

Abstract
Prokaryotes and eukaryotes respond to various environmental stimuli using the two-component system (TCS). Essentially, it consists of membrane-bound histidine kinase (HK) which senses the stimuli and further transfers the signal to the response regulator, which in turn, regulates expression of various target genes. Recently, sequence-based genome wide analysis has been carried out in Arabidopsis and rice to identify all the putative members of TCS family. One of the members of this family i.e. AtHK1, (a putative osmosensor, hybrid-type sensory histidine kinase) is known to interact with AtHPt1 (phosphotransfer proteins) in Arabidopsis. Based on predicted rice interactome network (PRIN), the ortholog of AtHK1 in rice, OsHK3b, was found to be interacting with OsHPt2. The analysis of amino acid sequence of AtHK1 showed the presence of transmitter domain (TD) and receiver domain (RD), while OsHK3b showed presence of three conserved domains namely CHASE (signaling domain), TD, and RD. In order to elaborate on structural details of functional domains of hybrid-type HK and phosphotransfer proteins in both these genera, we have modeled them using homology modeling approach. The structural motifs present in various functional domains of the orthologous proteins were found to be highly conserved. Binding analysis of the RD domain of these sensory proteins in Arabidopsis and rice revealed the role of various residues such as histidine in HPt protein which are essential for their interaction.
Keywords: histidine kinase, histidine phosphotransfer protein, two component system, CHASE domain, transmitter domain, receiver domain
1. Background
Prokaryotes and Eukaryotes regulate their cellular mechanism in response to the environmental changes using ‘Two-Component Signal (TCS)’ machinery (Chang & Stewart, 1998). TCS regulatory systems were earlier found in Eubacteria (Hess, Oosawa, Kaplan, & Simon, 1988) and are considered as sophisticated signaling systems marked by a highly modular design that has been adapted and integrated into wide variety of cellular signaling targets (Stock, Robinson, & Goudreau, 2000). In plants, the signaling cascade generally consists of three protein elements; a sensory Histidine Kinase (HK), a Histidine-containing Phosphotransfer (HPt) protein, and a Response Regulator (RR). Upon sensing the environmental stimulus, HK is autophosphorylated at the conserved His residue. The phosphoryl group is transferred to a conserved Asp residue in RR, which modulates its activity. The phosphoryl group is further passed on to a His residue of the phosphotransfer protein (HPt) and an Asp residue of the receiver domain of RR, resulting in a signal output by interaction with target proteins or specific DNA motifs (Saito, 2001; Stock et al., 2000). In Arabidopsis, His kinase proteins play a crucial role in ethylene and cytokinin signal transduction (Hwang, Chen, & Sheen, 2002; Lohrmann & Harter, 2002). More complex phosphorelay systems include hybrid sensor kinases (Hybrid HK) which also contain phosphoaspartate RD. These hybrid-type HKs contain multiple phosphodonor and phosphoreceptor sites, thus providing advantage of having multiple regulatory checkpoints for signal cross talk and negative regulation by certain phosphatases (Urao, Yamaguchi-Shinozaki, & Shinozaki, 2000).
Analysis of hybrid-type HKs show the presence of three distinct domains namely, CHASE signaling domain, transmitter domain (TD) and RR domain (also known as RD). Abbreviated CHASE domain is known as cyclases/HKs associated sensory extracellular domain because of its presence in diverse receptor-like proteins with HK and nucleotide cyclase domains. CHASE domain has been characterized as an extracellular domain, found in transmembrane receptors from bacteria, lower eukaryotes, and plants. The domain always occurs N-terminally in extracellular or periplasmic locations, followed by an intracellular tail housing diverse enzymatic signaling domains such as HK, adenyl cyclase, etc.
The TD of hybrid HKs consists of a set of characteristic sequence motifs, labeled as H, N, G1, F, and G2 boxes (Parkinson & Kofoid, 1992). The TD consists of two distinct functional domains: an N-terminal dimerization-histidine phosphotransfer (DHp) domain and a C-terminal catalytic-ATP-binding (CA) domain. The N-terminal domain contains the autophosphorylation site and assists in formation of stable dimer and can also be phosphorylated in presence of ATP by the CA domain (Stock et al., 2000).
RRs are characterized by their possession of RD (Sakakibara, Taniguchi, & Sugiyama, 2000). The most common function of RRs is the regulation of gene expression. Some of the RRs exist as fusion proteins with a RD and an output domain, but some others have only a RD similar to CheY, a RR for chemotaxis in Escherichia coli (Bilwes, Alex, Crane, & Simon, 1999). Genome-wide analysis of RRs and related proteins in Arabidopsis thaliana shows the existence of 32 genes encoding putative RRs and related proteins that are not fused to HK (Hwang et al., 2002); Imamura et al., 1999). In contrast, a total of 32 genes that code for 44 putative RR proteins have been reported in Oryza sativa (Pareek et al., 2006). The majority of these RRs have two main domains: a conserved N-terminal regulatory domain and a variable C-terminal effector domain. The RRs can be classified into two subtypes, type-A and type-B, based on their structures (Riechmann et al., 2000). In A. thaliana, some of the type-A RRs have been characterized as negative feedback regulators of cytokinin signaling (Kiba et al., 1999; To et al., 2004).
HPt domains are ∼120 amino acids in length and contain a His residue which is capable of participating in phosphoryl transfer reactions (Stock et al., 2000). HPt domains are known to serve dual purpose, as a phosphoreceiver and phosphodonor, in order to shuttle phosphoryl groups between two or more RR domains (Janiak-spens, Sparling, & West, 2000; Suzuki, Imamura, Ueguchi, & Mizuno, 1998). In a generalized phosphorelay system, HPts act as phosphodonor to RD of RRs, although receivers are themselves capable of catalyzing autophosphorylation with phosphor-His as substrates (Mizuno, 1998).
In order to have an overview of the protein–protein interactions, several attempts have been made over the years to co-crystallize the phosphorelay partners. However, due to transient nature of these interactions and the chemical lability of the phosphotidinyl and phosphor-aspartyl linkages, these attempts have not been able to shed light on the structural aspect of these interactions. However, X-ray structure analysis of YPD1 and SLN1 RR (SLN1-R1) complex has been performed which highlights the structural details of HPt protein and RR complexes (Xu, Porter, & West, 2003). In another attempt, crystal structure of ArcB HPt domain and CheY RR complex in E. coli has clarified the molecular recognition of RR by the HPt domain at an atomic level (Kato, Shimizu, Mizunob, & Hakoshimaa, 1999).
Previously, the interaction of a sensory HK protein functioning as a putative ‘osmosensor’ in Arabidopsis (AtHK1) with a histidine phosphotransfer protein has been analyzed (Urao et al., 2000). In rice, a sensory HK protein namely OsHK3b has been observed to show interaction with OsHPt2 protein by yeast complementation assays (our unpublished results). Also, OsHK3b has been reported to interact with OsHPt2 protein based on predicted rice interactome network database (Gu, Zhu, Jiao, Meng, & Chen, 2011). With the availability of sequences of various HKs and HPts and the structure of various functional domains of these TCS members in plants (especially O. sativa and A. thaliana), it would be highly desirable to get an insight into the structural aspects of the various domains of these interacting proteins involved in the signal transmission across the membrane from the sensing domain to the kinase core. In the present study, we have analyzed the structures of various functional domains of related Histidine Kinases (sensory HKs) protein from rice and Arabidopsis. Further, considering the important roles played by interaction of RD in HK protein with phosphotransfer (HPt) protein in phosphorelay mechanism, we have made an attempt to present the structural features of protein–protein interaction.
2. Methodology
2.1. Identification of conserved domains and secondary structure
The sequences of OsHK3b (Os01g69920.2) (UniProt Accession: Q5JJP1) and OsHPt2 (Os08g44350.1) (Uni-Prot Accession: Q6VAK3) from TIGR rice database (ver. 7.0) and sequences of AtHK1 (At2g17820.1) (UniProt Accession: Q9SXL4) and AtHPt1 (At3g29350.1) (UniProt Accession: Q9ZNV8) in Arabidopsis (TAIR ver. 10) were used for modeling and analysis of various functional domains and their interactions. The conserved domains of the sequences were identified from Pfam (Finn et al., 2010). The conserved domain searched against CDD (Marchler-Bauer et al., 2009) also supported the Pfam results. The secondary structures of the domains and the protein were predicted using JNET prediction software (Cole, Barber, & Barton, 2008), PREDATOR (Frishman & Argos, 1996), STRIDE (Frishman & Argos, 1995), and PSIPRED (Jones, 1999a). The fold recognition analyses were performed using FUGUE (Shi, Blundell, & Mizuguchi, 2001), GenTHREADER (Jones, 1999b). The architectural motifs and the topology of proteins with known three-dimensional (3D) structure were analyzed according to SCOP (Murzin, Brenner, Hubbard, & Chothia, 1995) and CATH (Greene et al., 2007) classification.
2.2. Homology modeling of HK and HPt proteins
The 3D structure of all the domains of HK and HPt proteins in rice and Arabidopsis was modeled in a stepwise procedure, starting with the identification of template structures. The identified templates were obtained from PDB and aligned using structure alignment software STAMP (Russell & Barton, 1992). This alignment was used as a profile for aligning the target sequence using ClustalX (Larkin et al., 2007) for modeling various domains. The alignment was subjected to manual adjustments for the conserved structural motifs. For modeling various domains of HK and HPt proteins, automated comparative protein-modeling program Modeller (ver 9.10) (Eswar et al., 2006) was used to generate a 100 allatom model.
2.3. Validation of homology models of HK and HPt proteins
The best model was chosen on the basis of the stereochemistry quality report generated using PROCHECK (Laskowski, MacArthur, Moss, & Thornton, 1993) and side chains of the modeled protein were optimized using SCWRL 4 (Krivov, Shapovalov, & Dunbrack, 2009). G-factor score is considered as a parameter for obtaining the quality of the modeled domain, and was obtained using PROCHECK. G-factor is essentially a log odds score based on the observed distribution of stereochemical parameters such as main chain bond angles, bond length, and phi-psi torsion angles. The bond distances and dihedral angle restraints on the target sequences were derived from its alignment with the template three-dimensional structures. The spatial restraints and the energy minimization steps were performed within Modeler using the CHARMM22 force field for proper stereochemistry of proteins. Since the domain sequence used for the alignment and modeling was found to have well-conserved structural motifs and regions, and functional information was also available, the problem of low-sequence identity has been resolved, i.e. a multiple sequence alignment obtained from the known sequences can provide a reasonable approach to comparative structure modeling. Earlier, attempts have been made to model the protein sequence having low identity with the template sequence (Singh, Kushwaha, & Sharma, 2008). In order to verify the quality of the sequence alignment and optimize the position of gaps, corresponding positions from secondary structures were used. Further evaluation of the modeled domain and protein structures was done using the PROSA-web (Sippl, 1993; Wiederstein & Sippl, 2007). Ramachandran plots were generated for domain and protein structures in order to determine deviations from normal bond lengths, dihedrals, and nonbonded atom–atom distances.
2.4. Analysis of HK and HPt interaction
The protein–protein interaction was analyzed using PatchDock software (Duhovny, Nussinov, & Wolfson, 2002; Schneidman-Duhovny et al., 2003). PatchDock software uses the geometry-based molecular docking algorithm, which was used to find the docking transformations that yield good molecular shape complementarity (Duhovny et al., 2002; Schneidman-Duhovny et al., 2003). The docking was performed using default parameters. The best model for analysis of interaction was chosen on the basis of geometric shape complementarity score and minimum energy. Molecular visualization and analysis of the final model were carried out with VMD (Humphrey, Dalke, & Schulten, 1996).
3. Result
The sequences of OsHK3b (Os01g69920.2) and OsHPt2 (Os08g44350.1) from TIGR rice database (ver. 7.0) and sequences of AtHK1 (At2g17820.1) and AtHPt1 (At3g29350.1) from Arabidopsis (TAIR ver. 10) were used for modeling and analysis of various functional domains and their interactions. The sequence of OsHK3b was observed to contain three functionally conserved domains. The first signature domain is CHASE signaling domain (sensory domain), second is the TD which acts as ATP-binding domain, and third is the RD which binds to the HPt protein. Earlier, CHASE domain has been reported in AtHK1 protein (Grefen & Harter, 2004) in addition to other two well-defined domains i.e. TD and RD. However, in our analysis of AtHK1 protein sequence using Pfam database, we could not establish CHASE domain. Instead, this analysis showed only a long stretch of 498 amino acids towards the N-terminus without any functional domain. Also, due to the absence of template structures, we were not able to model the interdomain regions.
3.1. Modeling of sensory/CHASE domain in AtHK1 and OsHK3b protein
In order to model the sensory domain structure of AtHK1 (216 amino acid) and CHASE domain in OsHK3b protein (227 amino acid), BLAST searches were performed against the PDB for proteins with similar sequence and known 3D structures. The BLAST results revealed template structure of Arabidopsis HK 4-sensor domain (3T4J.pdb) as a suitable template for modeling CHASE domain in OsHK3b protein. The target CHASE domain sequence of OsHK3b showed 53% identity with 3T4J, template structure. The template structures possess sensor kinase like secondary structure folds. The BLAST results were unable to capture any possible template for modeling AtHK1 sensory domain sequence. Hence, the AtHK1 sensory domain sequence was modeled using threading approach using CPHmodels server (Nielsen, Lundegaard, Lund, & Petersen, 2010) and EsyPred3D (Lambert, Leonard, De Bolle, & Depiereux, 2002), while CHASE domain of OsHK3b was modeled using comparative modeling approach (see Method) (Supplementary Figure 1). The results obtained from CPH webserver and EsyPred3d consensually identified 3C8C.pdb (crystal structure of Mcp_N and cache domains of methyl-accepting chemotaxis protein from Vibrio cholera) as possible template for modeling the AtHK1 sensory domain sequence. The Ramachandran plot analysis showed that the modeled AtHK1 CHASE domain has 92.9% residues found in most favorable regions with the remaining 6.6% of residues occurring in generously allowed regions, while 0.5% residues were found in disallowed region. While in OsHK3b, CHASE domain has 98.5% residues in most favorable regions with the remaining 0.5% of residues occurring in generously allowed regions, 1.0% of the residues were found in disallowed region (Supplemental Figure 2(A) and (B)). The PROCHECK result summary showed 29 out of 212 residues labeled in CHASE domain structure of AtHK1, while in CHASE domain of OsHK3b, 10 out of 225 residues were found to be labeled. The torsion angles of the side chain designated by χ1–χ2 plots showed 3 labeled residues out of 121 in AtHK1 CHASE domain, while in OsHK3b CHASE domain, 3 out of 133 residues were found labeled. The G-factor score of the AtHK1 CHASE domain was found to be −0.35 for dihedral bonds, −0.55 for covalent bonds, and −0.41 overall, while the OsHK3b CHASE domain model was observed to be −0.04 for dihedral bonds, −0.31 for covalent bonds, and −0.13 overall. The distribution of the main chain bond lengths and bond angles were 96.9% and 86.7% within limits for the modeled AtHK1 CHASE domain. On the other hand, for OsHK3b CHASE domain, main chain bond lengths and bond angles were found to be 98.4 and 91.6% within the limit. The PROSA-web energy plots for CHASE domains of AtHK1 and OsHK3b protein showed z-score for pair, surface and combined energy which was found to be −5.93 and −5.69, respectively (Supplementary Figure 3(A) and (B)).
Figure 1.
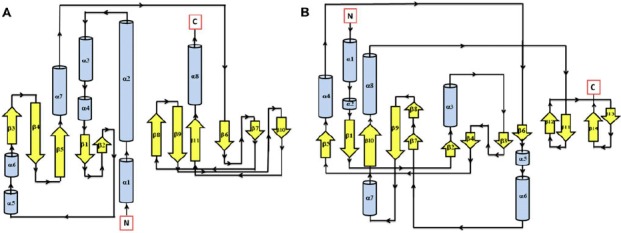
Secondary structure topology of CHASE domain of AtHK1 showing 11 β-sheets and 8 α-helices structures (A) and OsHK3b (B) showing 14 β-sheets and 8 α-helices.
Figure 2.
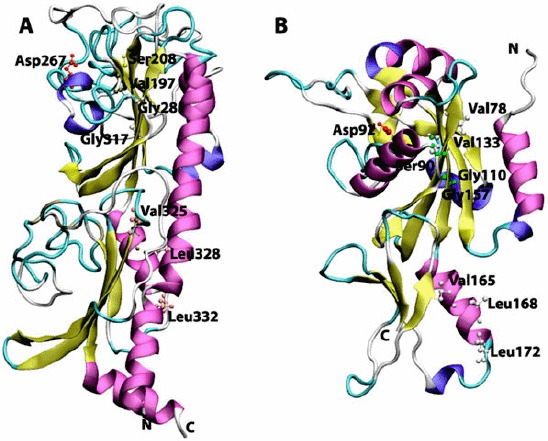
Cartoon view diagram of the secondary structure of CHASE domain of AtHK1 showing the arrangement of 11 β-sheets and 8 α-helices structures (A) and OsHK3b showing 14 β-sheets and 8 α-helices (B). The highlighted residues were found conserved in various sensory domains. The residues are numbered according to their respective position in the complete sequence of AtHK1 and OsHK3b.
Figure 3.
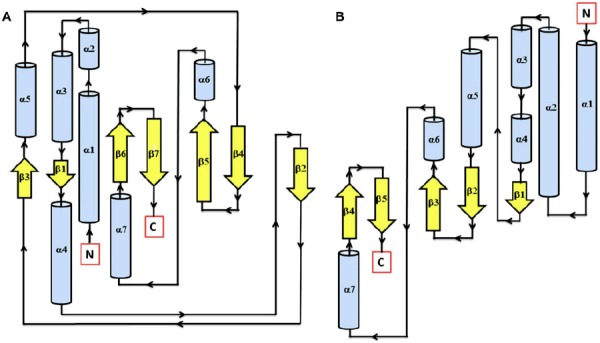
Secondary structure topology of TD domain of showing presence of 7 β-sheets and 7 α-helices in AtHK1 (A) and 5 β-sheets and 7 α-helices in OsHK3b (B).
3.2. Comparative modeling of TD in AtHK1 and OsHK3b
In order to model the TD structure of AtHK1 and OsHK3b proteins, BLAST searches were performed against the PDB for proteins with similar sequence and known 3D structures using the length of TD domain in AtHK1 – 264 amino acid and length of TD domain in OsHK3b – 280 amino acid. The search identified structures from Thermotoga meritima such as 2C2A.pdb (cytoplasmic portion of HK protein), 3DGE.pdb (HK-RR complex), and 3A0R.pdb (HK, ThkA in complex with RR, TrrA, ADP bound chain A); and 3D36.pdb (HK, KinB) from Geobacillus stearothermophilus (ADP bound) as possible templates for modeling both the domains using the threading approach (see Method) (Supplementary Figure 4(A) and (B)). The target sequence of TD in AtHK1 showed 32, 38, and 35% identity with 2C2A, 3DGE, and 3D36, respectively, while TD in OsHK3b showed 31, 34, and 30% identity with 3A0R, 2C2A, and 3D36, respectively. The Ramachandran plot for the modeled TD of AtHK1 showed 96.6% residues in most favorable regions with the remaining 2.5% of residues occurring in generously allowed regions while 0.8% residues were found in disallowed region (Supplemental Figure 5(A) and (B)). The modeled TD in OsHK3b showed 96.7% residues in most favorable regions with the remaining 2.1% of residues occurring in generously allowed regions, while only 1.2% residues were found in disallowed region. The PROCHECK result summary showed 14 out of 260 residues labeled in TD of AtHK1 while in OsHK3b TD, 20 out of 276 residues were found to be labeled. The torsion angles of the side chain designated by χ1–χ2 plots showed only 5 labeled residues out of 165 in AtHK1 TD while in OsHK3b TD, 6 out of 155 were found labeled. The observed G-factor score of the modeled TD of AtHK1 was found to be −0.11 for dihedral bonds, −0.38 for covalent bonds, and −0.21 overall. In OsHK3b, it was found to be −0.12 for dihedral bonds, −0.81 for covalent bonds, and −0.38 overall. The distribution of the main chain bond lengths and bond angles were 97.8 and 90.1%, within limits for the modeled TD of AtHK1, while for modeled OsHK3b TD, main chain bond lengths and bond angles were 95.7 and 88.7% within the limit. The PROSA-web energy plots for TD structures of AtHK1 and OsHK3b protein showed a z-score for pair, surface and combined energy which was found to be −4.3 and −5.56, respectively (Supplemental Figure 6(A) and (B)).
Figure 4.
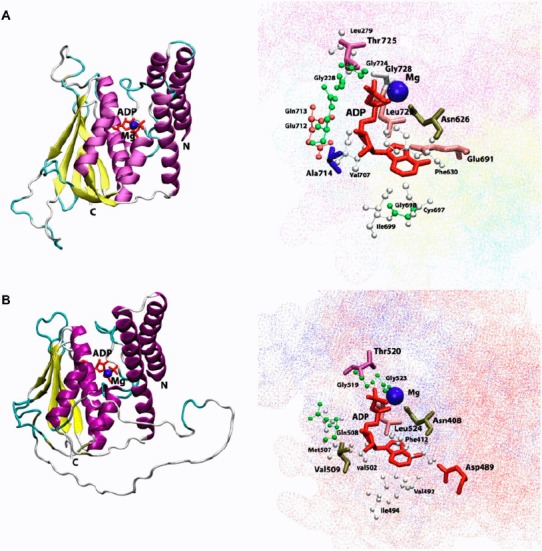
Cartoon view diagram of the secondary structure of TD domain showing the arrangement of 7 β-sheets and 7 α-helices in AtHK1 (A) and of 5 β-sheets and 7 α-helices in OsHK3b (B) with ADP and Mg along with the marked residues which were observed to form hydrogen bond with ADP. The residues are numbered according to their respective position in the complete sequence of AtHK1 and OsHK3b.
Figure 5.
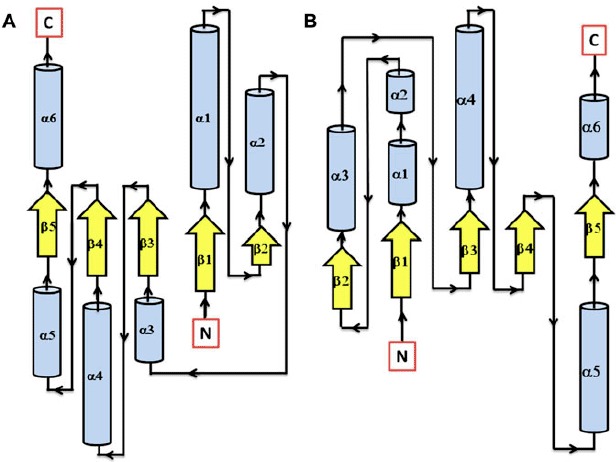
Secondary structure topology of RD showing the presence of 5 β-sheets and 6 α-helices in both AtHK1 (A) and OsHK3b (B).
Figure 6.
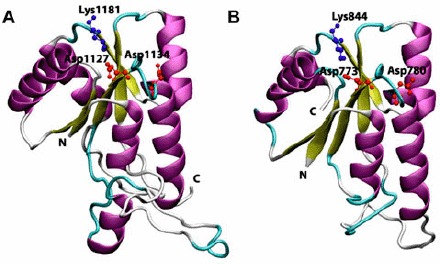
Cartoon view diagram of the secondary structure of RD showing the presence of 5 β-sheets and 6 α-helices arrangements in both AtHK1 (A) and OsHK3b (B) showing conserved residues. The residues are numbered according to their respective position in the complete sequence of AtHK1 and OsHK3b.
3.3. Comparative modeling of RD in AtHK1 and OsHK3b
To create a model of RD of AtHK1 and OsHK3b protein, searches were made against PDB database using BLAST for proteins with similar sequence and known 3D structures using the 162 amino acid and 132 amino acid domain sequences, respectively. The search resulted in identification of structures of RR in Arabidopsis (3MM4.pdb, RD of HK, CKI1 and 3MMN.pdb, RD of HK, CKI1 with Mg2+), and Saccharomyces cerevisiae (1OXB.pdb, YPD1 and SLN1 complex), as possible templates for modeling RD of AtHK1 and OsHK3b protein (Supplementary Figure 7(A) and (B)). The target sequence of RD in AtHK1 showed 32, 32, and 30% identity with template structures 3MM4, 3MMN, and 1OXB, respectively, while the target sequence of RD in OsHK3b showed 37, 37, and 34% identity with the template structures 3MM4, 3MMN, and 1OXB, respectively. Ramachandran plot for the modeled RD structure of AtHK1 showed 97.3% residues in allowed region, 1.4% in generously allowed region, and only 1.4% in disallowed region, while the modeled RD of OsHK3b showed 96.5% residues in allowed region, 2.6% in generously allowed regions, and only 0.9% in disallowed region (Supplemental Figure 8(A) and (B)). The PROCHECK result summary showed 7 out of 160 residues labeled in RD for AtHK1, while for that of OsHK3b, 8 residues out of 130 were labeled. The torsion angles of the side chain designated by χ1–χ2 plots showed 3 labeled residues out of 92 in RD for AtHK1, it was found to be 2 out of 82 residues in case of RD structure of OsHK3b. The G-factor scores of the model were −0.03 for dihedral bonds, −0.34 for covalent bonds, and −0.14 overall in RD structure in AtHK1, while in the model of RD in OsHK3b, the observed G-factor scores were 0.06 for dihedral bonds, −0.29 for covalent bonds, and −0.07 overall. The distribution of the main chain bond lengths and bond angles were 98.9 and 90.9% within limits for RD in AtHK1 and 98.5 and 90.8% within the limit for RD model of OsHK3b. The PROSA-web energy plots for RD of AtHK1 and OsHK3b showed a z-score for pair, surface and combined energy was found to be −3.97 and −4.42, respectively (Supplemental Figure 9(A) and (B)).
Figure 7.
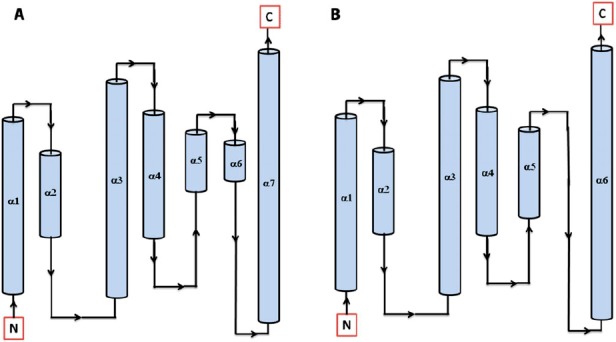
Secondary structure topology of HPt protein showing the presence of 7 α-helices in Arabidopsis (AtHPt1) (A) and 6 α-helices in O. sativa (OsHPt2) (B).
Figure 8.
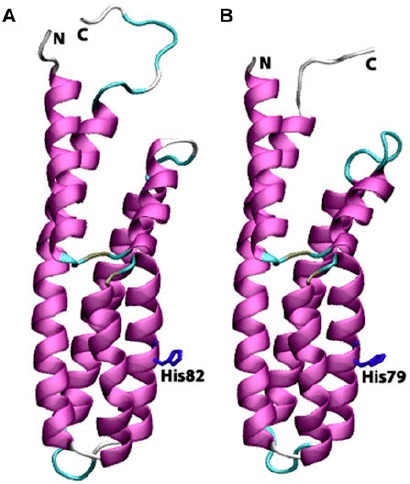
Cartoon view diagrams of the secondary structure arrangements of 7 α-helices in Arabidopsis (AtHPt1) (A) and 6 helices in O. sativa (OsHPt2) (B) with the conserved His residue. The residues are numbered according to their respective position in the complete sequence of AtHPt1 and OsHPt2.
Figure 9.
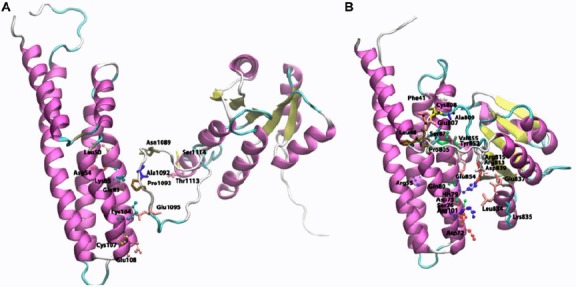
Cartoon view diagram of the docking of RD with HPt protein in Arabidopsis (A) and O. sativa (B) showing residues which plays role in protein–protein interactions. The residues are numbered according to their respective position in the complete sequence of AtHK1, OsHK3b, AtHPt1, and OsHPt2.
3.4. Comparative modeling of AtHPt1 and OsHPt2 protein
In order to model AtHPt1 and OsHPt2 proteins, BLAST searches were made against PDB for proteins with similar sequence and known 3D structures using the 156 amino acid and 147 amino acid sequences, respectively. The search resulted in identification of structures of phosphotransfer protein in Zea mays (1WN0.pdb, Phosphotransfer protein, ZmHP2) and O. sativa (1YVI. pdb, Phosphotransfer protein, AK104879) as possible templates for modeling AtHPt1 and OsHPt2 proteins (Supplementary Figure 10(A) and (B)). The template structures (1YVI and 1WN0) showed 43 and 45% identity with AtHPt1 and 73 and 72% identity with OsHPt2 protein. The Ramachandran plot analysis for the modeled AtHPt1 had 100% residues in allowed region, while in OsHPt2, 99.3% residues were found in allowed region and 0.7% were found in generously allowed region (Supplementary Figure 11(A) and (B)). The PROCHECK result summary showed none of the residue as labeled in both the modeled proteins. The torsion angles of the side chain designated by χ1–χ2 plots showed only 2 labeled residues out of 154 in AtHPt1, while in OsHPt2, 1 out of 145 residues were found to be in labeled region. The observed G-factor scores of the model were 0.22 for dihedral bonds, −0.07 for covalent bonds, and 0.11 overall in AtHPt1 protein structure, while in OsHPt2, observed G-factor scores of the model were 0.40 for dihedral bonds, −0.05 for covalent bonds, and 0.23 overall. The distribution of the main chain bond lengths and bond angles were 99.4 and 95.1% within limits for model of both AtHPt1, while for OsHPt2 protein structure, the main chain bond lengths and bond angles were 99.7 and 95.4% within the limit. The PROSA-web energy plots for AtHPt1 and OsHPt2 protein show a z-score for pair, surface and combined energy, which was found to be −6.81 and −6.02, respectively (Supplement Figure 12(A) and (B)).
4. Discussion
The absence of suitable template structure of the interdomain regions has forced us to analyze only the functional domains of the orthologous HK proteins from Arabidopsis and rice. However, details provided in the paper may assist future studies targeting the generation of a single plausible structure of the putative osmosensor proteins from two genera.
4.1. Analysis of CHASE domain AtHK1 and OsHK3b proteins
Based on sequence conservation, various types of CHASE domains (CHASE1 to CHASE6) have been reported (Anantharaman & Aravind, 2001). Till date, only one bacterial CHASE domain-containing HK i.e. VsrA has been analyzed with respect to its biological function. It is a CHASE3 domain-containing HK, which is required for the expression of virulence factors in Pseudomonas solanacearum (Schell, Denny, & Huang, 1994). Based on analysis of various signaling domains, it was hypothesized that sensing domains may have diverged during evolution but still possess the same folds (Aravind, Mazumder, Vasudevan, & Koonin, 2002). This may be due to the fact that protein–ligand interaction is carried out by a small number of closely related receptors, which are diverse in sequence due to the nature of ligands they bind (Zhulin, Nikolskaya & Galperin, 2003).
The AtHK1 sensory domain secondary structure consists of 11 β-sheets and 8 α-helices structures, while the secondary structure of OsHK3b CHASE domain consists of 14 β-sheets and 8 α-helices (Figure 1(A) and (B)). The sensory/CHASE domain structures of both AtHK1 and OsHK3b had α + β fold, with two extended helices on both boundaries and two central helices separated by sheets (Figure 2(A) and (B)). CHASE domains were reported to bind to diverse low molecular weight ligands, such as cytokinin-like adenine derivatives or peptides, and mediate signal transduction through the respective receptors (Zhulin et al., 2003). Earlier analysis of CHASE domain in CRE1a protein had shown the position of conserved Thr278 within the sensing domain (Zhulin et al., 2003). Alignment of AtHK1 and OsHK3b with CRE1a protein showed the presence of conserved Thr288 and Thr132, respectively. The mutation of this conserved residue leads to the loss of the function which is hypothesized to play a major role in ligand interaction or the formation of entry to the binding pocket (Zhulin et al., 2003). Recently, crystal structure of AtHK4 cytokinin receptor has been resolved using X-ray crystallography, which has shed light on the molecular basis of recognition of natural and synthetic cytokinins (Hothorn, Dabi, & Chory, 2011). Other residues which might play an important role in the sensing activity such as Val197, Ser208, Gly317, Val325, Leu328, and Leu332 in AtHK1 and Val78, Ser90, Gly157, Val165, Leu168, and Leu172 in OsHK3b were also found conserved within the sensing domain (Figure 2(A) and (B)).
4.2. Analysis of TD structure of AtHK1 and OsHK3b protein
The EnvZ TD structure showed a mixed α/β sandwich fold made from five β-strands and three α-helices (Tomomori et al., 1999). Analysis of TD of AtHK1 showed presence of seven β-sheets and seven α-helices in the modeled structure, while TD of OsHK3b showed the presence of five β-sheets and seven α-helices (Figure 3(A) and (B)). The kinase domain in both TDs was found structurally related to the ATP-binding domains of the GHL ATPase family (GyraseB, Hsp90 and MutL) and hence, these ATPase form the GHKL superfamily (Dutta & Inouye, 2000). The TD structure in Thermotoga maritima shows long α-helices at the N-terminal region with a kink due to presence of proline residue in the structural motif (Marina, Mott, Auyzenber, Hendrickson, & Waldburger, 2001). In TD of AtHK1 and OsHK3b, proline residue (Pro606 in AtHK1 TD and Pro388 in OsHK3b TD) was conserved in the secondary structure of the domain.
Analysis of ADP binding to the TD of AtHK1 and OsHK3b showed various hydrogen bond interactions with the active site residues (Tables 1 and 2) (Figure 4(A) and (B)). The conserved Thr725, Gly728 of TD in AtHK1 and Thr520, and Gly523 of TD in OsHK3b were observed to form hydrogen bond interaction. Analysis showed hydrogen bond interaction between conserved Leu726 and Leu524 residues of TD in both AtHK1 and OsHK3b. Sequence analysis of TD showed that the conserved Val492 in OsHK3b has been replaced with the Cys697 in TD of AtHK1. Analysis of binding in TD with ADP revealed that β5 and β6 contributes to the interaction with ADP in AtHK1 while in OsHK3b, α4, α5, and β5 contributes to the interaction with ADP (Figure 4(A) and (B)). The interaction of ADP with conserved Asn626 was observed in TD of AtHK1 as well as OsHK3b (Asn408). Analysis of Hsp90–ADP complex has shown that the Asn (residue 347 in E. coli and 37 in Hsp90) binds to the ADP. In an earlier study, the ATP-dependent autokinase activity was lost when Asn347 was mutated to Asp in EnvZ mutant. However, in a similar mutation study in OmpR, autokinase activity was retained (Yang & Inouye, 1993). Analysis of EnvZ NMR structures revealed the presence of Hsp90 and DNA gyrase B like folds responsible for the ATP binding. The motif G1 (DxGxGΦ) (694–699 in AtHK1 and 489–494 in OsHK3b) and G2 (GΦGΦ) (724–727 in AtHK1 and 521–524 in OsHK3b) were conserved in TD. These motifs were close to the ATP binding site, similar to Hsp90 and EnvZ structure. Previously identified N, G1, F, and G2 boxes were conserved in the TD of both AtHK1 and OsHK3b. Previous mutagenesis studies have also established that the glycine-rich regions, G1 and G2, are essential for kinase activity (Yang & Inouye, 1993).
Table 1.
Hydrogen bonds between ADP and modeled TD of AtHK1. Hydrogen bond distances between the residues were calculated using HBPLUS.
| ADP | TD residues | Distance (Å) | |
|---|---|---|---|
| O1A | Asn626 | ND2 | 2.76 |
| N7 | Glu691 | OE1 | 3.10 |
| N6 | Glu691 | OE1 | 3.22 |
| O2′ | Ala714 | N | 3.14 |
| O3′ | Ala714 | N | 3.28 |
| O3B | Thr725 | N | 2.89 |
| O1B | Gly728 | N | 3.38 |
| O2A | Leu729 | N | 2.72 |
Table 2.
Hydrogen bonds between ADP and modeled TD of OsHK3b. Hydrogen bond distances between the residues were calculated using HBPLUS.
| ADP | TD residues | Distance (Å) | |
|---|---|---|---|
| O1A | Asn408 | ND2 | 2.91 |
| N6 | Asp489 | OD2 | 3.01 |
| O2′ | Val509 | N | 3.19 |
| O3′ | Val509 | N | 3.11 |
| O1B | Thr520 | N | 3.19 |
| O3B | Thr520 | N | 2.61 |
| O2A | Leu524 | N | 2.85 |
4.3. Analysis of RD in AtHK1 and OsHK3b
A comparison of the amino acid sequence of RD of both AtHK1 and OsHK3b with other experimentally resolved RDs structures using various structural approaches revealed more than 30 conserved amino acid residues. Analysis of RD of AtHK1 and OsHK3b protein showed five β-sheets and six α-helices (Figure 5(A) and (B)). Earlier, analysis of crystal structure of RR from Streptococcus pneumoniae in both complex and native state has been performed (Bent, Isaac, Mitchell, & Tunnicliffe, 2004). The active site of RD showed the presence of conserved Asp773 in OsHK3b and Asp1127 in AtHK1 as compared to template Asp52 in S. pneumoniae. The conserved Lys1181 in RD of AtHK1 and Lys844 in RD of OsHK3b play a major role in interaction with the phosphate group, like Lys109 in CheY, a bacterial chemotaxis protein. In the analysis of the structure of CheY, RR showed that the fold is a doubly wound, five-stranded parallel sheet with topology β2-β1-β3-β4-β5, which was observed in the structure of RD of AtHK1 and OsHK3b (Figure 6(A) and (B)). Analysis of structure of RRs like NtrC (Volkman, Nohaile, Amy, Kustu, & Wemmer, 1995), NarL (Baikalov et al., 1998), CheB (Djordjevic, Goudreau, Xu, Stock, & West, 1998), ETR1 (Muller-Dieckmann, Grantz, & Kim, 1999), and Spo0A (Robinson, Buckler, & Stock, 2000) in other organisms had shown similar secondary structural features. Analysis of experimentally determined structures revealed that the activated regulatory domains show conformational changes that primarily involve small repositioning of secondary structure elements that appear to be linked to the rearrangement of a specific set of side chains. These structural changes were found responsible for the protein–protein interaction (Posas et al., 1996).
4.4. Analysis of HPt protein in Arabidopsis (AtHPt1) and O. sativa (OsHPt2)
Earlier, the structures of P1 domain of E. coli CheA (Posas et al., 1996), HPt domain of E. coli ArcB (Kato, Mizuno, Shimizu, & Hakoshima, 1997), Bacillus subtilis Spo0B protein (Varughese, Madhusudan, Zhou, Whiteley, & Hoch, 1998), and S. cerevisiae YPD1 protein (Xu & West, 1999; Song et al., 1998) have been analyzed experimentally. X-ray structure of ArcB revealed the presence of six α-helices (A–F) with four helices forming a bundle in which conserved His717 was found located at helix-D. Analysis of HPts shows that they share a common four-helix bundle motif despite their overall lack of sequence similarity. Both AtHPt1 and OsHPt2 proteins showed striking similarity in their secondary structures. The detailed analysis of the structure of the AtHPt1 protein showed seven α-helices namely α1(3–23), α2(26–38), α3(42–68), α4(73–92), α5 (93–102), α6(103–110), and α7(111–146), while OsHPt2 protein also showed presence of six helices, namely α1 (1–21), α2(24–37), α3(39–66), α4(70–89), α5(90–107), and α6(108–142) (Figure 7(A) and (B)). Topology analysis of AtHPt1 suggested that α5 and α6 are closely woven with slight break in the helix depicting a two-helix state rather than one, therefore, leading to the seven α-helices in AtHPt1. Similarly, four C-terminal helices form an antiparallel four-helix bundle in YPD1 (Song et al., 1998; Xu & West, 1999), and the HPt domain of ArcB (Varughese et al., 1998). Earlier reports have shown that the conserved histidine (His80) present in the center of helix D in Z. mays acts as a site for phosphorylation (Sakakibara et al., 1999). The histidine residue was also found conserved in AtHPt1 (His82) and OsHPt2 (His79). Sequence similarity of specific residues surrounding the His residue has prompted postulation of structural and functional roles of the conserved residues (Robinson et al., 2000). Despite the overall fold conservation, specific differences in helix length and orientation in each HPt domain provide structural features needed for individual functions (Figure 8(A) and (B)). These structural variations likely result in modification of surface properties designed to promote proper intermolecular contacts (Stock et al., 2000).
Structure alignment of templates, AtHPt1 and OsHPt2 protein showed the existence of conserved residues near the active site region. In comparison to earlier resolved HPt protein structure such as YPD1 (Song et al., 1998), the hydrophobic reverse turn between helix C and D was also conserved in AtHPt1 and OsHPt2 protein structures. The Asp67 and Asp73 in α3 were conserved in AtHPt1 and OsHPt2 and have been reported earlier to serve as a hydrogen bond acceptor in other resolved HPt proteins (Sakakibara et al., 1999). The residues surrounding the active site were arranged in such a manner that it can stabilize the accessibility of His. In OsHPt2, Gly83 residue and Gly86 in AtHPt1 located four residues away from the active-site histidine residue is highly conserved among the known HPt proteins. The absence of a side-chain at this position provides more exposure of the active-site histidine residue and increases the solvent accessibility of the histidine residue (Song et al., 1998). The structure of ArcB HPt domain possess kinks in the helix C and helix D of the four helix bundle because of the proline residue in the middle of one helix (Song et al., 1998). Hence, the structure of the HPt domain had a concave surface surrounding the active-site histidine residue. Analysis of YPD1 structural domain revealed that this structural feature was not common to all the HPt structures (Song et al., 1998). The structure of AtHPt1 and OsHPt2 proteins was also not observed to contain proline-induced kinks, but was found to maintain a flat molecular surface encompassing the active-site histidine residue.
4.5. Binding analysis of AtHK1 with AtHPt1 and OsHK3b with OsHPt2
Earlier reports have established that the bacterial RR CheY acts as a phosphoryl donor to the yeast YPD1 protein (Song et al., 1998). Docking studies performed earlier shows that there can be many possible conformations for the histidine residue (His64) of YPD1 to be a reasonable distance to the active-site aspartate residue of CheY (Asp57) (Varughese et al., 1998). Analysis of binding site of the RD of AtHK1 protein and AtHPt1 protein showed residues in α4 and α5 of HPt protein to be involved in hydrogen bond interactions. Docking analysis of RD of AtHK1 and AtHPt1 protein revealed the hydrogen bond interaction of Leu50, Gln83, Asp54, Lys85, Lys104, Cys107, and Glu108 of AtHPt1 protein with Ser1114, Thr1113, Asn1089, Pro1093, and Glu1095 of AtHK1 protein, respectively. Analysis of the docking study of OsHK3b and OsHPt2 showed hydrogen bond interaction between Glu65, Cys58, Ser61, Arg62, and Asp75 residues of OsHPt2 protein with Pro805, Glu854, Cys808, and Val855 of OsHK3b protein (Figure 9(A) and (B)) (Residues represented as observed in analysis). Docking analysis of modeled RD of OsHK3b and OsHPt2 protein showed nonbonded interaction between the active site His79 of OsHPt2 protein and Glu131 of the RD of the OsHK3b protein. In AtHPt1 and AtHK1 docking analysis, active-site residue Gln50 of RD of AtHK1 protein had an interaction with Cys107 of AtHPt1 protein. With the binding analysis, it can be postulated that the modeled complex between RD of AtHK1 with AtHPt1 protein and RD of OsHK3b with OsHPt2 protein suggests shape complementarity; hydrophobic interactions contribute to the interaction between the two proteins.
5. Conclusion
Sequence analysis of OsHK3b using Pfam and CDD revealed the presence of three conserved domains namely CHASE, TD, and RD while AtHK1 revealed only two conserved domains namely TD and RD. The structural analysis of the functional domains of HK and phosphotransfer protein indicated the conserved nature of the proteins in both the genera (Table 3).
Table 3.
Comparative analysis of the secondary structure elements in AtHK1, OsHK3b, AtHPt1, and OsHPt2 proteins.
| Proteins | Domains | Arabidopsis | Rice | ||
|---|---|---|---|---|---|
| HK | α | β | α | β | |
| CHASE | 8 | 11 | 8 | 14 | |
| TD | 7 | 7 | 7 | 5 | |
| RR | 6 | 5 | 6 | 5 | |
| HPt | 6 | 0 | 7 | 0 | |
The analysis of secondary structure of AtHK1 sensing domain showed 11 β-sheets and 8 α-helices structure while OsHK3b CHASE domain showed the presence of 14 β-sheets and 8 α-helices. The predicted structure of the sensory/CHASE domain of both AtHK1 and OsHK3b showed the presence of α + β fold. Analysis of TD of both AtHK1 showed the presence of seven β-sheets and seven α-helices and OsHK3b showed the presence of five β-sheets and seven α-helices. Analysis of the modeled domain showed the presence of conserved residues, which plays an important role in ADP binding. The conserved signature boxes such as N, G1, F, and G2 were found conserved in the TD of both AtHK1 and OsHK3b. The vast number of crystal structures for ATP binding domains of His-Kinase-like proteins have aided in the identification of conserved and varied functional features at their active sites subsequent to phosphorylation. Hence, the modeled structures of TD of AtHK1 and OsHK3b protein will enhance the current understanding of the interaction of ADP with the conserved active site residues. Analysis of amino acid sequence of RD of both AtHK1 and OsHK3b showed five β-sheets and six α-helices. Analysis of the modeled domain revealed the conserved five-stranded parallel sheet with topology β2-β1-β3-β4-β5. The residues, which play crucial role in the interaction with phosphotransfer protein, were found conserved in RD of AtHK1 and OsHK3b protein. Analysis of HPt protein showed common four-helix bundle motif. Structure of AtHPt1 showed seven α-helices, while OsHPt2 protein showed six α-helices. In AtHPt1 and OsHPt2, histidine (His82 in AtHPt1 and His79 in OsHPt2) was conserved along with other secondary structure motifs.
In order to analyze the interaction of RD of AtHK1 with AtHPt1 and of OsHK3b with OsHPt2 docking analysis was performed. Docking study showed residues in α4 and α5 of HPt protein to be involved in hydrogen bond interactions in both AtHK1 and OsHK3b, hence playing major role in the interactions. It was observed that the shape complementarity and hydrophobic interactions play crucial role in interaction between the RD of AtHK1 vs. AtHPt1 protein and RD of OsHK3b vs. OsHPt2 protein.
The insights provided in the paper may assist in understanding the mechanism of signal transduction using phosphorylation in TCS. The modeled structures of functional domains may further assist in the study of complete structure of HK proteins using various other experimental techniques.
Acknowledgements
Financial support received from Department of Biotechnology and Department of Science and Technology, Ministry of Science and Technology is acknowledged.
Supplementary material
The supplementary material for this paper is available online at http://dx.doi.[10.1080/07391102.2013.818576].
References
- Anantharaman V., Aravind L. The CHASE domain: A predicted ligand-binding module in plant cytokinin receptors and other eukaryotic and bacterial receptors. Trends in Biochemical Sciences. 2001;26:579–582. doi: 10.1016/S0968-0004(01)01968-51330. [DOI] [PubMed] [Google Scholar]
- Aravind L., Mazumder R., Vasudevan S., Koonin E. V. Trends in protein evolution inferred from sequence and structure analysis. Current Opinion in Structural Biology. 2002;12:329–392. doi: 10.1016/S0959-440X(02)00334-2. [DOI] [PubMed] [Google Scholar]
- Baikalov I., Schroder I., Kaczor-Grzeskowiak M., Cascio D., Gunsalus R. P., Dickerson R. E. NarL dimerization? Suggestive evidence from a new crystal form. Biochemistry. 1998;37:3665–3676. doi: 10.1021/bi972365a. [DOI] [PubMed] [Google Scholar]
- Bent C. J., Isaac N. W., Mitchell T. J., Tunnicliffe A. R. Crystal structure of the response regulator 02 receiver domain, the essential YycF two-component system of Streptococcus pneumonia in both complexed and native states. Journal of Bacteriology. 2004;186:2872–2879. doi: 10.1128/JB.186.9.2872-2879.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilwes A. M., Alex L. A., Crane B. R., Simon M. I. Structure of CheA, a signal transducing histidine kinase. Cell. 1999;96:131–141. doi: 10.1016/S0092-8674(00)80966-6. [DOI] [PubMed] [Google Scholar]
- Chang C., Stewart R. C. The two-component system: Regulation of diverse signaling pathways in prokaryotes and eukaryotes. Plant Physiology. 1998;117:723–731. doi: 10.1104/pp.117.3.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole C., Barber J. D., Barton G. J. The Jpred 3 secondary structure prediction server. Nucleic Acids Research. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djordjevic S., Goudreau P. N., Xu Q., Stock A. M., West A. H. Structural basis for methylesterase CheB regulation by a phosphorylation-activated domain. Proceedings of the National Academy of Sciences. 1998;95:1381–1386. doi: 10.1073/pnas.95.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhovny D., Nussinov R., Wolfson H. J. Efficient unbound docking of rigid molecules. Lecture Notes in Computer Science. 2002;2452:185–200. doi: 10.1007/3-540-45784-4_14. [DOI] [Google Scholar]
- Dutta R., Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends in Biochemical Sciences. 2000;25:24–28. doi: 10.1016/S0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- Eswar N., Marti-Renom M. A., Webb B., Madhusudhan M. S., Eramian D., Sali A. Comparative Protein Structure Modeling With MODELLER. Current Protocols in Bioinformatics. 2006;15:5.6.1–5.6.30. doi: 10.1002/0471250953.bi0506s15. Retrieved from http://onlinelibrary.wiley.com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D., Mistry J., Tate J., Coggill P., Heger A., Pollington J. E., Bateman A. The Pfam protein families database. Nucleic Acid Research. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. Retrieved from http://www.ncbi.nlm.nih.gov/pmc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frishman D., Argos P. Knowledge-based protein secondary structure assignment. Proteins: Structure, Function and Genetics. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- Frishman D., Argos P. Incorporation of long-distance interactions into a secondary structure prediction algorithm. Protein Engineering. 1996;9:133–142. doi: 10.1093/protein/9.2.133. Retrieved from http://peds.oxfordjournals.org. [DOI] [PubMed] [Google Scholar]
- Greene L. H., Lewis T. E., Addou S., Cuff A., Dallman T., Dibley M., Orengo C. A. The CATH domain structure database: New protocols and classification levels give a more comprehensive resource for exploring evolution. Nucleic Acids Research. 2007;35:D291–D297. doi: 10.1093/nar/gkl959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grefen C., Harter K. Plant two-component systems: Principles, functions, complexity and cross talk. Planta. 2004;219:733–742. doi: 10.1007/s00425-004-1316-4. [DOI] [PubMed] [Google Scholar]
- Gu H., Zhu P., Jiao Y., Meng Y., Chen M. PRIN, a predicted rice interactome network. BMC Bioinformatics. 2011;12:161. doi: 10.1186/1471-2105-12-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess J. F., Oosawa K., Kaplan N., Simon M. I. Phosphorylation of three proteins in the signaling pathway of bacterial chemotaxis. Cell. 1988;53:79–87. doi: 10.1016/0092-8674(88)90489-8. [DOI] [PubMed] [Google Scholar]
- Hothorn M., Dabi T., Chory J. Structural basis for cytokinin recognition by Arabidiopsis thaliana histidine kinase 4. Nature Chemical Biology. 2011;7:766–768. doi: 10.1038/nchembio.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD – Visual molecular dynamics. Journal of Molecular Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Hwang I., Chen H. C., Sheen J. Two-component signal transduction pathways in Arabidopsis. Plant Physiology. 2002;129:500–515. doi: 10.1104/pp.005504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura A., Hanaki N., Nakamura A., Suzuki T., Taniguchi M., Kiba T., Mizuno T. Compilation and characterization of Arabidopsis thaliana response regulators implicated in His-Asp phosphorelay signal transduction. Plant and Cell Physiology. 1999;40:733–742. doi: 10.1093/oxfordjournals.pcp.a029600. [DOI] [PubMed] [Google Scholar]
- Janiak-spens F., Sparling D. P., West A. H. Novel role for an HPt domain in stabilizing the phosphorylated state of a response regulator domain. Journal of Bacteriology. 2000;182:6673–6678. doi: 10.1128/JB.182.23.6673-6678.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. T. Protein secondary structure prediction based on position-specific scoring matrices. Journal of Molecular Biology. 1999a;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Jones D. T. GenTHREADER: An efficient and reliable protein fold recognition method for genomic sequences. Journal of Molecular Biology. 1999b;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- Kato M., Mizuno T., Shimizu T., Hakoshima T. Insights into multistep phosphorelay from the crystal structure of the C-terminal HPt domain of ArcB. Cell. 1997;88:717–723. doi: 10.1016/S0092-8674(00)81914-5. [DOI] [PubMed] [Google Scholar]
- Kato M., Shimizu T., Mizunob T., Hakoshimaa T. Structure of the histidine-containing phosphotransfer (HPt) domain of the anaerobic sensor protein ArcB complexed with the chemotaxis response regulator CheY. Acta Crystallographica Section D: Biological Crystallography. 1999;55:1257–1263. doi: 10.1107/s0907444999005053. Retrieved from http://scripts.iucr.org. [DOI] [PubMed] [Google Scholar]
- Kiba T., Taniguchi M., Imamura A., Ueguchi C., Mizuno T., Sugiyama T. Differential expression of genes for response regulators in response to cytokinins and nitrate in Arabidopsis thaliana. Plant and Cell Physiology. 1999;40:767–771. doi: 10.1093/oxfordjournals.pcp.a029604. [DOI] [PubMed] [Google Scholar]
- Krivov G. G., Shapovalov M. V., Dunbrack R. L. Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert C., Leonard N., De Bolle X., Depiereux E. ESyPred3D: Prediction of proteins 3D structures. Bioinformatics. 2002;18:1250–1256. doi: 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Higgins D. G. ESyPred3D: Prediction of proteins 3D structures. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. PROCHECK: A program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Lohrmann J., Harter K. Plant two-component signaling systems and the role of response regulators. Plant Physiology. 2002;128:363–369. doi: 10.1104/pp.0109071331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A., Zheng C., Chitsaz F., Derbyshire M. K., Geer L. Y., Geer R. C., Bryant S. H. CDD: Specific functional annotation with the conserved domain database. Nucleic Acids Research. 2009;37:D205–D210. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina A., Mott C., Auyzenber A., Hendrickson W. A., Waldburger C. D. Structural and mutational analysis of PhoQ histidine kinase catalytic domain. Journal of Biological Chemistry. 2001;276:41182–41190. doi: 10.1074/jbc.M106080200. [DOI] [PubMed] [Google Scholar]
- Mizuno T. His-Asp phosphotransfer signal transduction. Journal of Biochemistry. 1998;123:555–563. doi: 10.1093/oxfordjournals.jbchem.a021972. [DOI] [PubMed] [Google Scholar]
- Muller-Dieckmann H. J., Grantz A. A., Kim S. H. The structure of the signal receiver domain of the Arabidopsis thaliana ethylene receptor ETR1. Structure with Folding & Design. 1999;7:1547–15569. doi: 10.1016/S0969-2126(00)88345-8. [DOI] [PubMed] [Google Scholar]
- Murzin A. G., Brenner S. E., Hubbard T., Chothia C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. Journal of Molecular Biology. 1995;247:536–540. doi: 10.1016/S0022-2836(05)80134-2. [DOI] [PubMed] [Google Scholar]
- Nielsen M., Lundegaard C., Lund O., Petersen T. N. CPHmodels-3.0-remote homology modeling using structure guided sequence profiles. Nucleic Acids Research. 2010;38:W576–W581. doi: 10.1093/nar/gkq535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareek A., Singh A., Kumar M., Kushwaha H. R., Lynn A. M., Singla-Pareek S. L. Whole-genome analysis of Oryza sativa reveals similar architecture of two-component signaling machinery with Arabidopsis. Plant Physiology. 2006;142:380–397. doi: 10.1104/pp.106.086371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson J. S., Kofoid E. C. Communication modules in bacterial signaling proteins. Annual Review of Genetics. 1992;26:71–112. doi: 10.1146/annurev.ge.26.120192.000443. [DOI] [PubMed] [Google Scholar]
- Posas F., Wurgler-Murphy S. M., Maeda T., Witten E. A., Thai T. C., Saito H. Yeast HOG1 MAP kinase cascade is regulated by a multistep phosphorelay mechanism in the SLN1-YPD1-SSK1 “two-component” Osmosensor. Cell. 1996;86:865–875. doi: 10.1016/S0092-8674(00)80162-2. [DOI] [PubMed] [Google Scholar]
- Riechmann J. L., Heard J., Martin G., Reuber L., Jiang C., Keddie J., Yu G. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science. 2000;290:2105–2110. doi: 10.1126/science.290.5499.2105. [DOI] [PubMed] [Google Scholar]
- Robinson V. L., Buckler D. R., Stock A. M. A tale of two components: A novel kinase and a regulatory switch. Natural Structural Biology. 2000;7:626–633. doi: 10.1038/77915. [DOI] [PubMed] [Google Scholar]
- Russell R. B., Barton G. J. Multiple protein sequence alignment from tertiary structure comparison: Assignment of global and residue confidence levels. Proteins. 1992;14:309–323. doi: 10.1002/prot.340140216. [DOI] [PubMed] [Google Scholar]
- Saito H. Histidine phosphorylation and two-component signaling in eukaryotic cells. Chemical Reviews. 2001;101:2497–2509. doi: 10.1021/cr000243+. [DOI] [PubMed] [Google Scholar]
- Sakakibara H., Hayakawa A., Deji A., Gawronski S., Sugiyama T. His-Asp phosphotransfer possibly involved in a nitrogen signal transduction mediated by cytokinin in maize: molecular cloning of cDNAs for two-component regulatory factors and demonstration of phosphotransfer activity in vitro. Plant Molecular Biology. 1999;41:63–573. doi: 10.1023/a:1006391304881. Retrieved from http://link.springer.com/article. [DOI] [PubMed] [Google Scholar]
- Sakakibara H., Taniguchi M., Sugiyama T. His-Asp phosphotransfer possibly involved in a nitrogen signal transduction mediated by cytokinin in maize: molecular cloning of cDNAs for two-component regulatory factors and demonstration of phosphotransfer activity in vitro. Plant Molecular Biology. 2000;42:273–278. doi: 10.1023/a:1006391304881. Retrieved from http://link.springer.com/article. [DOI] [PubMed] [Google Scholar]
- Schell M. A., Denny T. P., Huang J. VsrA, Lt second two-component system regulating virulence genes of Preudomrmas solanacearum. Molecular Microbiology. 1994;11:489–500. doi: 10.1111/j.1365-2958.1994.tb00330.x. Retrieved from http://onlinelibrary.wiley.com. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D., Inbar Y., Polak V., Shatsky M., Halperin I., Benyamini H., Wolfson H. J. Taking geometry to its edge: Fast unbound rigid (and hinge-bent) docking. Proteins. 2003;52:107–112. doi: 10.1002/prot.10397. [DOI] [PubMed] [Google Scholar]
- Shi J., Blundell T. L., Mizuguchi K. FUGUE: Sequence-structure homology recognition using environment-specific substitution tables and structure- dependent gap penalties. Journal of Molecular Biology. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- Singh A., Kushwaha H. R., Sharma P. Molecular modelling and comparative structural account of aspartyl beta-semialdehyde dehydrogenase of Mycobacterium tuberculosis (H37Rv) Journal of Molecular Modeling. 2008;14:249–263. doi: 10.1007/s00894-008-0267-2. [DOI] [PubMed] [Google Scholar]
- Sippl M. J. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- Song H. K., Lee J. Y., Lee L. G., Moon J., Min K., Yang J. K., Suh S. W. Insights into eukaryotic multistep phosphorelay signal transduction revealed by the crystal structure of Ypd1p from Saccharomyces cerevisiae. Journal of Molecular Biology. 1998;293:753–761. doi: 10.1006/jmbi.1999.3215. [DOI] [PubMed] [Google Scholar]
- Stock A. M., Robinson V. L., Goudreau P. N. Two-component signal transduction. Annual Review of Biochemistry. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- Suzuki T., Imamura A., Ueguchi C., Mizuno T. Histidine-containing phosphotransfer (HPt) signal transducers implicated in His-to-Asp phosphorelay in Arabidopsis. Plant and Cell Physiology. 1998;39:1258–1268. doi: 10.1093/oxfordjournals.pcp.a029329. [DOI] [PubMed] [Google Scholar]
- To J. P., Haberer G., Ferreira F. J., Deruere J., Mason M. G., Schaller G. E., Kieber J. J. Type-A Arabidopsis response regulators are partially redundant negative regulators of cytokinin signaling. Plant Cell. 2004;16:658–671. doi: 10.1105/tpc.018978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomomori C., Toshiyuki T., Dutta R., Park H., Saha S. K., Zhu Y., Ikura M. Solution structure of the homodimeric core domain of Escherichia coli histidine kinase EnvZ. Natural Structural Biology. 1999;6:729–734. doi: 10.1038/11495. [DOI] [PubMed] [Google Scholar]
- Urao T., Yamaguchi-Shinozaki K., Shinozaki K. Two-component systems in plant signal transduction. Trends in Plant Science. 2000;5:67–74. doi: 10.1016/S1360-1385(99)01542-3. [DOI] [PubMed] [Google Scholar]
- Varughese K. I., Madhusudan L., Zhou X. Z., Whiteley J. M., Hoch J. A. Formation of a novel four-helix bundle and molecular recognition sites by dimerization of a response regulator phosphotransferase. Molecular Cell. 1998;2:485–493. doi: 10.1016/S1097-2765(00)80148-3. [DOI] [PubMed] [Google Scholar]
- Volkman B. F., Nohaile N. J., Amy N. K., Kustu S., Wemmer D. E. Three dimensional solution structure of the N-terminal receiver domain of NtrC. Biochemistry. 1995;34:413–1424. doi: 10.1021/bi00004a036. [DOI] [PubMed] [Google Scholar]
- Wiederstein M., Sippl M. J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research. 2007;35:W407–W410. doi: 10.1093/nar/gkm2901332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Porter S. W., West A. H. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Structure. 2003;11:1569–1581. doi: 10.1016/j.str.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Xu Q., West A. H. Conservation of structure and function among histidine-containing phosphotransfer (HPt) domains as revealed by the crystal structure of YPD1. Journal of Molecular Biology. 1999;292:1039–1050. doi: 10.1006/jmbi.1999.3143. [DOI] [PubMed] [Google Scholar]
- Yang Y., Inouye M. Requirement of both kinase and phosphatase activities of an Escherichia coli receptor (Tazl) for ligand dependent signal transduction. Journal of Molecular Biology. 1993;231:335–342. doi: 10.1006/jmbi.1993.1286. [DOI] [PubMed] [Google Scholar]
- Zhulin I. B., Nikolskaya A. N., Galperin M. Y. Common extracellular sensory domains in transmembrane receptors for diverse signal transduction pathways in bacteria and archaea. Journal of Bacteriology. 2003;185:285–294. doi: 10.1128/JB.185.1.285-294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]