher and Niemann-Pick disease with chitotriosidase deficiency. Blood Cells Mol Dis. 2010;44:38–40. doi: 10.1016/j.bcmd.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 61.Baldellou A., Andria G., Campbell P.E. Paediatric non-neuronopathic Gaucher disease: recommendations for treatment and monitoring. Eur J Pediatr. 2004;163:67–75. doi: 10.1007/s00431-003-1363-z. [DOI] [PubMed] [Google Scholar]
- 62.Chabás A., Cormand B., Balcells S. Neuronopathic and non-neuronopathic presentation of Gaucher disease in patients with the third most common mutation (D409H) in Spain. J Inherit Metab Dis. 1996;19:798–800. doi: 10.1007/BF01799179. [DOI] [PubMed] [Google Scholar]
- 63.Bohlega S., Kambouris M., Shahid M. Gaucher disease with oculomotor apraxia and cardiovascular calcification (Gaucher type IIIC) Neurology. 2000;54:261–263. doi: 10.1212/wnl.54.1.261. [DOI] [PubMed] [Google Scholar]
- 64.Svennerholm L., Håkansson G., Lindsten J., Wahlström J., Dreborg S. Prenatal diagnosis of Gaucher disease. Assay of the beta-glucosidase activity in amniotic fluid cells cultivated in two laboratories with different cultivation conditions. Clin Genet. 1981;19:16–22. doi: 10.1111/j.1399-0004.1981.tb00661.x. [DOI] [PubMed] [Google Scholar]
- 65.Altarescu G., Renbaum P., Eldar-Geva T. Preimplantation genetic diagnosis (PGD) for a treatable disorder: Gaucher disease type 1 as a model. Blood Cells Mol Dis. 2011;46:15–18. doi: 10.1016/j.bcmd.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 66.Lo S.M., McNamara J., Seashore M.R., Mistry P.K. Misdiagnosis of Niemann-Pick disease type C as Gaucher disease. J Inherit Metab Dis. 2010;33(suppl 3):S429. doi: 10.1007/s10545-010-9214-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schnabel D., Schröder M., Sandhoff K. Mutation in the sphingolipid activator protein 2 in a patient with a variant of Gaucher disease. FEBS Lett. 1991;284:57–59. doi: 10.1016/0014-5793(91)80760-z. [DOI] [PubMed] [Google Scholar]
- 68.Barton N.W., Brady R.O., Dambrosia J.M. Replacement therapy for inherited enzyme deficiency–macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 1991;324:1464–1470. doi: 10.1056/NEJM199105233242104. [DOI] [PubMed] [Google Scholar]
- 69.Kaplan P., Baris H., De Meirleir L. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013;172:447–458. doi: 10.1007/s00431-012-1771-z. [DOI] [PubMed] [Google Scholar]
- 70.Hollak C.E., Aerts J.M., Goudsmit R. Individualised low-dose alglucerase therapy for type 1 Gaucher's disease. Lancet. 1995 Jun 10;345:1474–1478. doi: 10.1016/s0140-6736(95)91037-9. [DOI] [PubMed] [Google Scholar]
- 71.Kaplan P., Mazur A., Manor O. Acceleration of retarded growth in children with Gaucher disease after treatment with alglucerase. J Pediatr. 1996;129:149–153. doi: 10.1016/s0022-3476(96)70203-2. [DOI] [PubMed] [Google Scholar]
- 72.Mistry P.K., Deegan P., Vellodi A., Cole J.A., Yeh M., Weinreb N.J. Timing of initiation of enzyme replacement therapy after diagnosis of type 1 Gaucher disease: effect on incidence of avascular necrosis. Br J Haematol. 2009;147:561–570. doi: 10.1111/j.1365-2141.2009.07872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Richards S.M., Olson T.A., McPherson J.M. Antibody response in patients with Gaucher disease after repeated infusion with macrophage-targeted glucocerebrosidase. Blood. 1993;82(5):1402. [PubMed] [Google Scholar]
- 74.Pastores G.M., Weinreb N.J., Aerts H. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol. 2004;41(4 suppl 5):4. doi: 10.1053/j.seminhematol.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 75.Weinreb N.J., Goldblatt J., Villalobos J. Long-term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. J Inherit Metab Dis. 2013;36:543–553. doi: 10.1007/s10545-012-9528-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zimran A., Altarescu G., Philips M. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood. 2010;115:4651. doi: 10.1182/blood-2010-02-268649. [DOI] [PubMed] [Google Scholar]
- 77.Zimran A. Velaglucerase alfa: a new option for Gaucher disease treatment. Drugs Today (Barc) 2011;47:515–529. doi: 10.1358/dot.2011.47.7.1608922. [DOI] [PubMed] [Google Scholar]
- 78.Zimran A., Brill-Almon E., Chertkoff R. Pivotal trial with plant cell-expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood. 2011;118:5767–5773. doi: 10.1182/blood-2011-07-366955. [DOI] [PubMed] [Google Scholar]
- 79.Cox T.M., Aerts J.M., Andria G. The role of the iminosugar N-butyldeoxynojirimycin (miglustat) in the management of type I (non-neuronopathic) Gaucher disease: a position statement. J Inherit Metab Dis. 2003;26:513–526. doi: 10.1023/a:1025902113005. Advisory Council to the European Working Group on Gaucher Disease SO. [DOI] [PubMed] [Google Scholar]
- 80.Elstein D., Dweck A., Attias D. Oral maintenance clinical trial with miglustat for type I Gaucher disease: switch from or combination with intravenous enzyme replacement. Blood. 2007;110:2296–2301. doi: 10.1182/blood-2007-02-075960. [DOI] [PubMed] [Google Scholar]
- 81.Belmatoug N., Burlina A., Giraldo P. Gastrointestinal disturbances and their management in miglustat-treated patients. J Inherit Metab Dis. 2011;34:991–1001. doi: 10.1007/s10545-011-9368-7. [DOI] [PubMed] [Google Scholar]
- 82.Cox T.M. Eliglustat tartrate, an orally active glucocerebroside synthase inhibitor for the potential treatment of Gaucher disease and other lysosomal storage diseases. Curr Opin Investig Drugs. 2010;11:1169–1181. [PubMed] [Google Scholar]
- 83.Ringden O., Groth C.G., Erikson A., Granqvist S., Mansson J.E., Sparrelid E. Ten years’ experience of bone marrow transplantation for Gaucher disease. Transplantation. 1995;59:864–870. [PubMed] [Google Scholar]
- 84.Ayto R.M., Hughes D.A., Jeevaratnam P. Long-term outcomes of liver transplantation in type 1 Gaucher disease. Am J Transplant. 2010;10:1934–1939. doi: 10.1111/j.1600-6143.2010.03168.x. [DOI] [PubMed] [Google Scholar]
- 85.Cohen I.J., Kornreich L., Mekhmandarov S., Katz K., Zaizov R. Effective treatment of painful bone crises in type I Gaucher's disease with high dose prednisolone. Arch Dis Child. 1996;75:218–222. doi: 10.1136/adc.75.3.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wenstrup R.J., Bailey L., Grabowski G.A. Gaucher disease: alendronate disodium improves bone mineral density in adults receiving enzyme therapy. Blood. 2004;104:1253–1257. doi: 10.1182/blood-2003-11-3854. [DOI] [PubMed] [Google Scholar]
- 87.Bell R.S., Mankin H.J., Doppelt S.H. Osteomyelitis in Gaucher disease. J Bone Jt Surg Am. 1986;68:1380–1388. [PubMed] [Google Scholar]
- 88.Khan A., Hangartner T., Weinreb N.J., Taylor J.S., Mistry P.K. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res. 2012;27:1839–1848. doi: 10.1002/jbmr.1680. [DOI] [PubMed] [Google Scholar]
- 89.Lo S.M., Liu J., Chen F. Pulmonary vascular disease in Gaucher disease: clinical spectrum, determinants of phenotype and long-term outcomes of therapy. J Inherit Metab Dis. 2011;34:643–650. doi: 10.1007/s10545-011-9313-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stein P., Yu H., Jain D., Mistry P.K. Hyperferritinemia and iron overload in type 1 Gaucher disease. Am J Hematol. 2010;85:472–476. doi: 10.1002/ajh.21721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Taddei T.H., Dziura J., Chen S. High incidence of cholesterol gallstone disease in type 1 Gaucher disease: characterizing the biliary phenotype of type 1 Gaucher disease. J Inherit Metab Dis. 2010;33:291–300. doi: 10.1007/s10545-010-9070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Suzuki Y. Chaperone therapy update: Fabry disease, GM1-gangliosidosis and Gaucher disease. Brain Dev. 2013;35:515–523. doi: 10.1016/j.braindev.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 93.Bendikov-Bar I., Maor G., Filocamo M., Horowitz M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol Dis. 2013;50:141–145. doi: 10.1016/j.bcmd.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fink J.K., Correll P.H., Perry L.K. Correction of glucocerebrosidase deficiency after retroviral-mediated gene transfer into hematopoietic progenitor cells from patients with Gaucher disease. Proc Natl Acad Sci U S A. 1990;87:2334–2338. doi: 10.1073/pnas.87.6.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Enquist I.B., Nilsson E., Ooka A. Effective cell and gene therapy in a murine model of Gaucher disease. Proc Natl Acad Sci U S A. 2006;103:13819–13824. doi: 10.1073/pnas.0606016103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Verma P.K., Ranganath P., Dalal A.B., Phadke S.R. Spectrum of lysosomal storage disorders at a medical genetics center in northern India. Indian Pediatr. 2012;49:799–804. doi: 10.1007/s13312-012-0192-4. [DOI] [PubMed] [Google Scholar]