

Abstract
The dyadic organization of ventricular myocytes ensures synchronized activation of sarcoplasmic reticulum (SR) Ca2+ release during systole. However, it remains obscure how the dyadic organization affects SR Ca2+ handling during diastole. By measuring intraluminal SR Ca2+ ([Ca2+]SR) decline during rest in rabbit ventricular myocytes, we found that ∼76% of leaked SR Ca2+ is extruded from the cytosol and only ∼24% is pumped back into the SR. Thus, the majority of Ca2+ that leaks from the SR is removed from the cytosol before it can be sequestered back into the SR by the SR Ca2+-ATPase (SERCA). Detubulation decreased [Ca2+]SR decline during rest, thus making the leaked SR Ca2+ more accessible for SERCA. These results suggest that Ca2+ extrusion systems are localized in T-tubules. Inhibition of Na+-Ca2+ exchanger (NCX) slowed [Ca2+]SR decline during rest by threefold, however did not prevent it. Depolarization of mitochondrial membrane potential during NCX inhibition completely prevented the rest-dependent [Ca2+]SR decline. Despite a significant SR Ca2+ leak, Ca2+ sparks were very rare events in control conditions. NCX inhibition or detubulation increased Ca2+ spark activity independent of SR Ca2+ load. Overall, these results indicate that during rest NCX effectively competes with SERCA for cytosolic Ca2+ that leaks from the SR. This can be explained if the majority of SR Ca2+ leak occurs through ryanodine receptors in the junctional SR that are located closely to NCX in the dyadic cleft. Such control of the dyadic [Ca2+] by NCX play a critical role in suppressing Ca2+ sparks during rest.
Introduction
During an action potential (AP), Ca2+ influx via L-type Ca2+ channels (LTCCs) activate ryanodine receptor (RyR) Ca2+ release channels on the sarcoplasmic reticulum (SR). This process, known as Ca2+-induced Ca2+ release (CICR), generates a global increase in cytosolic Ca2+ ([Ca2+]i) (1). In adult ventricular myocytes, CICR occurs at specialized cellular microdomains called dyads. In these domains, LTCCs in the membrane of the T-tubule come into close contact with a cluster of RyRs in the junctional SR (2). The narrow space between the junctional SR and T-tubule membranes (known as the dyadic cleft) ensures a high fidelity of RyR activation by L-type Ca2+ current. The simultaneous opening of RyRs within a single release cluster generates a local increase in [Ca2+]i, or a Ca2+ spark (3). The spatiotemporal summation of thousands of Ca2+ sparks during an AP produces the global Ca2+ transient that activates contraction. It is well accepted that the dyadic organization of ventricular myocytes provides the necessary local control of SR Ca2+ release by L-type Ca2+ current during systole (4). However, it remains less clear how the dyadic organization affects SR Ca2+ handling during diastole.
In resting ventricular myocytes, spontaneous openings of RyRs generate SR Ca2+ leak (5–7). This leak causes depletion of SR Ca2+ content and reduction of contractile force. Logically, a longer period of rest (at slow heart rates) would cause larger loss of the intra-SR [Ca2+] ([Ca2+]SR). However, the degree of the postrest decay of SR Ca2+ content depends also on the activity of Ca2+ transporters such as the SR Ca2+-ATPase (SERCA), the Na+-Ca2+ exchanger (NCX), and the plasmalemmal Ca2+-ATPase (PMCA). Depending on the animal species, the fraction of the leaked Ca2+ that is resequestered into the SR by SERCA or extruded from the cell by NCX and PMCA may vary significantly (8). Species in which Ca2+ removal mechanisms predominantly rely on SERCA activity (e.g., rat, mouse) have a minimal loss of SR Ca2+ content during rest, whereas species with a significant contribution of NCX to [Ca2+]i regulation (e.g., rabbit, human) are more prone to the postrest decay of SR Ca2+ content. It has been estimated that in rabbit ventricular myocytes the contribution of NCX and SERCA to the cytosolic Ca2+ removal average 40% and 60%, respectively; whereas the role of PMCA is very limited (9).
Because activity of Ca2+ transporters highly depends on local [Ca2+]i, SR Ca2+ leak would preferentially activate nearby Ca2+ pumps and exchangers. Thus, the localization of Ca2+ pumps and leak channels within the myocyte should also have a significant impact of SR Ca2+ balance during rest. Previous work has shown that in adult ventricular myocytes several important components of Ca2+ transport systems are located at the dyads. RyRs are mainly concentrated as large clusters in the junctional SR, facing the dyadic cleft (2,10,11). Studies of detubulated ventricular myocytes revealed that the major sarcolemmal Ca2+ extrusion systems, such as NCX and PMCA, are located in the T-tubule membrane (12,13). As a result, a significant fraction of the junctional RyRs is located in close proximity to NCX (14). Such colocalization can explain the mechanism by which Ca2+ influx via the reverse mode of NCX enhances CICR during systole (15–17). During diastole, however, Ca2+ that leaks via junctional RyRs would be extruded from the dyadic cleft by NCX, working in the direct mode. Moreover, mitochondria that occupy space around junctional SR and the T-tubule would restrict diffusion of the leaked Ca2+ into the cytosol (11). Thus, if the majority of SR Ca2+ leak occurs via junctional RyRs, the leaked Ca2+ would then be preferentially extruded from the cell. This would also imply that RyR-mediated SR Ca2+ leak would have a rather limited effect on cytosolic [Ca2+] then previously suggested.
However, the mechanisms of SR Ca2+ handling during rest have not been critically evaluated because it is notoriously difficult to dissect SR Ca2+ leak in an intact cellular environment. In this work, we used a novel, to our knowledge, approach to directly measure changes of [Ca2+]SR during rest after inhibition of different Ca2+ transporters or disruption of the T-tubule system (TTS). We found that in rabbit ventricular myocytes a main portion of SR Ca2+ leak is extruded from the cell by NCX before it can be sequestered back into the SR by SERCA. This implies that the majority of SR Ca2+ leak occurs via junctional RyRs that are colocalized with NCXs at dyads. This mechanism plays an important role in decreasing contractile force at low heart rate. Furthermore, an effective Ca2+ extrusion from the dyadic cleft by NCX would limit local CICR within a RyR cluster, thus preventing Ca2+ spark activation. Thus, in addition to local positive control of SR Ca2+ release by LTCC (4), we proposed what we believe to be a novel negative control of local CICR by NCX. These two mechanisms of local control of CICR are not mutually exclusive as they work at different periods of the cardiac cycle. The first mechanism works during systole to recruit Ca2+ sparks; the second mechanism works during diastole to prevent Ca2+ sparks.
Materials and Methods
Myocyte isolation
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of Loyola University, and comply with United States regulations on animal experimentation. Ventricular myocytes were isolated from hearts of New Zealand White rabbits (2–2.5 kg) according to the procedure described previously (18). Rabbits were anesthetized with sodium pentobarbital (50 mg/kg I.V.). Next, thoracotomy hearts were quickly excised, mounted on a Langendorff apparatus, and retrogradely perfused with Liberase (Roche Applied Science, Indianapolis, IN) Blendzyme-containing solution at 37°C. Digested tissue was then minced, filtered, and washed in a minimum essential medium solution containing Ca2+ (50 μM) and bovine serum albumin (10 mg/ml). Chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. All experiments were performed at room temperature (20–24°C).
Intra-SR [Ca2+] measurements
[Ca2+]SR was recorded with the low affinity Ca2+ indicator Fluo-5N (Molecular Probes/Invitrogen, Carlsbad, CA). To load the SR with Ca2+ indicator, myocytes were incubated with 5 μM Fluo-5N/AM for 2.5 h at 37°C in Tyrode solution (in mM: NaCl 140; KCl 4; CaCl2 2; MgCl2 1; glucose 10; HEPES 10; pH 7.4) as described before (18,19). [Ca2+]SR was measured using scanning confocal microscopy (Radiance 2000 MP, Bio-Rad, UK and LSM 410, Zeiss, Germany) according to the protocol described previously (7,20). Fluo-5N was excited with the 488 nm line of an argon laser and fluorescence was collected at >515 nm. To improve the signal/noise ratio of the low intensity Fluo-5N signal, the fluorescence was recorded with an open pinhole and averaged over the entire cellular width of the two-dimensional image (pixel size 0.2 μm). Action potentials were induced by electrical field stimulation using a pair of platinum electrodes, which were connected to a Grass stimulator (Astro-Med.).
Changes in [Ca2+]SR were calculated by the equation (21): [Ca2+]SR = Kd × R/(Kd/[Ca2+]SR diast - R + 1), where R was the normalized Fluo-5N fluorescence (R = [F-Fmin]/[F0-Fmin]); F0 and Fmin were the fluorescence level at rest and after depletion of the SR with caffeine, respectively; Kd (Fluo-5N Ca2+ dissociation constant) was 390 μM based on in situ calibrations (7), and diastolic [Ca2+]SR at 1 Hz was 1000 μM (18,22). To confirm that in our conditions diastolic [Ca]SR is also 1000 μM, Fluo-5N was calibrated by previously described protocol (22). Fmin was measured after depletion of the SR with caffeine (10 mM) and Fmax was measured during a Fluo-5N saturation induced by adrenergic stimulation in low [Na+] Tyrode solution. To prevent irreversible cell contraction, myocytes were pretreated with blebbistatin (10 μM). The Fluo-5N signal was converted to [Ca] using the formula: [Ca]SR = Kd × (F-Fmin)/(Fmax-F). From analysis of 10 cells, we estimated that [Ca]SR at 1 Hz was 988 ± 51 μM. The rate of [Ca2+]SR decline during rest was measured as the changes of total [Ca2+]SR ([Ca2+]SRT) over time (d[Ca2+]SRT/dt). [Ca2+]SRT was calculated as [Ca2+]SRT = Bmax/(1+Kd/[Ca2+]SR) + [Ca2+]SR; where Bmax and Kd were 2700 and 630 μM, respectively (23). The rate of [Ca2+]SR decline was plotted as a function of [Ca2+]SR for each time point (15 s).
Cytosolic [Ca2+] measurements
To record [Ca2+]i we used the high affinity Ca2+ indicator Fluo-4 (Molecular Probes/Invitrogen). To load the cytosol with Ca2+ indicator, cells were incubated at room temperature with 10 μM Fluo-4/AM for 15 min in Tyrode solution, followed by a 20 min wash. Fluo-4 was excited with the 488 nm line of an argon laser and fluorescence was measured at >515 nm. Fluo-4 images were acquired in line scan mode (3 ms per scan; pixel size 0.12 μm). Action potentials were induced by electrical field. Ca2+ sparks were detected and analyzed during a period of rest using SparkMaster (24). SR Ca2+ load was measured from the peak amplitude of the [Ca2+]i transient induced by the rapid application of 10 mM caffeine. This concentration of caffeine fully activates RyRs (25) and leads to the synchronized release of the total Ca2+ stored in the SR.
[Mg2+]i measurements
Ventricular myocytes were loaded with Mag-Fluo-4/AM (20 μM; Molecular Probes) for 20 min in Tyrode solution (26). Mag-Fluo-4 was excited at 488 nm and emitted fluorescence was collected at wavelengths >515 nm. Two-dimensional images were acquired at 15 s intervals. Changes of [Mg2+]i are presented as background-subtracted normalized fluorescence (F/F0). In our previous study, we found that changes in diastolic [Ca2+]i did not affect the measured Mag-Fluo-4 signal, because Ca2+ waves with a small amplitude (∼3 ΔF/F0) or Ca2+ sparks could not be detected with this indicator (26).
Myocyte detubulation
Detubulation was induced by osmotic shock according to the previously described protocol (27). This protocol has been used extensively to study the role of the TTS in regulation of [Ca2+]i (12,13,28,29). Myocytes were incubated in a solution containing (in mM): NaCl 113; KCl 5; Mg2SO4 1; CaCl2 1; Na2HPO4 1; sodium acetate 20; glucose 10; HEPES 10 and insulin 5 U/l; pH was adjusted to 7.4 by NaOH. Osmotic shock was induced by adding formamide (1.5 M) to this solution. After exposure for 15 min to the formamide-containing solution, the cells were resuspended in normal Tyrode solution. The level of detubulation was evaluated after staining the surface membrane with the voltage-sensitive fluorescent dye Di-8-ANEPPS. Control and detubulated myocytes were incubated with Tyrode solution containing 5 μM of Di-8-ANEPPS for 15 min at room temperature. The Di-8-ANEPPS fluorescence was excited at 488 nm and the emitted signal was recorded at >600 nm. The density of TTS was measured by integrating the Di-8-ANEPPS signal (excluding the sarcolemma).
Statistics
Data are presented as mean ± SE of n measured cells. Statistical comparisons between two groups were performed by the Student’s t-test. Significance between multiple groups was determined by two-way analysis of variance followed by a Newman-Keuls post-hoc test. Differences were considered statistically significant at P < 0.05.
Results
Rest-dependent [Ca2+]SR decline during SERCA inhibition
Changes in [Ca2+]SR were measured with Fluo-5N in control conditions and after SERCA inhibition with thapsigargin (TG). In control cells, cessation of electrical pacing (1 Hz) caused a gradual decline of [Ca2+]SR to full depletion (Fig. 1 A). After depletion, the SR could be replenished with Ca2+ to the prerest level by a brief period of electrical stimulation (5–7 s). These results indicate that SERCA is highly active during electrical stimulation. When the experiments were performed in the presence of TG (10 μM), [Ca2+]SR decline during rest only moderately increased (Fig. 1 B). In control conditions the full [Ca2+]SR depletion occurred after 6.9 ± 0.8 min (n = 12), whereas in TG-treated cells after 5.1 ± 0.8 min (n = 12). After SR Ca2+ depletion, electrical stimulation failed to increase [Ca2+]SR indicating complete SERCA inhibition by TG (Fig. 1 B). To accurately estimate SERCA contribution to SR Ca2+ balance during rest, changes of total [Ca2+]SR ([Ca2+]SRT) over time (d[Ca2+]SRT/dt) were analyzed as a function of the corresponding [Ca2+]SR (Fig. 1 C). SERCA inhibition increased the rate of [Ca2+]SR decline by 24 ± 3% (measured at 760 μM [Ca2+]SR).
Figure 1.
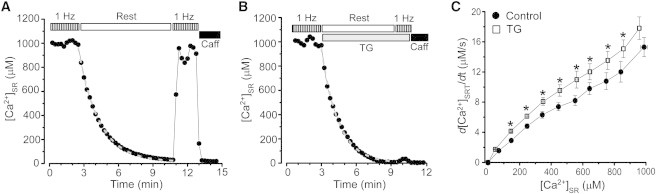
Effect of SERCA inhibition on rest-dependent [Ca2+]SR decline. The decline of [Ca2+]SR during rest in control conditions (A) and after SERCA inhibition with TG (10 μM; B). Before rest, myocytes were electrically stimulated at the constant rate of 1 Hz. Application of 10 mM caffeine at the end of the experiment caused complete depletion of [Ca2+]SR. During the period of rest, the experimental points were fitted with a single exponential function (dashed gray lines). (C) The rate of [Ca2+]SR decline during rest as a function of diastolic [Ca2+]SR measured in control conditions (●) and after SERCA inhibition (□). ∗P < 0.05 vs. control.
We obtained similar results by measuring SR Ca2+ load from the amplitude of cytosolic Ca2+ transient during caffeine (10 mM) application. Because TG irreversibly inhibits SERCA, changes in SR Ca2+ load can be measured only at one time point in the presence of TG. Fig. 2, A and B, show changes in SR Ca2+ load after 3 min of rest in control conditions and after SERCA inhibition, correspondingly. At this time point, SR Ca2+ load decreased to 43 ± 3% (n = 12) of the prerest level in control conditions and to 32 ± 3% (n = 9; P < 0.05) in the presence of TG (Fig. 2 C). The application of caffeine after 8 min did not cause any increase in [Ca2+]i, indicating that SR Ca2+ load was fully depleted (data not shown). These results suggest that SERCA inhibition accelerates the decline of SR Ca2+ load during rest by 24%. SERCA inhibition caused an increase of [Ca2+]i during rest by 16 ± 5% (n = 9). Changes in [Ca2+]i were compared before the first and second caffeine transients. A relatively small contribution of SERCA to [Ca2+]SR balance during rest can be explained if the majority of Ca2+ that leaks from the SR is extruded from the cytosol before it can be sequestered back into the SR by SERCA. This can occur if SR Ca2+ predominantly leaks into a relatively restricted space of the dyadic cleft where Ca2+ extrusion systems are located in the T-tubule membrane.
Figure 2.
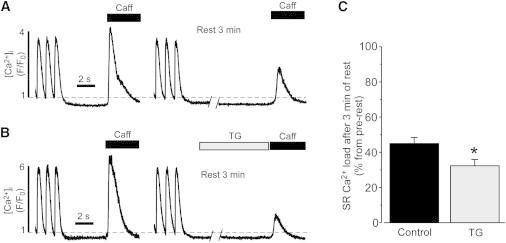
Effect of SERCA inhibition on the loss of SR Ca2+ load during rest. Decreases in SR Ca2+ load after 3 min of rest in control conditions (A) and after SERCA inhibition with TG (10 μM; B). SR Ca2+ load was measured from the amplitude of the caffeine-induced Ca2+ transient. Before rest, myocytes were electrically stimulated at the constant rate of 1 Hz. C, Summary results of decreases in SR Ca2+ load after 3 min of rest in control conditions and after SERCA inhibition with TG. ∗P < 0.05 vs. control.
Rest-dependent [Ca2+]SR decline in detubulated myocytes
It has been shown that in adult ventricular myocytes Ca2+ extrusion mechanisms such as NCX or PMCA are located in the TTS (12,13). Thus, we tested whether detubulation of myocytes would decrease [Ca2+]SR decline during rest. Ventricular myocytes were detubulated by an osmotic shock (27). This approach has been successfully used to study the role of the TTS in regulation of SR Ca2+ release in ventricular myocytes (12,13,27–29). Because the majority of LTCCs are also located in the TTS (30), complete removal of T-tubules would prevent loading of the SR with Ca2+. Thus, we studied SR Ca2+ handling only in partially detubulated cells. Changes in the density of the TTS were measured with Di-8-ANEPPS (Fig. 3 A). After applying the detubulation protocol for 15 min, the cell size (length and width) did not significantly change but the Di-8-ANEPPS signal (excluding the sarcolemmal signal) decreased by 53 ± 7% (n = 27; P < 0.05). Measurements of caffeine-induced Ca2+ release revealed that SR Ca2+ load is 10% (n = 7; P < 0.05) lower in detubulated than in control myocytes. This 10% value was subsequently used to adjust Fluo-5N fluorescence in detubulated myocytes. Similar to control myocytes, detubulated cells were able to maintain steady-state SR Ca2+ load during electrical stimulation (Fig. 3 B). When electrical stimulation was interrupted, [Ca2+]SR in detubulated myocytes declined at a much slower rate (black symbols) compared to control myocytes (gray dashed line). On average, the rate of [Ca2+]SR decline during rest decreased by 61 ± 8% (n = 10) after detubulation. However, a decrease in the TTS did not affect SR Ca2+ leak (e.g. [Ca2+]SR decline during SERCA inhibition). The Ca2+ leak rate measured at [Ca2+]SR = 760 μM was 10.7 ± 1.1 μM/s (n = 12) in control and 11.9 ± 1.4 μM/s (n = 6) after myocyte detubulation. Thus, disruption of the TTS increases the SERCA contribution in [Ca2+]SR balance, presumably by decreasing the competition of Ca2+ extrusion mechanisms for cytosolic Ca2+.
Figure 3.
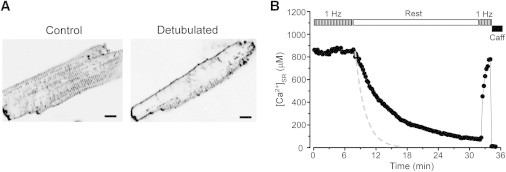
Effect of myocyte detubulation on rest-dependent [Ca2+]SR decline. (A) Images of control and detubulated myocytes stained with Di-8-ANEPPS. Calibration bar corresponds to 10 μm. (B) The decline of [Ca2+]SR during rest in detubulated myocytes. Before rest, myocytes were electrically stimulated at the constant rate of 1 Hz. Application of 10 mM caffeine at the end of the experiment caused complete depletion of the SR. For comparison, the dashed line shows the rate of [Ca2+]SR decline in control myocytes.
Rest-dependent [Ca2+]SR decline during NCX inhibition
In the following experiments we studied whether inhibition of NCX can prevent [Ca2+]SR decline during rest. NCX was inhibited by applying Tyrode solution where Ca2+ was omitted and Na+ was replaced by Li+ (0[Na+]/0[Ca2+] solution). The myocytes were electrically stimulated at 1 Hz until NCX was inhibited by 0[Na+]/0[Ca2+] solution. Inhibition of NCX significantly slowed [Ca2+]SR decline during rest, but did not prevent the overall [Ca2+]SR loss (Fig. 4 A). Relationships between the rate of [Ca2+]SR decline and SR Ca2+ load in control conditions and during NCX inhibition are illustrated in Fig. 4 B. The inhibition of NCX decreased the rest-dependent [Ca]SR decline by 69 ± 6% (n = 15). Similar results were obtained after inhibition of NCX with Ni2+ (10 mM; data not shown). NCX inhibition also increased [Ca2+]i during rest by 46 ± 5% (n = 12). Interestingly, the rate of rest-dependent [Ca]SR decline during NCX inhibition was similar to [Ca2+]SR decline recorded in detubulated myocytes (Fig. 3 B). These results suggest that NCX located in T-tubules effectively competes with SERCA for Ca2+ that leaks from the SR during rest. These findings also indicate that other mechanisms besides NCX participate in [Ca2+]i removal.
Figure 4.
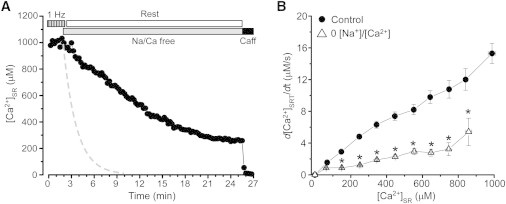
Effect of NCX inhibition on rest-dependent [Ca2+]SR decline. (A) The decline of [Ca2+]SR during rest after inhibition of NCX with 0[Na+]/0[Ca2+] solution (Na/Ca free). Before rest, myocytes were electrically stimulated at the constant rate of 1 Hz. Application of 10 mM caffeine at the end of the experiment caused complete depletion of [Ca2+]SR. The dashed line shows the rate of [Ca2+]SR decline in control myocytes. (B) The rate of [Ca2+]SR decline during rest as a function of diastolic [Ca2+]SR measured in control conditions (●) and after NCX inhibition (▵).∗P < 0.05 vs. control.
We tested to what extent PMCA contributes to [Ca2+]i extrusion in rabbit ventricular myocytes. The PMCA inhibitor eosin cannot be used because this compound has strong fluorescence at the Fluo-5N excitation settings. Thus, we used several different approaches to inhibit PMCA. We measured changes of [Ca2+]SR during rest after combined inhibition of NXC by 0[Na+]/0[Ca2+] solution and PMCA by La3+. We did not observe any additional inhibitory effect on rest-mediated [Ca2+]SR decline when La3+ (500 μM) was added to 0[Na+]/0[Ca2+] solution (data not shown). Moreover, a PMCA inhibitory peptide caloxin 2A1 (500 μM) did not affect [Ca2+]SR decline during rest (data not shown). These data suggest that PMCA plays a very minor role in Ca2+ handling in rabbit ventricular myocytes.
Rest-dependent [Ca2+]SR decline during mitochondrial depolarization
In the following experiments we determined the role of mitochondria in SR Ca2+ handling during rest. To eliminate mitochondrial Ca2+ buffering, membrane potential on the inner membrane (ΔΨ) was depolarized by the protonophore carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (2 μM). Collapse of ΔΨ can cause accumulation of cytosolic Mg2+ as a result of ATP hydrolysis by the F0F1-ATPase working in reverse mode (26). Because Mg2+ can directly inhibit RyR-mediated Ca2+ leak and prevent [Ca2+]SR decline during rest, FCCP was applied after pre-treatment of myocytes with the F0F1-ATPase inhibitor oligomycin (1 μg/ml) for 6 min. Using a similar approach to measure intracellular [Mg2+] (26), we found that oligomycin effectively prevents cytosolic Mg2+ accumulation during FCCP application (Fig. 5 A). We found that oligomycin did not change diastolic [Ca2+]SR during electrical stimulation (Fig. 5 B). This can be explained by the fact that ventricular myocytes contain a sufficient cytosolic energy reserve to maintain SERCA activity for the duration of the experiment. The subsequent application of FCCP increased [Ca2+]SR by 19 ± 4% (n = 8), suggesting that Ca2+ released from depolarized mitochondria is pumped into the SR by SERCA. The consecutive inhibition of NXC prevented [Ca2+]SR decline during rest (Fig. 5 B). [Ca2+]SR decline as a function of SR Ca2+ load at different experimental conditions are shown in Fig. 5 C. The simultaneous inhibition of NCX and the mitochondrial function increased [Ca2+]i during rest by 105 ± 8% (n = 7). These results indicate that the combined inhibition of mitochondrial Ca2+ buffering and Ca2+ extrusion by NCX prevents the rest-dependent [Ca2+]SR decline.
Figure 5.
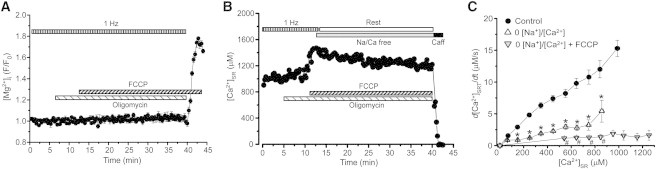
Effects of mitochondria depolarization and NCX inhibition on [Mg2+]i and rest-dependent [Ca2+]SR decline. (A) Effect of FCCP on [Mg2+]i in the presence of oligomycin (1 μg/ml). Myocyte was electrically stimulated at the constant rate of 1 Hz. [Mg2+]i was measured during diastole. (B) The decline of [Ca2+]SR during rest after depolarization of the mitochondrial membrane potential by the protonophore FCCP (2 μM) and inhibition of NCX by 0[Na+]/0[Ca2+] solution (Na/Ca free). Before the FCCP application, myocytes were pretreated with the F0F1-ATPase inhibitor oligomycin (1 μg/ml) for 6 min. Application of 10 mM caffeine at the end of the experiment caused complete depletion of [Ca2+]SR. (C) The rate of [Ca2+]SR decline during rest as a function of diastolic [Ca2+]SR measured in control conditions (●), after NCX inhibition (▵), and simultaneous NCX inhibition and mitochondria depolarization (▿).∗P < 0.05 vs. control. #P < 0.05 vs. NCX inhibition.
Ca2+ sparks during NCX inhibition and in detubulated myocytes
We found that during rest Ca2+ sparks are very rare events in control conditions (Fig. 6, A and B). At the same time, the SR exhibits a substantial Ca2+ leak rate (Fig. 1 C). These results suggest that in control conditions the majority of SR Ca2+ leak in intact myocytes occurs as nonspark-mediated Ca2+ leak. Low Ca2+ spark frequency can be explained by an effective Ca2+ extrusion from the dyadic cleft by NCX. This mechanism keeps dyadic [Ca2+] at low level, suppressing local CICR within a RyR cluster. To prove this idea, we measured Ca2+ spark activity during NCX inhibition. We found that inhibition of NCX by 0[Na+]/0[Ca2+] solution promotes spontaneous SR Ca2+ release events in forms of sparks and waves (Fig. 6 A). The occurrence of spontaneous SR Ca release events were not due to an increase in SR Ca2+ load, because the switching from normal Tyrode to 0[Na+]/0[Ca2+] solution did not increase [Ca2+]SR during rest (Fig. 4 A). Detubulation also promoted Ca2+ sparks and waves in normal Tyrode solution during rest (Fig. 6, B and C).
Figure 6.
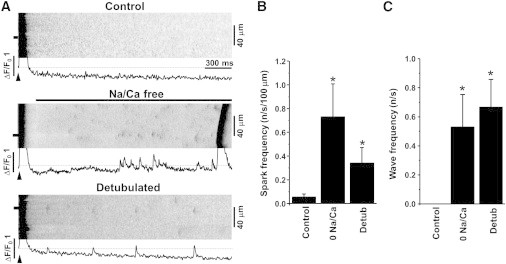
Effects of NCX inhibition and detubulation on Ca2+ sparks and waves. (A) Example line scan images and F/F0 profiles of Ca2+ transients, sparks, and waves recorded in control conditions, after inhibition of NCX with 0[Na+]/0[Ca2+] solution (Na/Ca free), and in detubulated myocytes. Action potentials were induced at time marked by arrowheads. The Ca2+ spark and wave profiles were obtained by averaging fluorescence from the 3 μm wide region marked by the black boxes. Summary data of Ca2+ spark (B) and Ca2+ wave frequency (C). ∗P < 0.05 vs. control.
Discussion
This study was directed at understanding mechanisms that control Ca2+ handling during rest in rabbit ventricular myocytes. These mechanisms play a key role in setting SR Ca2+ load and thus the amplitude of Ca2+ transient that initiate contraction. By measuring [Ca2+]SR decline during rest in control conditions and during SERCA inhibition, we found that ∼76% of leaked SR Ca2+ is extruded from the cytosol and only ∼24% is pumped back into the SR (Fig. 1). These results can be explained if the majority of SR Ca2+ leak occurs via junctional RyRs that are located closely to NCX at dyads. Thus, Ca2+ that leaks from the SR would be preferentially extruded from the dyadic cleft by NCX before it can diffuse into the cytosol (Fig. 7 A). Supporting this idea, high-resolution optical imaging has revealed that a significant portion of RyR clusters are colocalized with NCXs (14). We also found that physical dissociation of T-tubules from the junctional SR increases the contribution of SERCA to [Ca2+]SR balance during rest (Fig. 3 B), suggesting that after detubulation the leaked Ca2+ becomes more accessible for SERCA.
Figure 7.
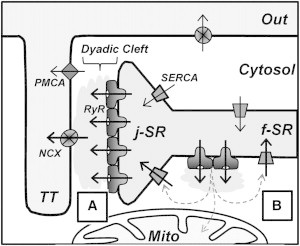
Mechanisms of SR Ca2+ leak and cytosolic Ca2+ removal in a ventricular myocyte. The diagram illustrates the local organization of different components of Ca2+ regulation in the T-tubule and in the SR at the dyad, with respect to the RyR, the NCX, the SERCA, and the PMCA. LTCCs, which are predominantly localized in the TT are not shown in this diagram. RyRs are concentrated as large clusters in the junctional SR (j-SR), facing the dyadic cleft. Ca2+ extrusion systems, such as NCX and PMCA, are located in the T-tubule membrane. SERCA and nonjunctional RyRs are distributed throughout the network of free SR (f-SR). Mitochondria (Mito) predominantly occupy the cytosolic space around the SR network. (A) SR Ca2+ that leaks via junctional RyRs would be preferentially extruded from the dyadic cleft by NCX. Such control of dyadic [Ca2+] would limit local CICR within a RyR cluster, thus preventing Ca2+ sparks. (B) SR Ca2+ that leaks via nonjunctional RyRs would be preferentially pumped back into the SR by SERCA.
If the majority of SR Ca2+ leak arises via junctional RyRs that are responsible for Ca2+ spark generation, why are sparks rare events during rest in rabbit ventricular myocytes? The absence of Ca2+ sparks can be explained if SR Ca2+ leak is mainly composed of unsynchronized openings of individual RyRs in a release cluster (31). This component of Ca2+ leak has been previously described as nonspark SR Ca2+ leak (7,32). A recent mathematical model proposed that the entire SR Ca2+ leak can be explained by RyR activity organized in junctional release clusters (33). At high diastolic [Ca2+]SR (>500 μM), a single opening of the RyR can increase [Ca2+]i in the dyadic cleft to the level that can trigger a spark. At low diastolic [Ca2+]SR (>500 μM), Ca2+ flux during RyR openings is insufficient to activate neighboring channels. However, results of this study revealed that other mechanisms than SR Ca2+ load should play a role in suppressing Ca2+ sparks, because in rabbit ventricular myocytes diastolic [Ca2+]SR is ∼1000 μM (18,22). Measurements of [Ca2+]SR decline during rest suggest that Ca2+ extrusion by NCX would effectively limit [Ca2+] accumulation in the dyadic cleft, thus preventing local CICR and Ca2+ spark generation. To prove this idea, we measured Ca2+ sparks during NCX inhibition. We found that 0[Na+]/0[Ca2+] solution promotes spontaneous SR Ca2+ release events in forms of sparks and waves (Fig. 6). The occurrence of spontaneous SR Ca2+ release was not due to an increase in SR Ca2+ load, because inhibition of NCX did not increase diastolic [Ca2+]SR during rest (Fig. 4 A). Because a significant fraction of NCX is localized in T-tubules (12,13), we tested whether Ca2+ spark frequency is higher in detubulated myocytes. We found that detubulation significantly increased Ca2+ spark activity. Based on these results, we proposed a new, to our knowledge, mechanism that controls local CICR. In addition to a positive control of junctional RyRs by LTCC (4), we proposed a novel, to our knowledge, mechanism of negative control by NCX. These two mechanisms work at different periods of the cardiac cycle. The positive control recruits Ca2+ sparks during systole; the negative control prevents Ca2+ sparks during diastole. In support of our results, a recent mathematical model showed that NCX located in the dyadic cleft can effectively suppress Ca2+ sparks even at high diastolic [Ca2+]SR (34).
We found that only 24% of SR Ca2+ that leaks during rest is resequestered into the SR by SERCA. This can still be explained if the entire SR Ca2+ leak occurs via junctional RyRs, but NCX in the dyadic cleft is able to extrude only 76% of the leaked Ca2+. The remaining Ca2+ diffuses into the cytosol where it is pumped into the SR by SERCA. However, we found that augmentation of NCX activity by increasing the Na+ gradient (after depletion of [Na+]i) did not increase the rate of [Ca2+]SR decline during rest (data not shown). This implies that even in control conditions the rate of Ca2+ extrusion by NCX is significantly higher than SR Ca2+ leak. Thus, during rest NCX can effectively prevent a spillover of Ca2+ from the dyadic cleft into the cytosol. It can be that a smaller fraction of SR Ca2+ leak that is sensitive to SERCA inhibition arises from nonjunctional RyRs (Fig. 7 B). It has been shown that in ventricular myocytes a certain fraction of RyRs is distributed outside of dyads as small clusters (14,35) or isolated channels (36). Based on the results of this work and previous structural studies (11,14,35), we suggest that in adult ventricular myocytes there are two distinct pathways of SR Ca2+ leak that affect Ca2+ homeostasis in different ways. Ca2+ that leaks via the junctional RyRs tunnels from the cell via the dyadic cleft, without affecting [Ca2+]i. It seems that this SR Ca2+ leak component controls only [Ca2+]SR and thus serves as an important safety mechanism that can unload SR without causing cytosolic Ca2+ overload. In contrast, the nonjunctional Ca2+ leak affects both diastolic [Ca2+]SR and [Ca2+]i. Thus, this Ca2+ leak pathway may participate in regulation of Ca2+-dependent mechanisms in the cytosol, including mitochondrial metabolism. Mitochondria occupy the cytosolic space around the free SR network (11,37), therefore they can compete for the leaked Ca2+ with SERCA. Indeed, we found that simultaneous inhibition of mitochondrial Ca2+ buffering and Ca2+ extrusion by NCX prevents [Ca2+]SR decline during rest (Fig. 5), thus Ca2+ that leaks from the SR can be effectively pumped back by SERCA.
The mechanisms of Ca2+ handling described in this study are relevant to a diastolic phase when [Ca2+]i and [Ca2+]SR return to presystolic levels. At low diastolic [Ca2+]i, Ca2+ that leaks via RyRs would preferentially stimulate closely located Ca2+ transporters. Because the majority of SR Ca2+ leak occurs via junctional RyRs that are colocalized with NCXs at dyads, the exchange would be more efficient to remove cytosolic Ca2+ during rest than SERCA. As a result of this dyadic organization, Ca2+ that leaks from the SR would preferentially leave the cell (causing total cellular Ca2+ loss). This mechanism plays an important role in decreasing contractile force at low heart rate. Furthermore, effective Ca2+ extrusion from the dyadic cleft by NCX would limit local CICR within a RyR cluster, thus preventing Ca2+ sparks and arrhythmogenic Ca2+ waves. During systole, however, membrane depolarization during the plateau phase of the AP would not only prevent Ca2+ extrusion but stimulate Ca2+ entry into the dyadic cleft via the reverse mode of NCX (16). This mechanism would further facilitate CICR initiated by LTCC Ca2+ current. Thus, depending on the membrane potential during the cardiac cycle, the NCX can either promote or prevent SR Ca2+ release.
Acknowledgments
This work was supported by the National Institutes of Health grants HL62426 and HL75494 to P.d.T. and The Research Career Development Award from The Schweppe Foundation to A.V.Z.
References
- 1.Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983;245:C1–C14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- 2.Franzini-Armstrong C., Protasi F., Ramesh V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys. J. 1999;77:1528–1539. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng H., Lederer W.J., Cannell M.B. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 4.Stern M.D. Theory of excitation-contraction coupling in cardiac muscle. Biophys. J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neary P., Duncan A.M., Smith G.L. Assessment of sarcoplasmic reticulum Ca2+ flux pathways in cardiomyocytes from rabbits with infarct-induced left-ventricular dysfunction. Pflugers Arch. 2002;444:360–371. doi: 10.1007/s00424-002-0794-0. [DOI] [PubMed] [Google Scholar]
- 6.Shannon T.R., Ginsburg K.S., Bers D.M. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ. Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- 7.Zima A.V., Bovo E., Blatter L.A. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J. Physiol. 2010;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bers D.M. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2001. Excitation-Contraction Coupling and Cardiac Contractile Force. [Google Scholar]
- 9.Bassani J.W., Bassani R.A., Bers D.M. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J. Physiol. 1994;476:279–293. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baddeley D., Jayasinghe I.D., Soeller C. Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc. Natl. Acad. Sci. USA. 2009;106:22275–22280. doi: 10.1073/pnas.0908971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayashi T., Martone M.E., Hoshijima M. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J. Cell Sci. 2009;122:1005–1013. doi: 10.1242/jcs.028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chase A., Orchard C.H. Ca efflux via the sarcolemmal Ca ATPase occurs only in the t-tubules of rat ventricular myocytes. J. Mol. Cell. Cardiol. 2011;50:187–193. doi: 10.1016/j.yjmcc.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 13.Yang Z., Pascarel C., Orchard C.H. Na+-Ca2+ exchange activity is localized in the T-tubules of rat ventricular myocytes. Circ. Res. 2002;91:315–322. doi: 10.1161/01.res.0000030180.06028.23. [DOI] [PubMed] [Google Scholar]
- 14.Jayasinghe I.D., Cannell M.B., Soeller C. Organization of ryanodine receptors, transverse tubules, and sodium-calcium exchanger in rat myocytes. Biophys. J. 2009;97:2664–2673. doi: 10.1016/j.bpj.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lines G.T., Sande J.B., Sejersted O.M. Contribution of the Na+/Ca2+ exchanger to rapid Ca2+ release in cardiomyocytes. Biophys. J. 2006;91:779–792. doi: 10.1529/biophysj.105.072447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Litwin S.E., Li J., Bridge J.H. Na-Ca exchange and the trigger for sarcoplasmic reticulum Ca release: studies in adult rabbit ventricular myocytes. Biophys. J. 1998;75:359–371. doi: 10.1016/S0006-3495(98)77520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Viatchenko-Karpinski S., Terentyev D., Györke S. Synergistic interactions between Ca2+ entries through L-type Ca2+ channels and Na+-Ca2+ exchanger in normal and failing rat heart. J. Physiol. 2005;567:493–504. doi: 10.1113/jphysiol.2005.091280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Domeier T.L., Blatter L.A., Zima A.V. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J. Physiol. 2009;587:5197–5209. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zima A.V., Picht E., Blatter L.A. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ. Res. 2008;103:e105–e115. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bovo E., Lipsius S.L., Zima A.V. Reactive oxygen species contribute to the development of arrhythmogenic Ca²+ waves during β-adrenergic receptor stimulation in rabbit cardiomyocytes. J. Physiol. 2012;590:3291–3304. doi: 10.1113/jphysiol.2012.230748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cannell M.B., Cheng H., Lederer W.J. Spatial non-uniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Biophys. J. 1994;67:1942–1956. doi: 10.1016/S0006-3495(94)80677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shannon T.R., Guo T., Bers D.M. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ. Res. 2003;93:40–45. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- 23.Shannon T.R., Ginsburg K.S., Bers D.M. Reverse mode of the sarcoplasmic reticulum calcium pump and load-dependent cytosolic calcium decline in voltage-clamped cardiac ventricular myocytes. Biophys. J. 2000;78:322–333. doi: 10.1016/S0006-3495(00)76595-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Picht E., Zima A.V., Bers D.M. SparkMaster: automated calcium spark analysis with ImageJ. Am. J. Physiol. Cell Physiol. 2007;293:C1073–C1081. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- 25.Rousseau E., Meissner G. Single cardiac sarcoplasmic reticulum Ca2+-release channel: activation by caffeine. Am. J. Physiol. 1989;256:H328–H333. doi: 10.1152/ajpheart.1989.256.2.H328. [DOI] [PubMed] [Google Scholar]
- 26.Zima A.V., Pabbidi M.R., Blatter L.A. Effects of mitochondrial uncoupling on Ca2+ signaling during excitation-contraction coupling in atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 2013;304:H983–H993. doi: 10.1152/ajpheart.00932.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brette F., Komukai K., Orchard C.H. Validation of formamide as a detubulation agent in isolated rat cardiac cells. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H1720–H1728. doi: 10.1152/ajpheart.00347.2002. [DOI] [PubMed] [Google Scholar]
- 28.Brette F., Despa S., Orchard C.H. Spatiotemporal characteristics of SR Ca2+ uptake and release in detubulated rat ventricular myocytes. J. Mol. Cell. Cardiol. 2005;39:804–812. doi: 10.1016/j.yjmcc.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 29.Fowler M.R., Dobson R.S., Harrison S.M. Functional consequences of detubulation of isolated rat ventricular myocytes. Cardiovasc. Res. 2004;62:529–537. doi: 10.1016/j.cardiores.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 30.Kawai M., Hussain M., Orchard C.H. Excitation-contraction coupling in rat ventricular myocytes after formamide-induced detubulation. Am. J. Physiol. 1999;277:H603–H609. doi: 10.1152/ajpheart.1999.277.2.H603. [DOI] [PubMed] [Google Scholar]
- 31.Porta M., Zima A.V., Fill M. Single ryanodine receptor channel basis of caffeine’s action on Ca2+ sparks. Biophys. J. 2011;100:931–938. doi: 10.1016/j.bpj.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santiago D.J., Curran J.W., Shannon T.R. Ca sparks do not explain all ryanodine receptor-mediated SR Ca leak in mouse ventricular myocytes. Biophys. J. 2010;98:2111–2120. doi: 10.1016/j.bpj.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato D., Bers D.M. How does stochastic ryanodine receptor-mediated Ca leak fail to initiate a Ca spark? Biophys. J. 2011;101:2370–2379. doi: 10.1016/j.bpj.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sato D., Despa S., Bers D.M. Can the sodium-calcium exchanger initiate or suppress calcium sparks in cardiac myocytes? Biophys. J. 2012;102:L31–L33. doi: 10.1016/j.bpj.2012.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lukyanenko V., Ziman A., Lederer W.J. Functional groups of ryanodine receptors in rat ventricular cells. J. Physiol. 2007;583:251–269. doi: 10.1113/jphysiol.2007.136549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sobie E.A., Guatimosim S., Lederer W.J. The Ca 2+ leak paradox and rogue ryanodine receptors: SR Ca 2+ efflux theory and practice. Prog. Biophys. Mol. Biol. 2006;90:172–185. doi: 10.1016/j.pbiomolbio.2005.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramesh V., Sharma V.K., Franzini-Armstrong C. Structural proximity of mitochondria to calcium release units in rat ventricular myocardium may suggest a role in Ca2+ sequestration. Ann. N. Y. Acad. Sci. 1998;853:341–344. doi: 10.1111/j.1749-6632.1998.tb08295.x. [DOI] [PubMed] [Google Scholar]