

Abstract
Progressive familial intrahepatic cholestasis (PFIC) is a group of rare disorders which are caused by defect in bile secretion and present with intrahepatic cholestasis, usually in infancy and childhood. These are autosomal recessive in inheritance. The estimated incidence is about 1 per 50,000 to 1 per 100,000 births, although exact prevalence is not known. These diseases affect both the genders equally and have been reported from all geographical areas. Based on clinical presentation, laboratory findings, liver histology and genetic defect, these are broadly divided into three types—PFIC type 1, PFIC type 2 and PFIC type 3. The defect is in ATP8B1 gene encoding the FIC1 protein, ABCB 11 gene encoding BSEP protein and ABCB4 gene encoding MDR3 protein in PFIC1, 2 and 3 respectively. The basic defect is impaired bile salt secretion in PFIC1/2 whereas in PFIC3, it is reduced biliary phospholipid secretion. The main clinical presentation is in the form of cholestatic jaundice and pruritus. Serum gamma glutamyl transpeptidase (GGT) is normal in patients with PFIC1/2 while it is raised in patients with PFIC3. Treatment includes nutritional support (adequate calories, supplementation of fat soluble vitamins and medium chain triglycerides) and use of medications to relieve pruritus as initial therapy followed by biliary diversion procedures in selected patients. Ultimately liver transplantation is needed in most patients as they develop progressive liver fibrosis, cirrhosis and end stage liver disease. Due to the high risk of developing liver tumors in PFIC2 patients, monitoring is recommended from infancy. Mutation targeted pharmacotherapy, gene therapy and hepatocyte transplantation are being explored as future therapeutic options.
Keywords: cholestasis, familial, bile secretion, pruritus, children
Abbreviations: ABC, ATP binding cassette; ASBT, apical sodium bile salt transporter; ATP, adenosine triphosphate; ATPase, adenosine triphosphatase; BRIC, benign recurrent intrahepatic cholestasis; BSEP, bile salt exporter protein; CFTR, cystic fibrosis transmembrane conductance regulator; CYP, cytochrome P; DNA, deoxyribonucleic acid; ERAD, endoplasmic reticulum associated degradation; ESLD, end stage liver disease; FIC1, familial intrahepatic cholestasis protein 1; FXR, farnesoid X receptor; HCC, hepatocellular carcinoma; IB, ileal bypass; ICP, intrahepatic cholestasis of pregnancy; LT, liver transplant; MARS, Molecular Adsorbent Recirculating System; MDR, multidrug resistance protein; MRCP, magnetic resonance cholangiopancreaticography; mRNA, messenger ribonucleic acid; PBD, partial biliary drainage; PEBD, partial external biliary drainage; PFIC, progressive familial intrahepatic cholestasis; PIBD, partial internal biliary drainage; pGp, p-glycoprotein; PPAR, peroxisome proliferator activator receptor; UDCA, ursodeoxycholic acid
Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of liver disorders of autosomal recessive inheritance, presenting as intrahepatic cholestasis in infancy or early childhood and resulting in end stage liver disease (ESLD) and death or liver transplantation in infancy to adulthood.1–3 Clayton et al first described this disease in 1965 as Byler disease in a population of Amish kindred.4 The disease has been classified into three types (types 1, 2 and 3) based on the genetic defect involved in bile transport.
PFIC accounts for 10–15% cases of neonatal cholestasis syndrome2,3 and 10–15% of children requiring liver transplantation.2,3 It is a rare disease with an estimated incidence of 1 per 50,000 to 1 per 100,000 births although the exact prevalence is not known. The disease affects both genders equally and has been reported from around the world.5–9
Etiopathophysiology
All the three types of PFIC are caused by defects in bile secretion from hepatocyte to canaliculi (Figure 1). The defects are in form of penetrant mutations in genes encoding proteins associated with hepatocellular transport system.
Figure 1.
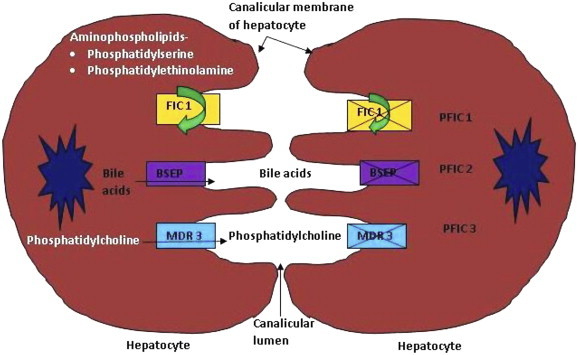
Etiopathogenesis of PFIC (PFIC: progressive familial intrahepatic cholestasis; FIC1: familial intrahepatic cholestasis protein 1; BSEP: bile salt exporter pump; MDR3: multidrug resistance protein 3).
PFIC1: It is also known as Byler disease and is associated with defects in ATP8B1 gene on chromosome 18 (18q21-22) which encodes for familial intrahepatic cholestasis 1 (FIC1) protein.10–12 FIC1 protein is a member of the type 4 subfamily of P type adenosine triphosphatase (ATPase). Type 4 ATPases are multispan transmembrane proteins that are involved in phospholipid translocation (flippase activity) from the exoplasmic (outer) to the cytoplasmic (inner) leaflet of the biological bilayer membrane.13 FIC1 is located on canalicular membrane of hepatocytes. It acts as a flippase for aminophospholipid transport and leads to movement of phosphatidylserine and phosphatidylethanolamine from the outer to inner leaflet of plasma membrane of hepatocyte. This flippase activity of FIC1 helps in maintaining asymmetric distribution of phospholipids in the membrane bilayer (higher concentration of phosphatidylserine and phosphatidylethanolamine in inner layer) which helps to protect the membrane from high bile salt concentration in canalicular lumen14–16 and maintain its integrity.17–19
Exact mechanism of cholestasis and other symptoms in PFIC1 is not fully elucidated. The proposed mechanisms include:
-
•
Overload of bile acid in hepatocyte due to reduced bile salt secretion and increased ileal bile salt reabsorption. Disturbed biliary secretion of bile salts occurs due to downregulation of farnesoid X receptor (FXR), a nuclear receptor related to regulation of metabolism of bile acids.1,2 This in turn results in downregulation of bile salt exporter pump (BSEP) protein and upregulation of synthesis of bile acid in the hepatocytes. There is also an upregulation of apical sodium bile salt transporter (ASBT) in microvilli of small intestine20–25 which increases the intestinal uptake. It is not clear if downregulation of FXR is primarily due to gene defect or is secondary to increased bile salt concentration.26
-
•
Increased secretion of cholesterol from apical (canalicular) membrane of hepatocyte in atp8b1 (capital letters denote human gene while small letters denote mouse gene) deficient mice has been shown.27 Cholesterol content of the membrane is an essential determinant of BSEP activity. Impaired BSEP activity leads to cholestasis as explained in pathogenesis of PFIC2.
-
•
Down regulation of cystic fibrosis transmembrane conductance regulator (CFTR) in cholangiocytes of patients with PFIC1 has been described which may explain extrahepatic features of the disease as well as contribute to the impaired bile secretion.1
-
•
ATP8B1 is also expressed in the membrane of cells of small intestine, kidney and pancreas.1,2 This might explain extrahepatic manifestations of PFIC1 viz. pancreatic insufficiency, sweat electrolyte abnormalities and diarrhea. FIC1 probably also has a general biological cell function and therefore results in features like short stature, and sensorineural deafness.1
Genotype–phenotype associations are complicated in patients with ATP8B1 mutations as these mutations are also present in patients with milder presentations like benign recurrent intrahepatic cholestasis 1 (BRIC1), transient neonatal cholestasis and intrahepatic cholestasis of pregnancy 1 (ICP1).28 These diseases are taken as continuum of FIC1 deficiency and the protein function is only partially impaired in them. In approximately 10% patients with PFIC1, only one mutated allele or no mutation is seen. In these patients, possible disease mechanisms include either the presence of mutations in regulatory sequences of the gene, or in the other genes involved in the transcription of PFIC1 gene or control of protein trafficking of FIC1 protein.29
PFIC2: This disease was previously known as Byler's syndrome6 and is a result of mutation in the ABCB 11 (ATP binding cassette [ABC] family B, member 11)30 gene encoding BSEP, located on chromosome 2 (2q24).
BSEP is a transporter protein, expressed at the canalicular membrane of hepatocyte.31 It is the main exporter of bile acids from hepatocyte to canaliculi against a concentration gradient.1 Genetic mutations (insertion, deletion, nonsense and splicing) result in either premature truncation of protein or total failure of protein production.32–37 These children usually have no detectable BSEP on immunostaining.33,37–40 Sometimes missense mutations result in defective processing or trafficking of protein or disruption of the structure or functional domain of BSEP. The immunostaining of liver in these patients may show presence of BSEP on canalicular membrane but it is not functional. These defects in BSEP synthesis and/or function lead to reduced bile salt secretion followed by decreased bile flow, accumulation of bile salts in hepatocytes and hepatocellular damage.
As with ATP8B1 mutations, milder mutations are associated with milder forms of cholestasis like BRIC2,34 ICP2,41 drug induced cholestasis42 and transient neonatal cholestasis.1 The main mutations in these milder variants are missense type and are located in less conserved regions of the gene.43
PFIC3: This disorder is different from PFIC1 and 2 in clinical presentation and is associated with high gamma glutamyl transpeptisae (GGT) as compared to normal/low GGT in patients with type 1 and 2.1,44,45 It is caused by defects in Adenosine triphosphate-binding cassette, subfamily B, member 4 (ABCB4) gene encoding multidrug resistance class III (MDR3) protein,46 located on chromosome 7 (7q21). MDR3 is a p-glycoprotein (pGp) which is a phospholipid translocator. It is expressed in canalicular membrane of hepatocytes10,38,47 and is a type of floppase, responsible for biliary secretion of phospholipids, predominantly phosphatidylcholine.48,49 As phospholipids are responsible for neutralizing the detergent effect of hydrophobic bile salts, defects in MDR3 protein result in injury of biliary epithelium and bile canaliculi, ultimately leading to cholestasis.45 Absence of phospholipids in bile also destabilizes micelles and promotes cholesterol crystallization resulting in increased biliary lithogenicity. This further increases liver damage by obstruction of small bile ducts.2
The mutations on ABCB4 gene are present on both alleles in most patients. In one third of cases, mutations lead to no expression of MDR3 pGp on canalicular membrane on immunohistochemistry. This complete loss of MDR3 protein has been attributed to quick destruction of truncated protein or a premature stop codon causing instability or decay of mRNA.3,50 Remaining two third of cases have missense mutations which may cause either defective transport function or intracellular misprocessing of MDR3 protein.51,52 These missense mutations are usually seen in highly conserved amino acid sequences of Walker A and B motifs.53 Immunohistochemistry shows a faint or normal MDR3 staining in these cases.
Milder phenotypes of PFIC3 present as ICP3,38 cholesterol gall stone disease,54 drug induced cholestasis,42 adult idiopathic cirrhosis,55,56 and transient neonatal cholestasis.57 In some patients the disease may present as a clinical continuum, starting with gall stone disease going on to cholestasis and then biliary cirrhosis.58,59
Clinical features
“Cholestasis”, which is characterized by jaundice and pruritus, is the hallmark presentation of PFIC. The age of onset and severity is variable, ranging from neonatal period in PFIC2 to adulthood/late adolescence in PFIC3. The main clinical, laboratory and histological features differentiating the various types of PFIC are shown in Table 1. Family history of affected sibling and consanguinity between parents can give clue to the diagnosis.
Table 1.
Clinical, Biochemical and Histological Features of Different Types of PFIC.
| Feature | PFIC1 | PFIC2 | PFIC3 |
|---|---|---|---|
| Age at presentation | Infancy | Neonatal period-early infancy | Late Infancy (∼30%) to early adulthood |
| End stage liver disease | First decade | Rapid, first few years | 1st to 2nd decade of life |
| Course of disease | Moderately severe | Severe | Insidious |
| Pruritus | Severe | Very severe | Moderate |
| Extrahepatic manifestations (watery diarrhoea, pancreatitis, sensorineural deafness, short stature, abnormalities in sweat chloride) | Present | Absent | Absent |
| Risk of development of liver tumors | No | High | Mild increase |
| Risk of cholesterol stone disease | Absent | Increased | Increased |
| Serum ALT | Mild elevation | Moderate elevation | Mild elevation |
| Serum AFP | Normal | Raised | Normal |
| Serum GGT | Normal | Normal | Elevated |
| Serum bile acids | Raised ++ | Raised +++ | Raised + |
| Bile composition | |||
| Primary bile acids | Low (3–8 mM) | Very low (<1 mM) | Normal |
| Phospholipids | Normal | Normal | Low |
| Liver histology | Bland cholestasis, mild lobular fibrosis | Cholestasis, giant cell hepatitis, hepatocellular necrosis, portal fibrosis | Bile ductular proliferation, inflammatory infiltrate, and biliary fibrosis |
| Electron microscopy | Granular bile | Amorphous bile | – |
PFIC: progressive familial intrahepatic cholestasis; GGT: gamma glutamyl transpeptidase; AFP: alpha fetoprotein; ALT: alanine aminotransferase.
In PFIC patients with normal GGT (PFIC1 and 2), the cholestasis is more severe and presents with persistent jaundice in type 2 in comparison to type 1 which has recurrent jaundice initially and permanent later in the disease course. The infant has to be of at least 4–5 months of age to manifest pruritus. Pruritus is the most debilitating symptom, leading to cutaneous mutilation, loss of sleep, irritability, poor attention and impaired school performance in children with PFIC.
Examination reveals icterus, hepatomegaly, scratch marks with excoriation and hyperpigmentation of skin and shiny nails. The weight and height may be below normal centiles due to fat malabsorption along with signs of fat soluble vitamin (A, D, E, and K) deficiency. Portal hypertension and decompensation develops earlier in the first year of life in type 2 as compared to early childhood in type 1. One child with PFIC2 presenting with liver failure in neonatal period has also been reported.60
Two studies have compared the clinical presentation and disease progression of approximately 200 genetically proven type 1 and 2 PFIC children.61,62 Overall, type 2 PFIC patients have more severe hepatobiliary disease with greater impairment of bile salt handling whereas type 1 patients have extrahepatic disease. The percentage of subjects with disease onset by 3 months of age (65–85%), jaundice at presentation (70–80%) and an affected sibling (15–25%) was similar in PFIC1 and 2. PFIC1 patients had higher alkaline phosphatase and lower serum albumin than PFIC2 cases. Whereas, higher serum aminotransferase, bile acids and alpha fetoprotein along with increased prevalence of portal hypertension, gall stones and hepatocellular carcinoma (HCC) was seen in type 2 PFIC cases.
No definite genotype–phenotype correlation has been shown within the PFIC1/2 subtypes. However, in the type 2 PFIC, the patients with D482G mutation have a slowly progressive disease and develop cirrhosis at an older age as compared to other BSEP patients.61 The alanine or aspartate aminotransferase level (ALT or AST) was found to be the most reliable differentiator61 between type 1 and 2 with values of >2 times upper limit of normal suggesting PFIC2 with a sensitivity of 88% (95% CI 73–96%) and specificity of 81% (95% CI 63–93%) in the study by Pawlikowska et al61 The higher risk of developing end stage liver disease (ESLD), HCC and cholangiocarcinoma63,64 in early life in PFIC2 requires close surveillance with regular (6–12 monthly) α-fetoprotein estimation and ultrasonography.
In contrast, patients with PFIC3 usually develop cholestasis in late infancy (one third of cases) to adolescent age group.2 Gastrointestinal bleeding due to cirrhosis and portal hypertension may be the first presentation in older children or young adults. The disease usually progresses from chronic cholestasis with or without jaundice to portal hypertension and ESLD. These children are also at increased risk of development of cholesterol stones in intrahepatic bile ducts or the gall bladder and drug induced cholestasis.46 Severe ICP may be seen in female patients who reach adulthood and pregnancy without requiring liver transplant. Hormonal changes (due to intake of oral contraceptives containing estrogen and progesterone or during pregnancy) may lead to precipitation of symptoms in patients with PFIC3. Thus it is important to remember that medical therapy of cholestasis should not be stopped in these females during pregnancy.65 The main differences between the three diseases i.e. PFIC, ICP and BRIC are shown in Table 2.66–70
Table 2.
Differences Between PFIC, ICP and BRIC.
| PFIC | BRIC | ICP | |
|---|---|---|---|
| Gene/types | |||
| ATP8B1 | PFIC 1 | BRIC 1 | ICP 1 |
| ABCB11 | PFIC2 | BRIC 2 | ICP 2 |
| ABCB4 | PFIC3 | ICP 3 | |
| Mutation | Homozygous Severe disruption of protein function | Homozygous Partial impairment of protein function | Heterozygous mutation |
| Age at presentation | Infancy–adulthood (depending on type) | Mostly after first decade | During second half of pregnancy |
| Disease | Permanent and usually progressive | Usually limited to attacks (variable duration of few weeks to months) Complete normalcy between two episodes | Transient cholestasis limited to pregnancy with complete resolution after delivery/during intake of OCP |
| Treatment | Drugs: UDCA, rifampicin, cholestyramine Partial biliary diversion Liver transplantation | Drugs: UDCA rifampicin, cholestyramine ENBD66,67 Plasmapharesis68 | Drugs: UDCA Elective delivery at 37–38 weeks of gestation69,70 |
| Complications | Cirrhosis, HCC and cholangiocarcinoma | – | Poor foetal outcome: prematurity, foetal distress, intrauterine death |
| Differential diagnosis | Depends on age of presentation Newborn and first 6 months: all causes of neonatal cholestasis Late infancy with pruritus: Alagille syndrome, non syndromic bile ductular paucity, sclerosing cholangitis (primary/secondary), AAT, cystic fibrosis, BASD | Extrahepatic biliary obstruction, Intrahepatic cholestasis: acute viral hepatitis, drug induced cholestasis, PSC, AIH, PBC, overlap syndrome, IgG4 associated disease, CHF and Caroli's syndrome, infiltrative disorders (lymphoma/histiocytosis) etc | Other causes of jaundice during pregnancy e.g. HELLP, acute fatty liver of pregnancy, cholestatic viral hepatitis, BCS, biliary disease etc |
(PFIC: progressive familial intrahepatic cholestasis, BRIC: benign recurrent intrahepatic cholestasis, ICP: intrahepatic cholestasis of pregnancy, OCP: oral contraceptive pill, UDCA: ursodeoxycholic acid, ENBD: endoscopic nasobiliary drainage, HCC: hepatocellular carcinoma, HELLP: hemolysis elevated liver enzymes and low platelet, BCS: Budd–Chiari syndrome, PSC: primary sclerosing cholangitis, AIH: autoimmune hepatitis, AAT: alpha 1 antitrypsin deficiency, BASD: bile acid synthesis defects, CHF: congenital hepatic fibrosis).
Investigations
The following investigations help in making a diagnosis of PFIC, its classification into type 1, 2 or 3 and differentiating it from other causes of cholestasis.
Liver Function Tests
These tests differ in three types of PFIC (Table 1). Prolonged international normalized ratio (INR) is common and correctable with injectable vitamin K in early stages of the disease. Poor synthetic functions (low serum albumin and uncorrectable coagulopathy) are seen in patients with advanced disease.
Radiology
Ultrasonography is the first test which is essentially normal except for the presence of cholelithiasis in some cases of PFIC 2/3. It also helps to exclude other causes of extrahepatic cholestasis. Cholangiography (magnetic resonance cholangiopancreaticography; MRCP) helps in excluding sclerosing cholangitis in patients with high GGT cholestasis. The role of hepatic scintigraphy71 and 31P MRS spectroscopy72 is being evaluated.
Liver Biopsy
Liver histology shows canalicular type of cholestasis, biliary plugs and lobular disarray and is often described as “bland” in PFIC1.73 Hepatocytes may show periportal biliary metaplasia. Ductular proliferation, giant cells and portal fibrosis are absent. Lobular fibrosis and cirrhosis develop later in the course of the disease. In PFIC2, canalicular cholestasis is associated with more marked hepatocellular disarray with lobular and portal fibrosis. Hepatocellular necrosis and giant cell hepatitis is predominant. Biliary metaplasia is also more pronounced. True ductular proliferation is absent. In PFIC3, portal fibrosis and true bile ductular proliferation is seen at disease onset. Most portal tracts demonstrate interlobular bile ducts. Giant cell hepatitis is mild. In later stages, there is marked portal fibrosis and biliary cirrhosis. Intraductal cholelithiasis may also be seen in some cases. Periductal fibrosis and biliary epithelial injury is not seen.
Immunohistochemistry
The biopsy specimen should be subjected to immunohistochemistry which shows mild or absent canalicular staining with MDR3 and BSEP antibodies in PFIC3 and PFIC2 respectively.40 Though absent/decreased immunostaining is diagnostic, normal immunostaining does not preclude the diagnosis of PFIC as some mutations are associated with only functional defect in protein which is otherwise normal in synthesis and expression. Standardized antibodies for immunohistochemistry for PFIC1 are yet not available.
Electron Microscopy
The bile in PFIC1 is coarse and granular (Byler's bile) in comparison to PFIC2 which has amorphous bile.
Bile Analysis
Bile can be obtained for analysis either by duodenal aspiration or gall bladder puncture.2,3 Low biliary phospholipid concentration (<15% of total biliary lipids; normal 19–24%) and normal biliary bile salt concentration is typical of PFIC3. The biliary phospholipids are very low (<2%) in subjects with severe mutations as compared to milder mutations which have >2% biliary phospholipids. The biliary bile salt to phospholipid ratio is increased (>5 fold) in PFIC3. A biliary phospholipid concentration of >7% of total biliary lipids predicts good response to ursodeoxycholic acid (UDCA). In contrast, reduced biliary bile salt and normal phospholipids is seen in PFIC1 and 2 with greater reduction in type 2 as compared to type 1.
Genetic Testing
This is the gold standard for diagnosis and involves DNA sequencing of the 27 coding exons and their splice junctions. A resequencing chip, dedicated to look for genetic syndromes of cholestasis has been developed and may facilitate diagnosis.74
As no phenotypic features can exclude PFIC1 or 2 in a patient with normal GGT PFIC, immunohistochemistry with BSEP staining followed by genetic analysis is recommended. In patients with negative BSEP staining one should first test for ABCB 11 whereas in patients with normal BSEP staining, ATB8B1 mutation should be looked for.
Differential diagnosis
The differential diagnosis depends on the age of presentation. In newborns and young infants, PFIC1/2 needs to be differentiated from other (obstructive, metabolic, infective, genetic and endocrinal) causes of cholestasis. GGT is very useful in this situation and should always be done in a patient with cholestasis. Normal GGT suggests PFIC1/2 while other common causes of neonatal cholestasis like biliary atresia, Alagille syndrome, alpha 1 antitrypsin deficiency, etc have high GGT values. Inborn errors of bile acid synthesis, also known as bile acid synthesis disorders (BASD) is a group of disorders of autosomal recessive inheritance which present with cholestasis and normal GGT.75 As the name suggests, the serum bile acid concentration is low or absent in BASD in comparison to high levels in PFIC1/2. Urinary bile acid analysis is required for diagnosis of BASD. The age of presentation in BASD is variable, pruritus is mild or absent, response to bile acid therapy is good and outcome is better in comparison to that of PFIC1/2 patients.
Other rare cholestatic conditions with normal GGT include Arthrogryposis-renal dysfunction syndrome (ARC syndrome)76 and familial Amish hypercholanemia.2 In subjects with cholestasis and high GGT (PFIC3), it is essential to rule out other causes of extrahepatic biliary obstruction and sclerosing cholangitis. The differential diagnosis for the different disease forms i.e. PFIC, BRIC and ICP are shown in Table 2.
Treatment
Medical Management
Medical therapy is the first line of treatment in patients with all types of PFIC. The objectives are to provide relief from pruritus, improve the nutritional status, correct vitamin deficiencies and treat complications of advanced liver disease like ascites and variceal bleeding if present. Simple measures like keeping the skin moisturized and trimming the fingernails are helpful to provide symptomatic relief. The total caloric intake should be around 125% of the recommended daily allowance (RDA). Dietary fat should be provided largely as medium chain triglycerides (MCT oil) as they do not require bile salts for absorption and help in improving nutrition. Water soluble vitamins are given at 1–2 times of the age appropriate RDA. The fat soluble vitamins are usually supplemented in the following dosage in children: vitamin A—5000–25,000 IU/day PO, vitamin D 400–800 IU/day PO, vitamin E 50–100 IU/day PO and vitamin K 2.5–5 mg/day PO or 2–5 mg intravenous every 3–4 weeks. Adequate sunlight exposure and dietary intake of calcium (800–2000 mg/day PO) is also essential. It is important to evaluate the child both clinically as well as biochemically (serum levels of vitamins) for signs of specific vitamin deficiencies and adjust the supplements accordingly.77
The most commonly used drug for pruritus is ursodeoxycholic acid (UDCA) which is a hydrophilic bile acid, non-toxic to hepatocytes.78 It replaces toxic hydrophobic bile salts and may amount to up to 40% of total serum bile salt concentration with long term therapy.79,80 Other postulated mechanisms of action include induction of BSEP and MDR3 expression with increased biliary secretion of bile acids and phospholipids. UDCA is a safe drug with no major side effects and has been shown to be effective in all forms of PFIC.78,81–84 Patients with total defect in MDR3 gene expression are usually non-responders to UDCA therapy.58 Overall, complete or partial response is seen in approximately 35–40% of low GGT PFIC and ∼70% cases of high GGT PFIC.10,79
Rifampicin induces the expression of CYP3A4 (enzyme of drug metabolism) which increases 6-α hydroxylation of bile salts. These bile salts are thereafter glucuronidated and excreted in urine. It also induces uridine diphosphate (UDP)-glucuronosyl transferase (UGT1A1) and leads to increased conjugation and excretion of bilirubin. Apart from reduced pruritus in some cases, use of rifampicin does not cause significant improvement in PFIC1/2.10 Also, potential hepatotoxicity should be kept in mind when using the drug.
Cholestyramine is a resin which binds bile salts in the intestinal lumen and thus reduces absorption and increases fecal bile salt excretion. It has not been found to be very useful in PFIC1/2 patients.10
Surgical Management
-
a.
Biliary diversion procedures: Decrease in the enterohepatic circulation with reduction of toxic bile salt accumulation is the basis of biliary diversion procedures. The serum bile acid concentration has been shown to be reduced in patients with successful biliary diversion.85 Two main types of procedures are commonly performed as shown in Figure 2.
-
i.
Partial biliary diversion (PBD)—This has been used successfully in many patients with PFIC1/2 who do not respond to medical therapy and are as yet not candidates for liver transplant. The best results are obtained in patients who have not developed cirrhosis at the time of surgery.2,86 PBD, along with UDCA may help in delaying the progression to ESLD.10 PBD is of two types –
Figure 2.
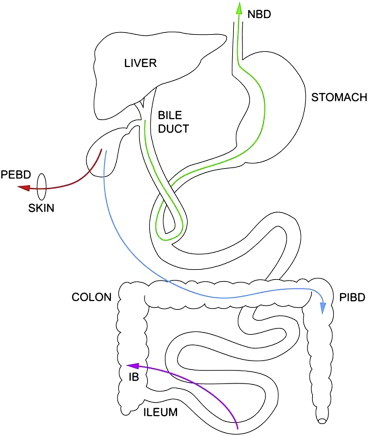
Biliary drainage procedures: a diagrammatic representation (IB: ileal bypass, NBD: nasobiliary drainage; PEBD: partial external biliary drainage; PIBD: partial internal biliary drainage).
Partial external biliary diversion (PEBD): this can be performed with open or laparoscopic techniques. The procedure, as originally described by Whittington et al,87 involves use of a 10–15 cm jejunal conduit between gall bladder and abdominal wall where a permanent stoma is created. Modifications of this procedure include use of a button of gall bladder wall88 or appendix89 as the conduit between gall bladder and skin. PEBD results in improved growth, improvement/normalization of liver function, significant reduction of serum bile acid and improvement in liver histology, largely in terms of reduced progression of fibrosis in approximately 80% patients with PFIC1/2.86 In a recent study of 24 patients (age 26 months [4mo-17y]) subjected to PEBD, 13 (54%) had a successful outcome with normalization of serum bile acids. None of these cases showed any progression of cholestasis over a long median follow-up of 9.8y (1.6 − 14.3y). In comparison, 11 (46%) cases failed to show normalization of bile acids and 9/11 of them required liver transplantation over a short follow-up period of 1.9 (0.5 − 3.8y). Amongst these 24 cases, 7/7 (100%) with cirrhosis required liver transplantation in comparison to 2/17 (12%) without cirrhosis. Thus, PEBD should be the first line of surgical therapy in PFIC patients and should be offered early before development of cirrhosis. Only patients with established cirrhosis should be taken for primary liver transplantation. Clinical response with normalization of serum bile acids at 1 year post PEBD is suggestive of a good long term outcome.85
Partial internal biliary drainage (PIBD): It involves use of a jejunal conduit between gall bladder and colon or anastomosis between gall bladder and anti-reflux loop of colon (cholecysto-colostomy).90–92 The main advantage is that there is no external fistula. It is a relatively newer technique with limited follow-up duration.
-
ii.
Ileal bypass—Some surgeons prefer ileal bypass (IB) in which ileocolic anastomosis is used to bypass distal 15% of small intestine, thus interrupting enterohepatic circulation of bile salts.93,94 This procedure was initially used for patients with previous cholecystectomy. The advantages include avoidance of external stoma and its associated fluid and electrolyte imbalance. Symptoms recur in almost half of the cases over 1 year of follow-up due to ileal adaption and thus IB is not as good as PEBD in low GGT PFIC.
Nasobiliary drainage (NBD) may help to select the patients who will respond to biliary diversion procedures.2,3 Comparative trials are not available for different types of PBD procedures for PFIC1/2. Genotype–phenotype markers to select subgroups of PFIC1/2 patients more likely to respond to medical therapy or PBD are also not yet available.
-
b.
Liver transplantation (LT): This is the last therapeutic option for patients with all types of PFIC and should be considered in patients with ESLD, HCC or those with poor quality of life due to refractory pruritus despite medical treatment and biliary diversion. LT improves cholestasis and its symptoms in 75–100% patients, irrespective of PFIC subtype over a short term follow-up of 3–5 years.2,3,95–97
For PFIC1 patients, LT should be offered after thoughtful consideration as extrahepatic manifestations like diarrhea, liver steatosis and short stature do not improve or even worsen after LT.98,99 Chronic diarrhea may become intractable after restoration of bile acid secretion post LT in some patients. This responds to therapy with bile salt sequestrating agents2,3,100 and biliary diversion.101 Liver steatosis may progress to cirrhosis and require retransplant.
Recurrence of PFIC after LT due to alloimmunization of the recipient against the affected protein (FIC1, BSEP or MDR3) is a possibility, especially in patients with severe mutations leading to absence of the protein and has been reported in two patients with PFIC2.102 Concerns of increased risk of immunosuppression related cholestasis/cholelithiasis in the post-transplant period due to the heterozygous state of donor liver (from father/mother) has not been proven to be true as yet.
Progressive familial intrahepatic cholestasis in India
The published data on PFIC from India is in the form of case reports103–105 and small case series.106 No data is available regarding the prevalence of PFIC in India. In the largest series of 7 children, one responded to medical therapy, 2 required biliary diversion and three of the four subjects with liver decompensation underwent liver transplantation.106 The biggest difficult faced by the gastroenterologist is of confirming the diagnosis of PFIC and its subtype due to lack of easy availability of genetic tests.
Future therapies
As there is no universally effective and non-invasive therapy for PFIC, newer therapeutic options are being explored. These include
-
1.
Hepatic support by albumin dialysis or Molecular Adsorbent Recirculating System (MARS) which has the potential to reverse symptoms and help tide over the crisis situations e.g. during pregnancy.107
-
2.
Hepatocyte transplantation and gene therapy with modified hepatocytes to correct the metabolic defect.108 The therapeutic use of these procedures remains to be established.
-
3.Mutation specific drug therapy—These include different approaches targeted towards increasing the expression of functional transporter proteins (FIC1, BSEP, MDR3) on the canalicular membrane (Figure 3). The different approaches towards achieving this include the following:
-
a.FXR receptor ligands like 6-α-ethyl-chenodeoxycholate, fibrates, statins etc.2,10 activate nuclear receptor FXR which in turn transactivates genes involved in bile secretion leading to reduced hepatic bile salt uptake and increased biliary secretion of bile salts and phospholipids. Ligands for other nuclear receptors e.g. peroxisome proliferator activator receptor alpha (PPAR α) which increases expression of ABCB4 and induces biliary phosphatidylcholine secretion.
-
b.Compounds that can suppress premature stop codons e.g. aminoglycosides and PTC124 may help skip and read through the stop codon. These agents are useful in patients in whom specific premature stop codon due to mutations result in premature truncation of transporter protein.
-
c.Drugs inhibiting endoplasmic reticulum associated degradation (ERAD) like MG132 decrease degradation of misfolded/truncated protein.
-
d.Pharmacological chaperone drugs (e.g. 4-phenylbutyrate) are small molecular weight compounds which correct misfolding and prevent mistrafficking of proteins.
-
a.
Figure 3.
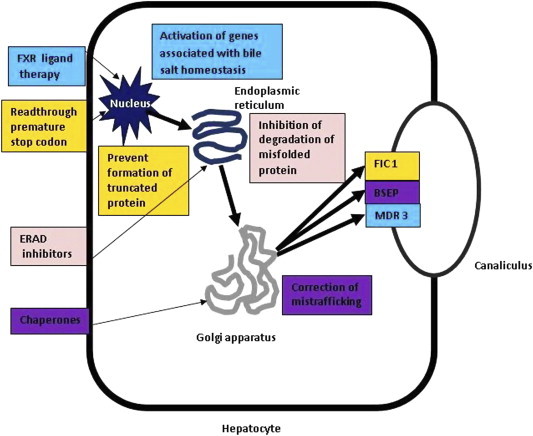
Future therapies for PFIC (black arrows denote steps in transcription of transporter protein (FIC1, BSEP, MDR 3) from nucleus to endoplasmic reticulum to Golgi apparatus to canalicular membrane. Farnesoid R receptor (FXR) ligand therapy activates a number of genes like BSEP associated with bile salt homeostasis. Agents acting by read through premature stop codon help mRNA to skip a stop codon; endoplasmic reticulum associated degradation (ERAD) inhibitors act on endoplasmic reticulum to decrease degradation of misfolded/truncated protein and chaperone agents act on Golgi apparatus to correct misfolding/mistrafficking of proteins).
Role of genetic counseling and antenatal diagnosis
The current level of understanding of genetics of PFIC should be utilized to test all affected patients for the genetic defect. The patients and their parents should be offered genetic counseling. The parents should also be tested for heterozygosity. Role of prenatal diagnosis is being explored as it requires clinical and biochemical expertise, available only at select centres.109,110
Prognosis
PFIC patients have a variable prognosis depending on the type of PFIC and severity of genetic defect within each type. Approximately 30% children respond to UDCA therapy and about 70–80% to PBD if offered early in course of disease, before development of cirrhosis. Patients with cirrhosis and end stage liver disease require liver transplant. The data on long term outcome is limited. In a study of PFIC with normal GGT,84 out of 33 children, 7 patients were 16 years or older at last follow-up. These children (n = 7) had symptoms in form of poor growth (5/7 below 5th centile for height), pruritus (6/7), vitamin deficiency rickets and vitamin E neuropathy (2/7) and gall stones (5/7). Another study of 62 children with normal GGT PFIC showed that nearly 87% subjects were alive at a median age of 10.5 (1–36) years with therapy. LT was required in 50% cases in this series.62
Conclusions
This review focuses on the etiopathogenesis, clinical features, diagnosis and therapy of patients with PFIC. Further studies are required to ascertain role of phenotype-genotype variations and efficacy of one therapeutic option over another. Trials are needed to find out the best biliary diversion procedure as well. Liver transplantation remains the treatment of choice for patients with end stage liver disease but there are issues of long term efficacy in PFIC1 patients.
Conflicts of interest
The author has none to declare.
References
- 1.Davit-Spraul A., Gonzales E., Baussan C., Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009;4:1. doi: 10.1186/1750-1172-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2012;36(suppl 1):S26–S35. doi: 10.1016/S2210-7401(12)70018-9. [DOI] [PubMed] [Google Scholar]
- 3.Hori T., Nguyen J.H., Uemoto S. Progressive familial intrahepatic cholestasis. Hepatobiliary Pancreat Dis Int. 2010;9:570–578. [PubMed] [Google Scholar]
- 4.Clayton R.J., Iber F.I., Ruebner B.H. Byler disease: fatal familial intrahepatic cholestasis in an Amish kindred. Am J Dis Child. 1969;117:112–124. [PubMed] [Google Scholar]
- 5.Bull L.N., Carlton V.E., Stricker N.L. Genetic and morphological findings in progressive familial intrahepatic cholestasis (Byler disease [PFIC 1] and Byler syndrome): evidence for heterogenicity. Hepatology. 1997;26:155–164. doi: 10.1002/hep.510260121. [DOI] [PubMed] [Google Scholar]
- 6.Bourke B., Goggin N., Walsh D., Kennedy S., Setchell K.D., Drumm B. Byler like familial cholestasis in an extended kindred. Arch Dis Child. 1996;75:223–227. doi: 10.1136/adc.75.3.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnell H., Nemeth A., Anneren G., Dahl N. Progressive familial intrahepatic cholestasis (PFIC): evidence for genetic heterogenicity by exclusion of linkage to chromosome 18q21-q22. Hum Genet. 1997;100:378–381. doi: 10.1007/s004390050519. [DOI] [PubMed] [Google Scholar]
- 8.Naveh Y., Bassan L., Rosenthal E. Progressive familial intrahepatic cholestasis among the Arab population in Israel. J Pediatr Gastroenterol Nutr. 1997;24:548–554. doi: 10.1097/00005176-199705000-00011. [DOI] [PubMed] [Google Scholar]
- 9.Kagalwalla A.F., Al Amir A.R., Khalipha A., Sylven M., Al Ajaji S., Kagalwalla Y.A. Progressive familial intrahepatic cholestasis (Byler’s disease) in Arab children. Ann Trop Paediatr. 1995;15:321–327. doi: 10.1080/02724936.1995.11747792. [DOI] [PubMed] [Google Scholar]
- 10.Stapelbroek J.M., van Erpercum K.J., Klomp L.W., Houwen R.H. Liver disease associated with canalicular transport defects: current and future therapies. J Hepatol. 2010;52:258–271. doi: 10.1016/j.jhep.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 11.Bull L.N., van Ejik M.J., Pawlikowska L. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18:219–224. doi: 10.1038/ng0398-219. [DOI] [PubMed] [Google Scholar]
- 12.Van Mil S.W., Klomp L.W., Bull L.N., Houwen R.H. FIC1 disease: a spectrum of intrahepatic cholestatic disorders. Semin Liver Dis. 2001;21:535–544. doi: 10.1055/s-2001-19034. [DOI] [PubMed] [Google Scholar]
- 13.Paulusma C.C., Oude Elferink R.P.J., Jansen P.L.M. Progressive familial intrahepatic cholestasis type 1. Semin Liver Dis. 2010;30:117–124. doi: 10.1055/s-0030-1253221. [DOI] [PubMed] [Google Scholar]
- 14.Devaux P.F., Lopez-Montero I., Bryde S. Proteins involved in lipid translocation in eukaryotic cells. Chem Phys Lipids. 2006;141:119–132. doi: 10.1016/j.chemphyslip.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Paulusma C.C., Oude Elferink R.P. Diseases of intra-membranous lipid transport. FEBS Lett. 2006;580:5500–5509. doi: 10.1016/j.febslet.2006.06.067. [DOI] [PubMed] [Google Scholar]
- 16.Oude Elferink R.P.J., Paulusma C.C., Groen A.K. Hepatocanalicular transport defects: pathophysiologic mechanisms of rare diseases. Gastroenterology. 2006;130:908–925. doi: 10.1053/j.gastro.2005.08.052. [DOI] [PubMed] [Google Scholar]
- 17.Paulusma M.L., Groen A., Kunne C. Atp8b1 deficiency in mice induces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile acid transport. Hepatology. 2006;44:195–204. doi: 10.1002/hep.21212. [DOI] [PubMed] [Google Scholar]
- 18.Verhulst P.M., van der Velden L.M., Oorschot V. A flippase-independent function of ATP8B1, the protein affected in familial intrahepatic cholestasis type 1, is required for apical protein expression and microvillus formation in polarized epithelial cells. Hepatology. 2010;51:2049–2060. doi: 10.1002/hep.23586. [DOI] [PubMed] [Google Scholar]
- 19.Groen A., Romero M.R., Kunne C. Complementary functions of the flippase ATP8B1 and the floppase ABCB4 in maintaining canalicular membrane integrity. Gastroenterology. 2011;141:1927–1937. doi: 10.1053/j.gastro.2011.07.042. [DOI] [PubMed] [Google Scholar]
- 20.Alvarez L., Jara P., Sanchez-Sabate E. Reduced hepatic expression of farnesoid X receptor in hereditary cholestasis associated to mutation in ATP8B1. Hum Mol Genet. 2004;13:2451–2460. doi: 10.1093/hmg/ddh261. [DOI] [PubMed] [Google Scholar]
- 21.Chen F., Ananthanarayanan M., Emre S. Progressive familial intrahepatic cholestasis, type 1, is associated with decreased farnesoid X receptor activity. Gastroenterology. 2004;126:756–764. doi: 10.1053/j.gastro.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 22.Chen F., Ellis E., Strom S.C., Shneider B.L. ATPase class 1 type 8B member 1 and protein kinase C zeta induce the expression of the canalicular bile salt transport pump in human hepatocyte. Pediatr Res. 2010;67:183–187. doi: 10.1203/PDR.0b013e3181c2df16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frankenberg T., Miloh T., Chen F.Y. The membrane protein ATPase class 1 type 8B member 1 signals through protein kinase C zeta to activate the farnesoid X receptor. Hepatology. 2008;48:1896–1905. doi: 10.1002/hep.22431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koh S., Takada T., Kukku I., Suzuki H. FIC1-mediated stimulation of FXR activity is decreased with PFIC1 mutations in HepG2 cells. J Gastroenterol. 2009;44:592–600. doi: 10.1007/s00535-009-0041-y. [DOI] [PubMed] [Google Scholar]
- 25.Martinez-Fernandez P., Hierro L., Jara P., Alvarez L. Knockdown of ATP8B1 expression leads to specific downregulation of the bile acid sensor FXR in HepG2 cells; effect of FXR agonist GW4064. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1119–G1129. doi: 10.1152/ajpgi.90371.2008. [DOI] [PubMed] [Google Scholar]
- 26.Cai S.Y., Gautam S., Nguyen T., Soroka C.J., Rahner C., Boyer J.L. ATP8B1 deficiency disrupts the bile canalicular membrane bilayer structure in hepatocytes, but FXR expression and activity are maintained. Gastroenterology. 2009;136:1060–1069. doi: 10.1053/j.gastro.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groen A., Kunne C., Jongsma G. Abcg5/8 independent biliary cholesterol excretion in Atp8b1 deficient mice. Gastroenterology. 2008;134:2091–2100. doi: 10.1053/j.gastro.2008.02.097. [DOI] [PubMed] [Google Scholar]
- 28.Klomp L.W., Vargas J.C., van Mil S.W. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 2004;40:27–38. doi: 10.1002/hep.20285. [DOI] [PubMed] [Google Scholar]
- 29.Paulusma C.C., Folmer D.E., Ho-Mok K.S. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology. 2008;47:268–278. doi: 10.1002/hep.21950. [DOI] [PubMed] [Google Scholar]
- 30.Strautnieks S.S., Kagalwalla A.F., Tanner M.S. Identification of a locus for progressive familial intrahepatic cholestasis PFIC2 on chromosome 2q24. Am J Hum Genet. 1997;61:630–633. doi: 10.1086/515501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kubitz R., Dröge C., Stindt J., Weissenberger K., Häussinger D. The bile salt export pump (BSEP) in health and disease. Clin Res Hepatol Gastroenterol. 2012;36:536–553. doi: 10.1016/j.clinre.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 32.Strautnieks S.S., Bull L.N., Knisely A.S. A gene encoding a liver specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–238. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 33.Jansen P.L., Strautnieks S.S., Jacquemin E. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology. 1999;117:1370–1379. doi: 10.1016/s0016-5085(99)70287-8. [DOI] [PubMed] [Google Scholar]
- 34.Van Mil S.W., van der Woerd W.L., van der Brugge G. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology. 2004;127:379–384. doi: 10.1053/j.gastro.2004.04.065. [DOI] [PubMed] [Google Scholar]
- 35.Lam C.W., Cheung K.M., Tsui M.S., Yan M.S., Lee C.Y., Tong S.F. A patient with novel ABCB11 gene mutation with phenotypic transition between BRIC2 and PFIC2. J Hepatol. 2006;44:240–242. doi: 10.1016/j.jhep.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 36.Keitel V., Vogt C., Haussinger D., Kubitz R. Combined mutations of canalicular transporter proteins cause severe intrahepatic cholestasis of pregnancy. Gastroenterology. 2006;131:624–629. doi: 10.1053/j.gastro.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 37.Strautnieks S.S., Byrne J.A., Pawlikowska L.N. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134:1203–1214. doi: 10.1053/j.gastro.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 38.Keitel V., Burdelski M., Warskulat U. Expression and localization of hepatobiliary transport proteins in progressive familial intrahepatic cholestasis. Hepatology. 2005;41:1160–1172. doi: 10.1002/hep.20682. [DOI] [PubMed] [Google Scholar]
- 39.Wang L., Soroka C.J., Boyer J.L. The role of bile salt export pump mutations in progressive familial intrahepatic cholestasis type II. J Clin Invest. 2002;110:965–972. doi: 10.1172/JCI15968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evason K., Bove K.E., Finegold M.J. Morphologic findings in progressive familial intrahepatic cholestasis 2 (PFIC2): correlation with genetic and immunohistochemical studies. Am J Surg Pathol. 2011;35:687–696. doi: 10.1097/PAS.0b013e318212ec87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pauli-Magnus C., Lang T., Meier Y. Sequence analysis of bile salt export pump (ABCB11) and multidrug resistance p-glycoprotein 2 (ABCB4, MDR 3) in patients with intrahepatic cholestasis of pregnancy. Pharmacogenetics. 2004;14:91–102. doi: 10.1097/00008571-200402000-00003. [DOI] [PubMed] [Google Scholar]
- 42.Lang C., Meier Y., Stieger B. Mutations and polymorphisms in the bile salt export pump and the multidrug resistance protein 3 associated with drug-induced liver injury. Pharmacogenet Genomics. 2007;17:47–60. doi: 10.1097/01.fpc.0000230418.28091.76. [DOI] [PubMed] [Google Scholar]
- 43.Lam P., Soroka C.J., Boyer J.L. The bile salt export pump: clinical and experimental aspects of genetic and acquired cholestatic disease. Semin Liver Dis. 2010;30:125–133. doi: 10.1055/s-0030-1253222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jansen P.L.M., Muller M. Genetic cholestasis: lessons from the molecular physiology of bile formation. Can J Gastroenterol. 2000;14:233–238. doi: 10.1155/2000/514172. [DOI] [PubMed] [Google Scholar]
- 45.De vree J.M., Jacquemin E., Sturm E. Mutations in MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A. 1998;95:282–287. doi: 10.1073/pnas.95.1.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davit Spraul A., Gonzales E., Baussan C., Jacquemin E. The spectrum of liver disease related to ABCB4 gene mutations: pathophysiology and clinical aspects. Semin Liver Dis. 2010;30:134–146. doi: 10.1055/s-0030-1253223. [DOI] [PubMed] [Google Scholar]
- 47.Smit J.J., Schinkel A.H., Mol C.A. Tissue distribution of the human MDR3 P-glycoprotein. Lab Invest. 1994;71:638–649. [PubMed] [Google Scholar]
- 48.Smit A.J., Timmermans-Hereijgers J.L., Roelofsen B. The human MDR3 P-glycoprotein promotes translocation of phosphatidylcholine through the plasma membrane of fibroblasts from transgenic mice. FEBS Lett. 1994;354:263–266. doi: 10.1016/0014-5793(94)01135-4. [DOI] [PubMed] [Google Scholar]
- 49.van Helvoort A., Smith A.J., Sprong H. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell. 1996;87:507–517. doi: 10.1016/s0092-8674(00)81370-7. [DOI] [PubMed] [Google Scholar]
- 50.Deleuze J.F., Jacquemin E., Dubuisson C. Defect of multidrug-resistance 3 gene expression in a subtype of progressive familial intrahepatic cholestasis. Hepatology. 1996;23:904–908. doi: 10.1002/hep.510230435. [DOI] [PubMed] [Google Scholar]
- 51.Dixon P.H., Weerasekera N., Linton K.J. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Hum Mol Genet. 2000;9:1209–1217. doi: 10.1093/hmg/9.8.1209. [DOI] [PubMed] [Google Scholar]
- 52.Delaunay J.L., Durand-Schneider A.M., Delautier D. A missense mutation in ABCB4 gene involved in progressive familial intrahepatic cholestasis type 3 leads to a folding defect that can be rescued by low temperature. Hepatology. 2009;49:1218–1227. doi: 10.1002/hep.22775. [DOI] [PubMed] [Google Scholar]
- 53.Gottesman M.M., Hrycyna C.A., Schoenlein P.V., Germann U.A., Pastan I. Genetic analysis of multidrug transporter. Annu Rev Genet. 1995;29:607–649. doi: 10.1146/annurev.ge.29.120195.003135. [DOI] [PubMed] [Google Scholar]
- 54.Rosmorduc O., Hermelin B., Poupon R. MDR3 gene defect in adults with symptomatic intrahepatic and gallbladder cholesterol cholelithiasis. Gastroenterology. 2001;120:1459–1467. doi: 10.1053/gast.2001.23947. [DOI] [PubMed] [Google Scholar]
- 55.Ziol M., Barbu V., Rosmorduc O. ABCB4 heterozygous mutations associated with fibrosing cholestatic liver disease in adults. Gastroenterology. 2008;135:131–141. doi: 10.1053/j.gastro.2008.03.044. [DOI] [PubMed] [Google Scholar]
- 56.Wendum D., Barbu V., Rosmorduc O., Arrive L., Flejou J.F., Poupon R. Aspects of liver pathology in adult patients with MDR3/ABCB4 gene mutations. Virchows Arch. 2012;460:291–298. doi: 10.1007/s00428-012-1202-6. [DOI] [PubMed] [Google Scholar]
- 57.Jacquemin E. Role of multidrug resistance 3 deficiency in pediatric and adult liver disease: one gene for three diseases. Semin Liver Dis. 2001;21:551–562. doi: 10.1055/s-2001-19033. [DOI] [PubMed] [Google Scholar]
- 58.Jacquemin E., De Vree J.M., Cresteil D. The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology. 2001;120:1448–1458. doi: 10.1053/gast.2001.23984. [DOI] [PubMed] [Google Scholar]
- 59.Lucena J.F., Herrero J.I., Quiroga J. A multidrug resistance 3 gene mutation causing cholelithiasis, cholestasis of pregnancy, and adulthood biliary cirrhosis. Gastroenterology. 2003;124:1037–1042. doi: 10.1053/gast.2003.50144. [DOI] [PubMed] [Google Scholar]
- 60.Sangorrin Iranzo A., Iriondo Sanz M., Alvarez García L., Jara Vega P., Martín de Carpi J. Progressive familial intrahepatic cholestasis presenting as liver failure] Ann Pediatr (Barc) 2009;71:510–513. doi: 10.1016/j.anpedi.2009.08.005. [Article in Spanish] [DOI] [PubMed] [Google Scholar]
- 61.Pawlikowska L., Strautnieks S., Jankowska I. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J Hepatol. 2010;53:170–178. doi: 10.1016/j.jhep.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davit-Spraul A., Fabre M., Branchereau S. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology. 2010;51:1645–1655. doi: 10.1002/hep.23539. [DOI] [PubMed] [Google Scholar]
- 63.Scheimann A.O., Strautnieks S.S., Knisely A.S., Byrne J.A., Thompson R.J., Finegold M.J. Mutations in bile salt export pump (ABCB11) in two children with progressive familial intrahepatic cholestasis and cholangiocarcinoma. J Pediatr. 2007;150:556–559. doi: 10.1016/j.jpeds.2007.02.030. [DOI] [PubMed] [Google Scholar]
- 64.Knisely A.S., Strautnieks S.S., Meier Y. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology. 2006;44:478–486. doi: 10.1002/hep.21287. [DOI] [PubMed] [Google Scholar]
- 65.Ganne-Carrie N., Baussan C., Grando V., Gaudelus J., Cresteil D., Jacquemin E. Progressive familial intrahepatic cholestasis type 3 revealed by oral contraceptive pills. J Hepatol. 2003;38:693–694. doi: 10.1016/s0168-8278(03)00049-7. [DOI] [PubMed] [Google Scholar]
- 66.Stapelbroek J.M., van Erpecum K.J., Klomp L.W. Nasobiliary drainage induces long-lasting remission in benign recurrent intrahepatic cholestasis. Hepatology. 2006;43:51–53. doi: 10.1002/hep.20998. [DOI] [PubMed] [Google Scholar]
- 67.Luketic V.A., Shiffman M.L. Benign recurrent intrahepatic cholestasis. Clin Liver Dis. 2004;8:133–149. doi: 10.1016/S1089-3261(03)00133-8. [DOI] [PubMed] [Google Scholar]
- 68.Folvik G., Hilde O., Helge G.O. Benign recurrent intrahepatic cholestasis: review and long-term follow up of five cases. Scand J Gastroenterol. 2012;47:482–488. doi: 10.3109/00365521.2011.650191. [DOI] [PubMed] [Google Scholar]
- 69.Geenes V., Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15:2049–2066. doi: 10.3748/wjg.15.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pathak B., Sheibani L., Lee R.H. Cholestasis Pregnancy. Obstet Gynecol Clin North Am. 2010;37:269–282. doi: 10.1016/j.ogc.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 71.Arnell H., Fischler B., Bergdahl S., Schnell P.O., Jacobsson H., Nemeth A. Hepatobiliary scintigraphy during cholestatic and non-cholestatic periods in patients with progressive familial intrahepatic cholestasis after partial external biliary diversion. J Pediatr Surg. 2011;46:467–472. doi: 10.1016/j.jpedsurg.2010.09.042. [DOI] [PubMed] [Google Scholar]
- 72.Müllenbach R., Bennett A., Tetlow N. ATP8B1 mutations in British cases with intrahepatic cholestasis of pregnancy. Gut. 2005;54:829–834. doi: 10.1136/gut.2004.058115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morotti R.A., Suchy F.J., Magid M.S. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: a review of the liver pathology findings. Semin Liver Dis. 2011;31:3–10. doi: 10.1055/s-0031-1272831. [DOI] [PubMed] [Google Scholar]
- 74.Liu C., Aronow B.J., Jegga A.G. Novel resequencing chip customized to diagnose mutations in patients with inherited syndromes of intrahepatic cholestasis. Gastroenterology. 2007;132:119–126. doi: 10.1053/j.gastro.2006.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gonzales E., Gerhardt M.F., Fabre M. Oral cholic acid for hereditary defects of primary bile acid synthesis: a safe and effective long term therapy. Gastroenterology. 2009;137:1310–1320. doi: 10.1053/j.gastro.2009.07.043. [DOI] [PubMed] [Google Scholar]
- 76.Cullinane A.R., Stratman-Iwanowska A., Zaucker A. Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization. Nat Genet. 2010;42:303–312. doi: 10.1038/ng.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Feranchak A.P., Ramirez R.O., Sokal R.J. Medical and Nutritional Management of Cholestasis. In: Suchy F.J., Sokol R.J., Balistreri W.F., editors. Liver Disease in Children. 2nd ed. Lippincott, Williams and Wilkins; 2001. pp. 195–238. [Chapter 10] [Google Scholar]
- 78.Poupon R. Intrahepatic cholestasis of pregnancy: from bedside to bench to bedside. Liver Int. 2005;25:467–468. doi: 10.1111/j.1478-3231.2005.01000.x. [DOI] [PubMed] [Google Scholar]
- 79.Jacquemin E., Hermans D., Myara A. Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis. Hepatology. 1997;25:519–523. doi: 10.1002/hep.510250303. [DOI] [PubMed] [Google Scholar]
- 80.Lazaridis K.N., Gores G.J., Lindor K.D. Ursodeoxycholic acid ‘mechanisms of action and clinical use in hepatobiliary disorders'. J Hepatol. 2001;35:134–146. doi: 10.1016/s0168-8278(01)00092-7. [DOI] [PubMed] [Google Scholar]
- 81.Paumgartner G., Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology. 2002;36:525–531. doi: 10.1053/jhep.2002.36088. [DOI] [PubMed] [Google Scholar]
- 82.Dinler D., Kocak N., Ozen H., Yuce A., Gurakan F. Ursodeoxycholic acid in children with Byler disease. Pediatr Int. 1999;41:662–665. doi: 10.1046/j.1442-200x.1999.01143.x. [DOI] [PubMed] [Google Scholar]
- 83.Kubitz R., Brinkmeyer C., Sagir A., Herebian D., Haussinger D. Genetic variants of the bile salt exporter pump: inducers and modifiers of liver diseases. Dig Dis. 2011;29:89–92. doi: 10.1159/000324140. [DOI] [PubMed] [Google Scholar]
- 84.Whitington P.F., Freese D.K., Alonso E.M., Schwarzenberg S.J., Sharp H.L. Clinical and biochemical findings in progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 1994;18:134–141. doi: 10.1097/00005176-199402000-00003. [DOI] [PubMed] [Google Scholar]
- 85.Schukfeh N., Metzelder M.L., Petersen C. Normalization of serum bile acids after partial external biliary diversion indicates an excellent long-term outcome in children with progressive familial intrahepatic cholestasis. J Pediatr Surg. 2012;47:501–505. doi: 10.1016/j.jpedsurg.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 86.Davis A.R., Rosenthal P., Newman T.B. Nontransplant surgical interventions in progressive familial intrahepatic cholestasis. J Pediatr Surg. 2009;44:821–827. doi: 10.1016/j.jpedsurg.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 87.Whitington P.F., Whitington G.L. Partial external diversion of bile for the treatment of intractable pruritus associated with intrahepatic cholestasis. Gastroenterology. 1988;95:130–136. doi: 10.1016/0016-5085(88)90301-0. [DOI] [PubMed] [Google Scholar]
- 88.Clifton M.S., Romero R., Ricketts R.R. Button cholecystostomy for management of progressive familial intrahepatic cholestasis syndromes. J Pediatr Surg. 2011;46:304–307. doi: 10.1016/j.jpedsurg.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 89.Rebhandl W., Felberbauer F.X., Turnbull J. Biliary diversion by use of the appendix (cholecystoappendicostomy) in progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 1999;28:217–219. doi: 10.1097/00005176-199902000-00026. [DOI] [PubMed] [Google Scholar]
- 90.Bustorff-Silva J., Sbraggia Neto L., Olimpio H. Partial internal biliary diversion through a cholecystojejunocolonic anastomosis – a novel surgical approach for patients with progressive familial intrahepatic cholestasis: a preliminary report. J Pediatr Surg. 2007;42:1337–1340. doi: 10.1016/j.jpedsurg.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 91.Diao M., Li L., Zhang J.S., Ye M., Cheng W. Laparoscopic cholecystocolostomy: a novel surgical approach for the treatment of progressive familial intrahepatic cholestasis. Ann Surg. 2013;258:1028–1033. doi: 10.1097/SLA.0b013e31827905eb. [DOI] [PubMed] [Google Scholar]
- 92.Mochizuki K., Obatake M., Takatsuki M. Partial internal biliary diversion for patients with progressive familial intrahepatic cholestasis type 1. Pediatr Surg Int. 2012;28:51–54. doi: 10.1007/s00383-011-3018-x. [DOI] [PubMed] [Google Scholar]
- 93.Hollands C.M., Rivera-Pedro F.J., Gonzales-Vallina R. Ileal exclusion for Byler’s disease: an alternative surgical approach with promising early results for pruritus. J Pediatr Surg. 1998;33:220–224. doi: 10.1016/s0022-3468(98)90435-3. [DOI] [PubMed] [Google Scholar]
- 94.Kaliciński P.J., Ismail H., Jankowska I. Surgical treatment of progressive familial intrahepatic cholestasis: comparison of partial external biliary diversion and ileal bypass. Eur J Pediatr Surg. 2003;13:307–311. doi: 10.1055/s-2003-43570. [DOI] [PubMed] [Google Scholar]
- 95.Aydogdu S., Cakir M., Arikan C. Liver transplantation for progressive familial intrahepatic cholestasis: clinical and histopathological findings, outcome and impact on growth. Pediatr Transplant. 2007;11:634–640. doi: 10.1111/j.1399-3046.2007.00722.x. [DOI] [PubMed] [Google Scholar]
- 96.Englert C., Grabhorn E., Richter A., Rogiers X., Burdelski M., Ganschow R. Liver transplantation in children with progressive familial intrahepatic cholestasis. Transplantation. 2007;84:1361–1363. doi: 10.1097/01.tp.0000282869.94152.4f. [DOI] [PubMed] [Google Scholar]
- 97.Hori T., Egawa H., Takada Y. Progressive familial intrahepatic cholestasis: a single-centre experience of living-donor liver transplantation during two decades in Japan. Clin Transplant. 2011;25:776–785. doi: 10.1111/j.1399-0012.2010.01368.x. [DOI] [PubMed] [Google Scholar]
- 98.Lykavieris P., van Mil S., Cresteil D. Progressive familial intrahepatic cholestasis type 1 and extrahepatic features: no catch-up of stature growth, exacerbation of diarrhea, and appearance of liver steatosis after liver transplantation. J Hepatol. 2003;39:447–452. doi: 10.1016/s0168-8278(03)00286-1. [DOI] [PubMed] [Google Scholar]
- 99.Miyagawa-Hayashino A., Egawa H., Yorifuji T. Allograft steatohepatitis in progressive familial intrahepatic cholestasis type 1 after living donor liver transplantation. Liver Transpl. 2009;15:610–618. doi: 10.1002/lt.21686. [DOI] [PubMed] [Google Scholar]
- 100.Egawa H., Yorifuji T., Sumazaki R., Kimura A., Hasegawa M., Tanaka K. Intractable diarrhoea after liver transplantation for Byler's disease: successful treatment with bile adsorptive resin. Liver Transpl. 2002;8:714–716. doi: 10.1053/jlts.2002.34384. [DOI] [PubMed] [Google Scholar]
- 101.Nicastro E., Stephenne X., Smets F. Recovery of graft steatosis and protein-losing enteropathy after biliary diversion in a PFIC 1 liver transplanted child. Pediatr Transplant. 2012;16:E177–E182. doi: 10.1111/j.1399-3046.2011.01514.x. [DOI] [PubMed] [Google Scholar]
- 102.Maggiore G., Gonzales E., Sciveres M. Relapsing features of bile salt export pump deficiency after liver transplantation in two patients with progressive familial intrahepatic cholestasis type 2. J Hepatol. 2010;53:981–986. doi: 10.1016/j.jhep.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 103.Ganesh R., Suresh N., Sathiyasekeran M., Ramachandran P. Partial internal biliary diversion: a solution for intractable pruritus in progressive familial intrahepatic cholestasis type 1. Saudi J Gastroenterol. 2011;17:212–214. doi: 10.4103/1319-3767.80387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sharma D., Shah U.H., Sibal A., Chowdhary S.K. Cholecystoappendicostomy for progressive familial intrahepatic cholestasis. Indian Pediatr. 2010;47:626–628. doi: 10.1007/s13312-010-0122-2. [DOI] [PubMed] [Google Scholar]
- 105.Koshy A., Ramesh H., Mahadevan P. Progressive familial intrahepatic cholestasis: a case with improvement in liver tests and growth following partial external biliary diversion. Indian J Gastroenterol. 2009;28:107–108. doi: 10.1007/s12664-009-0038-8. [DOI] [PubMed] [Google Scholar]
- 106.Kaur S., Sharma D., Wadhwa N., Gupta S., Chowdhary S.K., Sibal A. Therapeutic interventions in progressive familial intrahepatic cholestasis: experience from a tertiary care centre in north India. Indian J Pediatr. 2012;79:270–273. doi: 10.1007/s12098-011-0516-8. [DOI] [PubMed] [Google Scholar]
- 107.Lemoine M., Revaux A., Francoz C. Albumin liver dialysis as pregnancy-saving procedure in cholestatic liver disease and intractable pruritus. World J Gastroenterol. 2008;14:6572–6574. doi: 10.3748/wjg.14.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dhawan A., Mitry R.R., Hughes R.D. Hepatocyte transplantation for liver-based metabolic disorders. J Inherit Metab Dis. 2006;29:431–435. doi: 10.1007/s10545-006-0245-8. [DOI] [PubMed] [Google Scholar]
- 109.Jung C., Driancourt C., Baussan C. Prenatal molecular diagnosis of inherited cholestatic diseases. J Pediatr Gastroenterol Nutr. 2007;44:453–458. doi: 10.1097/MPG.0b013e318036a569. [DOI] [PubMed] [Google Scholar]
- 110.Chen S.T., Chen H.L., Su Y.N. Prenatal diagnosis of progressive familial intrahepatic cholestasis type 2. J Gastroenterol Hepatol. 2008;23:1390–1393. doi: 10.1111/j.1440-1746.2008.05432.x. [DOI] [PubMed] [Google Scholar]