

Abstract
Work in heterologous systems has revealed that members of the Rad, Rem, Rem2, Gem/Kir (RGK) family of small GTP-binding proteins profoundly inhibit L-type Ca2+ channels via three mechanisms: 1), reduction of membrane expression; 2), immobilization of the voltage-sensors; and 3), reduction of Po without impaired voltage-sensor movement. However, the question of which mode is the critical one for inhibition of L-type channels in their native environments persists. To address this conundrum in skeletal muscle, we overexpressed Rad and Rem in flexor digitorum brevis (FDB) fibers via in vivo electroporation and examined the abilities of these two RGK isoforms to modulate the L-type Ca2+ channel (CaV1.1). We found that Rad and Rem both potently inhibit L-type current in FDB fibers. However, intramembrane charge movement was only reduced in fibers transfected with Rad; charge movement for Rem-expressing fibers was virtually identical to charge movement observed in naïve fibers. This result indicated that Rem supports inhibition solely through a mechanism that allows for translocation of CaV1.1’s voltage-sensors, whereas Rad utilizes at least one mode that limits voltage-sensor movement. Because Rad and Rem differ significantly only in their amino-termini, we constructed Rad-Rem chimeras to probe the structural basis for the distinct specificities of Rad- and Rem-mediated inhibition. Using this approach, a chimera composed of the amino-terminus of Rem and the core/carboxyl-terminus of Rad inhibited L-type current without reducing charge movement. Conversely, a chimera having the amino-terminus of Rad fused to the core/carboxyl-terminus of Rem inhibited L-type current with a concurrent reduction in charge movement. Thus, we have identified the amino-termini of Rad and Rem as the structural elements dictating the specific modes of inhibition of CaV1.1.
Introduction
In addition to its function as voltage-sensor for excitation-contraction (EC) coupling in skeletal muscle, the 1,4-dihydropyridine receptor (CaV1.1) also conducts L-type Ca2+ current (Tanabe et al. (1); for a review, see Bannister and Beam (2)). Although the physiological significance of Ca2+ flux via CaV1.1 has been the subject of much debate since the 1960s, work over the last 10 years has revealed that L-type Ca2+ entry maintains myoplasmic Ca2+ levels during repetitive activity (3,4), promotes development of neuromuscular junctions (5), and augments muscle contraction (6). Moreover, the reemergence of a high-conductance embryonic CaV1.1 splice variant in muscle of myotonic dystrophy patients indicates that alterations in L-type channel gating almost certainly have pathological consequences (7). Thus, modulation of CaV1.1 activity may profoundly affect skeletal muscle function.
Members of the Rad, Rem, Rem2, Gem/Kir (RGK) family of monomeric G proteins inhibit L-type Ca2+ channels in a variety of physiological systems via interactions with both channel α1 and β-subunits ((8–13); reviewed recently by Yang and Colecraft (14)). In collaboration with others, our group has documented that overexpression of Rem in normal developing myotubes substantially reduces L-type current and lessens charge movement (15). We have recently found that Rad and Gem also inhibit L-type currents in myotubes (16).
Although the earlier work with cultured primary myotubes has provided useful basic information regarding the function of RGK proteins, these cell types are not always suitable models for investigating mechanisms that regulate excitability in fully differentiated skeletal muscle. In this study, we have overcome this confounding factor by examining the impact of RGK proteins on CaV1.1 channel function in adult mouse flexor digitorum brevis (FDB) fibers overexpressing either Rad or Rem via in vivo electroporation (17). Using this approach, we have found that Rad and Rem modulate CaV1.1 via distinct mechanisms in mature fibers. Specifically, Rad inhibits L-type current by utilizing at least one mechanism that reduces charge movement (i.e., decreased channel membrane expression and/or voltage-sensor immobilization), whereas Rem exclusively employs a mode of inhibition that allows significant voltage-sensor translocation (i.e., low channel Po). In addition, we have employed a chimeric strategy to identify the amino-termini of Rad and Rem as the structural elements that dictate these distinct modes of CaV1.1 inhibition in differentiated skeletal muscle.
Materials and Methods
In vivo electroporation and dissociation of FDB fibers
All procedures involving mice were approved by the University of Colorado Denver-Anschutz Medical Campus Institutional Animal Care and Use Committee. cDNA plasmids encoding YFP (Clontech, Mountain View, CA) or a Venus-mouse RGK protein fusion construct (V-Rad and V-Rem; GenBank accession Nos. NP_062636 and NP_033073, respectively; both generous gifts from Drs. S. R. Ikeda and H. J. Puhl, III) were delivered to FDB fibers of anesthetized 2–3-month-old male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) via an in vivo electroporation protocol similar to that originally described by DiFranco et al. (17). Briefly, 10 μL of 2 mg/mL hyaluronidase solution was injected into the FDB muscle with a 30-gauge hypodermic needle. After 1 h, mice were reanesthetized and 20 μL of cDNA (3–5 μg/μL) was injected into the muscle. Then, after 5 min, two gold-plated acupuncture needle electrodes (Lhasa OMS, Weymouth, MA) coupled to an isolated pulse stimulator (A-M Systems, Sequim, WA) were placed subcutaneously near the proximal and distal tendons of the muscle (∼1 cm apart). cDNAs were then electroporated into the FDB muscle with twenty 100-V, 20-ms pulses delivered at 1 Hz.
Naïve and electroporated (9–10 days posttransfection) FDB muscles were dissected in cold Rodent Ringer’s solution (146 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, pH 7.4 with NaOH). Muscles were then digested in a mild collagenase solution (155 mM Cs-aspartate, 10 mM HEPES, 5 mM MgCl2, pH 7.4 with CsOH, supplemented with 1 mg/mL BSA and 1 mg/mL collagenase type IA; both from Sigma-Aldrich, St. Louis, MO) with agitation at 37°C for ∼60 min. Immediately after digestion, the collagenase solution was replaced with a dissociation solution (140 mM Cs-aspartate, 10 mM Cs2EGTA, 10 mM HEPES, 5 mM MgCl2, pH 7.4 with CsOH, supplemented with 1 mg/mL BSA) and muscles were triturated gently with a series of fire-polished glass pipettes of descending bore. Dissociated FDB fibers were then plated onto ECL-attachment-matrix (Millipore, Lake Placid, NY)-coated 35-mm plastic culture dishes (Falcon, San Jose, CA). Experiments were performed with FDB fibers 1–6 h after dissociation.
Measurement of L-type Ca2+ currents and charge movements
Patch pipettes were fabricated from borosilicate glass and had resistances of ≤1.0 MΩ when filled with internal solution, which consisted of 140 mM Cs-aspartate, 10 mM Cs2-EGTA, 5 mM MgCl2, and 10 mM HEPES, pH 7.4 with CsOH; fibers were dialyzed in the whole-cell configuration for >20 min before recording.
For recording of L-type Ca2+ currents, the external solution contained 145 mM tetraethylammonium (TEA)-methanesulfonic acid, 2 mM CaCl2, 10 mM HEPES, 2 mM MgCl2, 1 mM 4-aminopyridine, 0.1 mM anthracene-9-carboxylic acid, and 0.002 mM tetrodotoxin, pH 7.4 with TEA-OH.
For measurement of charge movements, the bath contained 145 mM TEA-methanesulfonic acid, 10 mM CaCl2, 10 mM HEPES, 2 mM MgCl2, 1 mM 4-aminopyridine, 0.1 mM anthracene-9-carboxylic acid, 0.002 mM tetrodotoxin, 1 mM LaCl3, and 0.5 mM CdCl2, pH 7.4 with TEA-OH.
Linear components of leak and capacitive current were corrected with −P/4 online subtraction protocols. Output filtering was at 2–5 kHz and digitization was either at 5 kHz (currents) or 10 kHz (charge movements). Cell capacitance was determined by integration of a transient from −80 mV to −70 mV using CLAMPEX 10.3 (Molecular Devices, Sunnyvale, CA) and was used to normalize charge movement (nC/μF) and current amplitude (pA/pF). The average value of Cm was 2.16 ± 0.06 nF (n = 193 fibers). To minimize voltage error, the time constant for decay of the whole-cell capacity transient (τm) was reduced as much as possible using the analog compensation circuit of the amplifier; the average values of τm and Ra were 1.08 ± 0.04 ms and 755 ± 43 kΩ, respectively. Current-voltage (I/V) curves were fitted according to
| (1) |
where I is the normalized current for the test potential V, Vrev is the reversal potential, Gmax is the maximum Ca2+ channel conductance, V1/2 is the half-maximal activation potential, and kG is the slope factor. QON was then normalized to Cm and plotted as a function of test potential (V) and the resultant Q-V relationships were fitted according to
| (2) |
where Qmax is the maximal QON, VQ is the potential causing movement of one-half the maximal charge, and kQ is a slope parameter. All experiments were performed at room temperature (∼25°C).
Molecular biology
V-Rad/Rem chimeras were created by swapping the regions encoding the amino-termini (NT) of the RGK proteins. Specifically, residues 1–85 of Rad were fused to residues 76–297 of Rem to make chimera V-RadNT-Rem and residues 1–75 of Rem were fused to residues 86–307 of Rad to make chimera V-RemNT-Rad (see Fig. 3, later in text). Both chimeras retained the Venus fluorescent protein affixed to their respective amino-termini.
Figure 3.
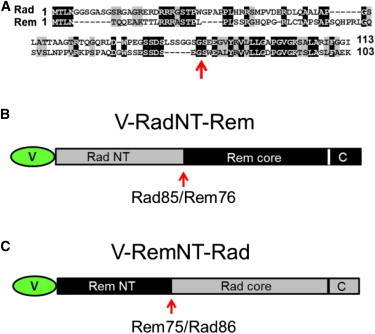
V-Rad/V-Rem chimeras. (A) Sequence comparison of the amino-terminal domains of Rad and Rem. (Black boxes) Residues of mouse Rad (GenBank Accession No. NP_062636) identical to those of mouse Rem (GenBank Accession No. NP_033073); (shaded boxes) residues conserved between the two RGK proteins. Please note the relative lack of conservation between the amino-termini. Schematic representations of V-RadNT-Rem (B) and V-RemNT-Rad (C). Both constructs were fused to Venus fluorescent protein via their amino-termini. (A–C, red arrow) Locus for the chimeric swap of the amino-termini (between residues G85 and S86 for Rad and residues G75 and S76 for Rem). To see this figure in color, go online.
To make the chimeras, a silent BamHI recognition site was introduced into both V-Rad and V-Rem parent vectors using the Change-IT site-directed mutagenesis kit (Affymetrix, Santa Clara, CA); a separate BamHI site was already present in either plasmid shortly after the Rad or Rem stop-codon. The sequences of the 5′phosphorylated mutagenesis primers were
for V-Rad and V-Rem, respectively. The reverse primer sequence was
for both RGK proteins. For V-RadNT-Rem, a 674-bp fragment was removed from the mutated V-Rem expression plasmid (5600 bp) via BamHI digestion. This segment of V-Rem replaced the corresponding 673-bp BamHI/BamHI fragment removed from the V-Rad expression plasmid. V-RemNT-Rad was constructed using a similar strategy where a 673-bp fragment was removed from the V-Rad expression plasmid (5629 bp) by BamHI digestion. This segment of V-Rad replaced the corresponding 674-bp BamHI/BamHI fragment removed from the V-Rem expression plasmid. The integrity of each construct was verified by restriction analysis and sequencing.
V-RemcoreCT was comprised of residues 76–297 of mouse Rem with Venus fluorescent protein affixed again to the amino-terminus. To make V-RemcoreCT, the V-Rem construct carrying the silent BamHI site (see above) was digested with BamHI and BglII, splitting the plasmid into three fragments. The 246-bp-long BglII-BamHI fragment, which encoded the V-Rem linker region and the first 75 residues of Rem was discarded. The 674 bp BamHI-BamHI fragment, corresponding to Rem residues 76–297, was ligated to the third fragment (4680 bp), which consisted of the vector backbone and the Venus coding sequence. Ligation was possible because the BglII restriction overhang of the third fragment was compatible to the ends of the 674 bp BamHI-BamHI fragment.
Analysis
All data are presented as mean ± SE. Statistical comparisons were made by one-way ANOVA followed by a Dunnett’s post hoc test with p < 0.05 considered significant. Figures were made using the software program SIGMAPLOT (Ver. 11.0; SSPS, San Jose, CA).
Results
Rad and Rem reduce L-type current density in FDB fibers
Rad and Rem are the two RGK protein isoforms known to be endogenously expressed in skeletal muscle (18–22). To investigate the influence of Rad and Rem on the biophysical properties of the L-type Ca2+ channel in differentiated skeletal muscle, we used in vivo electroporation (17) to transfect FDB muscles of otherwise normal 2–3-month-old mice with either YFP or a Venus-fused RGK protein construct (i.e., V-Rad and V-Rem). Live cell imaging showed successful transfection of FDB fibers with all three constructs (see Fig. S1 In the Supporting Material).
Subsequently, L-type Ca2+ currents were recorded in the whole-cell configuration from naïve and transfected FDB fibers to determine whether Rad and/or Rem also inhibit the channel function of CaV1.1 in adult skeletal muscle (23–25). In control experiments, naïve FDB fibers produced robust L-type currents that were not different than those observed in fibers expressing YFP (−5.8 ± 0.5 pA/pF; n = 19 and −5.3 ± 0.5 pA/pF; n = 12, respectively, at +10 mV; p > 0.05; Fig. 1 A). By comparison, L-type currents were reduced by ∼60% and ∼45% in fibers expressing V-Rad (−2.4 ± 0.4 pA/pF; n = 9; p < 0.001; Fig. 1 B) or V-Rem (−3.2 ± 0.2 pA/pF; n = 11; p < 0.001; Fig. 1 C), respectively.
Figure 1.
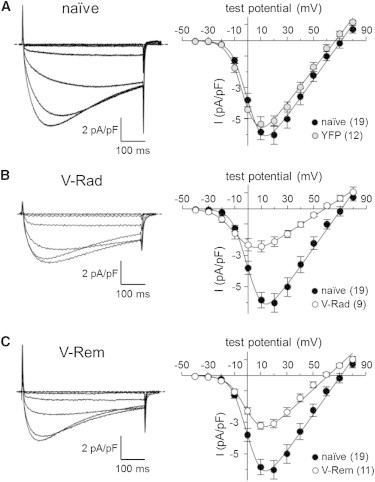
Rad and Rem reduce L-type Ca2+ current in FDB fibers. Representative recordings of skeletal muscle L-type currents elicited by 500-ms depolarizations from −50 mV to −30 mV, −20 mV, −10 mV, 0 mV, +10 mV, and +20 mV are shown for a naïve FDB fiber (A, left panel) and fibers expressing V-Rad (B, left panel) or V-Rem (C, left panel). (A–C, right panels) The peak I/V relationships corresponding to each current family (in A, right panel, the peak I/V relationship for fibers expressing unfused YFP is also shown). Currents were evoked at 0.1 Hz by test potentials ranging from −40 mV through +80 mV in 10-mV increments. (Smooth curves) Plotted according to Eq. 1 with fit parameters displayed in Table 1. Throughout, error bars represent ±SE.
Intramembrane charge movements in FDB fibers are reduced by Rad, but are not affected by Rem
Next, we recorded membrane-bound charge movements to investigate the possibility that the RGK protein-mediated reductions in L-type current amplitude were a consequence of impaired voltage-sensor function. Predictably, naïve and YFP-expressing FDB fibers both produced substantial and nearly equivalent charge movement (Qmax = 25.5 ± 1.7 nC/μF; n = 35 and 23.8 ± 2.1 nC/μF; n = 15, respectively; p > 0.05; Fig. 2 A; Table 1). In the case of fibers expressing V-Rad, charge movements were reduced by nearly 40% relative to control fibers (Qmax = 16.4 ± 1.0 nC/μF; n = 23; p < 0.001; Fig. 2 B; Table 1). In stark contrast, fibers expressing V-Rem displayed charge movement virtually identical to control and YFP-expressing fibers (Qmax = 23.5 ± 1.2 nC/μF; n = 22; p > 0.05; Fig. 2 C; Table 1). The unaltered Q-V relationship for fibers expressing V-Rem indicates that V-Rem inhibits CaV1.1 almost exclusively by a mechanism that allows movement of the voltage sensors. Importantly, this mechanism is fundamentally different from the inhibition produced by Rad, which is characterized by a reduction in charge movement.
Figure 2.
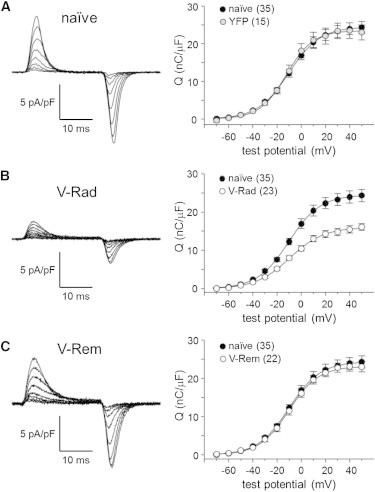
Intramembrane charge movements in FDB fibers are reduced by Rad, but are not affected by Rem. Representative recordings of intramembrane charge movements elicited by 25-ms depolarizations from −80 mV to −50 mV, −40 mV, −30 mV, −20 mV, −10 mV, 0 mV, +10 mV, and +20 mV are shown for a naïve FDB fiber (A, left) and transfected fibers expressing V-Rad (B, left panel) or V-Rem (C, left panel). (A–C, right panels) The Q-V relationships corresponding to each current family. (A, right panel) The Q-V relationship for fibers expressing nonfused YFP is also shown. Charge movements were evoked at 0.1 Hz by test potentials ranging from −70 mV through +50 mV in 10-mV increments. (Smooth curves) Plotted according to Eq. 2 with fit parameters displayed in Table 1.
Table 1.
L-type conductance and intramembrane charge movement
|
G-V |
Q-V |
||||||
|---|---|---|---|---|---|---|---|
| Gmax (nS/nF) | V1/2 (mV) | kG (mV) | Vrev (mV) | Qmax (nC/μF) | VQ (mV) | kQ (mV) | |
| Naïve | 124 ± 12 (19) | 0.7 ± 1.4 | 4.9 ± 0.4 | 69.1 ± 2.3 | 25.5 ± 1.7 (35) | −9.7 ± 2.5 | 11.8 ± 0.8 |
| YFP | 120 ± 11 (12) | −0.9 ± 24 | 5.2 ± 0.5 | 66 ± 1.1 | 23.8 ± 2.1 (15) | −11.4 ± 1.8 | 11.0 ± 0.7 |
| V-Rad | 63 ± 10a (9) | −4.2 ± 5.3 | 5.6 ± 0.4 | 60.6 ± 2.0b | 16.4 ± 1.0c (23) | −10.5 ± 2.3 | 12.5 ± 0.9 |
| V-Rem | 76 ± 7a (11) | −1.4 ± 1.8 | 5.7 ± 0.5 | 60.5 ± 2.1b | 23.5 ± 1.2 (22) | −10 ± 2.2 | 10.8 ± 0.6 |
| V-RadNT-Rem | 86 ± 5b (11) | 2.4 ± 1.8 | 6.5 ± 0.2b | 59.2 ± 1.5b | 14.8 ± 1.9a (8) | −3.6 ± 3.4 | 11.4 ± 1.0 |
| V-RemNT-Rad | 66 ± 6a (6) | 7.0 ± 5.0 | 7.3 ± 0.4b | 53.2 ± 2.6c | 22.0 ± 1.7 (9) | −8.4 ± 2.8 | 11.9 ± 1.0 |
| V-RemcoreCT | 77 ± 6b (7) | 8.6 ± 1.7 | 5.1 ± 1.5 | 60.2 ± 4.4 | 21.9 ± 2.8 (6) | −10.7 ± 1.8 | 11.6 ± 1.2 |
Data are given as mean ± SE, with the numbers in parentheses indicating the number of FDB fibers tested. Conductance and charge movement data were fit by Eqs. 1 and 2, respectively. Significant differences between naïve fibers and fibers expressing either YFP or a particular V-RGK construct are indicated.
p < 0.005; one-way ANOVA, followed by a Dunnett’s post hoc test.
p < 0.05; one-way ANOVA, followed by a Dunnett’s post hoc test.
p < 0.001; one-way ANOVA, followed by a Dunnett’s post hoc test.
The amino-terminus of Rad is a critical element for reducing charge movement
Multiple modes of RGK protein-mediated inhibition of L-type channels have been identified:
-
1.
Reduction of channel membrane expression;
-
2.
Immobilization of the voltage-sensors; and
-
3.
Reduced channel Po without impeded voltage-sensor movement (11,12,14).
Our unexpected results demonstrating that Rem inhibits CaV1.1 function in adult muscle by only the latter mechanism enabled the investigation of structures responsible for the differential modes of CaV1.1 inhibition by Rad and Rem. To do this, we adopted a believed-novel chimeric approach designed to convey Rad function to Rem, and vice versa.
Examination of the peptide sequences of Rad and Rem revealed little sequence similarity between the amino-termini (34.2% identity; Fig. 3 A), whereas the combined Ras-like core and carboxyl-termini domains were fairly conserved (68.2% identical; comparison not shown). We constructed two Venus-tagged chimeras based on this information (Fig. 3, B and C). The first chimera, V-RadNT-Rem, consisted of the amino-terminal 85 residues of Rad affixed to the core/carboxyl-terminus of Rem (residues 76–297). The other chimera, V-RemNT-Rad, constituted the near mirror image of the first chimera with the amino-terminal 75 residues of Rem fused to the core/carboxyl-terminus of Rad (residues 86–307). Like V-Rad and V-Rem, both chimeras were readily expressed in FDB fibers (see Fig. S1).
In electrophysiological experiments, both L-type currents (−3.1 ± 0.2 pA/pF; n = 12; Fig. 4 A) and charge movements (Qmax = 14.8 ± 1.9 nC/μF; n = 8; Fig. 4 B) were reduced in fibers expressing V-RadNT-Rem relative to naïve control fibers (p < 0.001 and p < 0.005, respectively; Table 1). In particular, the ∼45% reduction in charge movement was similar to that observed for wild-type V-Rad (p > 0.05; Table 1), indicating the amino-terminus of Rad as a critical element for Rad-mediated inhibition of CaV1.1 function in adult skeletal muscle. On the other hand, V-RemNT-Rad inhibited L-type current (−1.8 ± 0.2 pA/pF; n = 6; p < 0.001 versus naïve fibers; Fig. 5 A) without greatly affecting charge movement (Qmax = 22.0 ± 1.7 nC/μF; n = 9; p > 0.05 versus naïve fibers; Fig. 5 B). A Rem-based construct lacking the 75 amino-terminal residues of wild-type Rem (V-RemcoreCT) also had little effect on the magnitude of charge movement (Qmax = 21.9 ± 2.8 nC/μF; n = 6; p > 0.05 versus naïve fibers), indicating that amino-terminus of Rad actively participates in the mode(s) of channel inhibition that reduce voltage-sensor translocation in differentiated skeletal muscle (Table 1).
Figure 4.
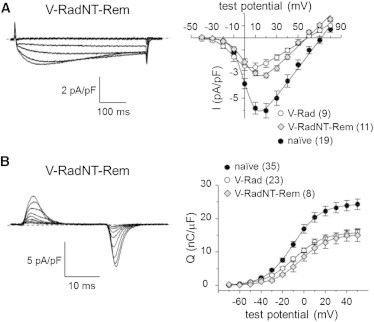
The amino-terminus of Rad supports modes of inhibition that reduce intramembrane charge movement. Representative recordings of (A, left panel) L-type currents and (B, left panel) intramembrane charge movements obtained from FDB fibers expressing V-RadNT-Rem. (A and B, right panels) The I-V and Q-V relationships corresponding to each current family. Peak I-V and Q-V data for naïve FDB fibers and fibers expressing V-Rad are replotted from Figs. 1 and 2, respectively.
Figure 5.
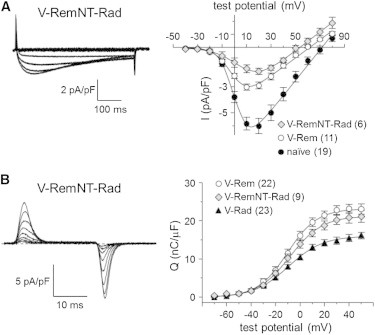
The amino-terminus of Rem allows the low Po mode of channel inhibition. Representative recordings of (A, left panel) L-type currents and (B, left panel) intramembrane charge movements obtained from FDB fibers expressing V-RemNT-Rad. (A and B, right panels) The I-V and Q-V relationships corresponding to each current family. Peak I/V data for naïve FDB fibers and fibers expressing V-Rem are replotted from Fig. 1. Q-V data for fibers expressing either V-Rem or V-Rad are replotted from Fig. 2.
Discussion
During this study, we found that both V-Rad and V-Rem reduced L-type Ca2+ flux when overexpressed in mature mouse FDB fibers via in vivo electroporation (Fig. 1). However, only V-Rad reduced maximal gating charge movement (Fig. 2). The ability to reduce charge movement was conferred to Rem by replacing its amino-terminus with that of Rad (Fig. 4). Conversely, Rad lost the ability to reduce charge movement when the Rem amino-terminus was fused to its core/carboxyl-terminus (Fig. 5). Thus, our results indicate that Rad and Rem both inhibit CaV1.1 function in adult skeletal muscle, but do so by utilizing different mechanisms.
RGK proteins are well known to inhibit L-type channels by three distinct mechanisms:
-
1.
Decreased channel membrane expression;
-
2.
Immobilization of the voltage-sensors; and
-
3.
Stabilization of a low Po gating mode (11,12,14).
Of these three modes of inhibition, only low Po mode, mechanism 3, is thought not to greatly affect intramembrane charge movement. Our findings indicate that Rad employs at least one mode of inhibition that reduces maximal charge movement. Unfortunately, our methods do not enable us to distinguish prudently whether Rad reduces charge movement by decreasing junctional channel expression, or does so by deterring translocation of the voltage-sensing particles of the channel. Even so, our results clearly indicate that Rem inhibits native CaV1.1 channels purely through the low Po mechanism.
The isolation of the low Po mode in Rem-expressing FDB fibers enabled us to probe the structural elements that determine the modes of action employed by Rem and Rad to inhibit native CaV1.1 channels. We focused on the amino-termini of Rem and Rad as the principal determinants for the divergent regulation of CaV1.1 charge movement because of the relatively low conservation between the amino-termini of the two RGK proteins (Fig. 3). Previous studies utilizing heterologous expression systems and cultured neurons have demonstrated that the RGK core region fused to the carboxyl-terminus is sufficient to support profound inhibition of high voltage-activated Ca2+ currents (11,26–29). By swapping the amino-termini of Rem and Rad, we were able to switch the mode of channel inhibition, thereby revealing what is believed to be a novel mechanism for RGK protein function (Figs. 4 and 5). In view of these results, one might suspect that the amino-terminus of Rad houses elements that are critical for the following:
-
1.
Intermolecular interactions with trafficking proteins that promote removal of the channel from the plasma membrane, or
-
2.
Interactions with the channel that limit charge movement by locking the voltage-sensors in a given position.
In regard to the second possibility, the amino-termini of Rad and other RGK protein isoforms are known to interact with scaffolding proteins that are involved in trafficking (e.g., Béguin et al. (30–32)).
Voltage-gated Na+ and K+ channels undoubtedly contributed some gating charge to our recordings. However, the collective contribution of these other channels to the total charge moved is very difficult to gauge because mice null for CaV1.1 do not live long enough for their fibers to differentiate (33). For this reason, we were unable to perform similar experiments in FDB fibers to those of Adams et al. (34), which revealed the non-CaV1.1-dependent component of charge movement in myotubes. Still, one may conclude that the non-CaV1.1 component of charge movement was equivalent for control (naïve or YFP-expressing fibers) and RGK protein-expressing fibers in our experiments because RGK proteins are specific inhibitors of HVA Ca2+ channels (8–10,35). Based on this reasonable assumption, the effect of Rad on charge movement attributable to CaV1.1 is probably underestimated. Thus, the abilities of the two RGK proteins to affect CaV1.1 charge movement may actually be more divergent than we report.
Rad and Rem are also expressed in the myocardium and both of these RGK protein isoforms potently inhibit cardiac EC coupling (35–40) which, unlike EC coupling in skeletal muscle, is directly dependent on Ca2+ current conducted by CaV1.2 L-type channels. The impact of Rad on charge movement of cardiomyocytes has not been described, but Rem inhibits endogenous CaV1.2 channels predominantly through the low Po mechanism in these cells (35). In this earlier work, L-type current and charge movement were both substantially reduced (∼90 and ∼33%, respectively) in cardiomyocytes infected with adenovirus encoding Rem. Yet, immunostaining indicated that channel density at the plasma membrane was similar to that of cardiomyocytes overexpressing a non-membrane-targeted, and therefore inert, Rem control construct. The latter observation largely ruled out diminished CaV1.2 membrane expression as the mechanism responsible for the reduction of L-type current density, leaving only the electrically silent, voltage-sensor immobile and the low-Po, voltage-sensor mobile populations of Rem-inhibited channels as the potential sources of current reduction. Because the dihydropyridine agonist Bay K 8644 restored L-type current density through its ability to promote higher Po gating modes, the low Po population of Rem-associated channels was judged to be predominantly responsible for inhibition of the current. Although such an interpretation provides a solid explanation for the Rem-mediated reduction in L-type current amplitude in cardiomyocytes, the source of the reduction in charge movement remains unresolved.
As noted above, only paltry charge movement has been reported in HEK293 cells coexpressing Rem and CaV1.2 channels (11,12) (R. A. Bannister, D. Beqollari, and U. Meza, unpublished). Because both Gem and Rem do not reduce charge movement as profoundly in rodent ventricular myocytes (10,35), the preservation of a component of charge movement in Rem-expressing FDB fibers would not have been unexpected (this was the case for Rad; see Fig. 2). Yet, we were astonished to see the total lack of effect of Rem on charge movement in differentiated fibers in view of our earlier results in which overexpression of a YFP-Rem clone in cultured myotubes reduced CaV1.1 charge movement by 44% (15). Although the reason(s) for the differential effects of Rem on charge movement in developing myotubes and fully differentiated FDB fibers is (are) not entirely clear, our results underscore the necessity of investigating the biophysical impact of RGK proteins and other muscle signaling molecules in their natural environment. In normal healthy muscle, RGK proteins are unlikely to have a great impact on basal CaV1.1 function because of low protein levels (21,22) (R. A. Bannister and C. F. Romberg, unpublished). However, Rad-mediated inhibition of CaV1.1 may contribute to excitability deficits in a wide spectrum of degenerative muscle disorders/conditions. Specifically, Rad expression has been found to be increased in skeletal muscle of the mdx mouse model of muscular dystrophy (21), in denervated mouse muscle (22), in muscle of amyotrophic lateral sclerosis patients (22), and in muscle of type II diabetics (Reynet and Kahn (18); but see also Paulik et al. (20)).
Acknowledgments
We thank Ms. W. A. H. Sumner and Mr. M. P. Scheele for technical assistance. We also thank Drs. S. R. Ikeda and H. L. Puhl, III for sharing the cadre of Venus-wild-type RGK protein expression plasmids and Dr. K. G. Beam for continued support.
This work was supported by startup funding from the University of Colorado Denver-AMC Department of Medicine-Cardiology Division and by grants from the National Institutes of Health (No. AG038778 to R.A.B.), the Colorado Clinical and Translational Sciences Institute (grant No. J-11-122 to R.A.B.), and the Consejo Nacional de Ciencia y Tecnología (grant No. 169006 to U.M.). D.B. received a stipend from grant No. 2T32AG000279-11 (to R. S. Schwartz, University of Colorado Denver-AMC Department of Medicine-Geriatrics Division). Confocal images were acquired in the University of Colorado Denver-AMC Advanced Light Microscopy Core (funded in part by National Institutes of Health/National Center for Research Resources Colorado Clinical and Translational Science Institute grant No. UL1 RR025780).
Supporting Material
References
- 1.Tanabe T., Beam K.G., Numa S. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 1988;336:134–139. doi: 10.1038/336134a0. [DOI] [PubMed] [Google Scholar]
- 2.Bannister R.A., Beam K.G. CaV1.1: the atypical prototypical voltage-gated Ca2+ channel. Biochem. Biophys. Acta Biomembr. 2013;1828:1587–1597. doi: 10.1016/j.bbamem.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cherednichenko G., Hurne A.M., Pessah I.N. Conformational activation of Ca2+ entry by depolarization of skeletal myotubes. Proc. Natl. Acad. Sci. USA. 2004;101:15793–15798. doi: 10.1073/pnas.0403485101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bannister R.A., Pessah I.N., Beam K.G. The skeletal L-type Ca2+ current is a major contributor to excitation-coupled Ca2+ entry. J. Gen. Physiol. 2009;133:79–91. doi: 10.1085/jgp.200810105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen F., Liu Y., Lin W. Neuromuscular synaptic patterning requires the function of skeletal muscle dihydropyridine receptors. Nat. Neurosci. 2011;14:570–577. doi: 10.1038/nn.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mosca B., Delbono O., Zorzato F. Enhanced dihydropyridine receptor calcium channel activity restores muscle strength in JP45/CASQ1 double knockout mice. Nat. Commun. 2013;4:1541. doi: 10.1038/ncomms2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang Z.Z., Yarotskyy V., Thornton C.A. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of CaV1.1 calcium channel. Hum. Mol. Genet. 2012;21:1312–1324. doi: 10.1093/hmg/ddr568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Béguin P., Nagashima K., Seino S. Regulation of Ca2+ channel expression at the cell surface by the small G-protein Kir/Gem. Nature. 2001;411:701–706. doi: 10.1038/35079621. [DOI] [PubMed] [Google Scholar]
- 9.Finlin B.S., Crump S.M., Andres D.A. Regulation of voltage-gated calcium channel activity by the Rem and Rad GTPases. Proc. Natl. Acad. Sci. USA. 2003;100:14469–14474. doi: 10.1073/pnas.2437756100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murata M., Cingolani E., Marbán E. Creation of a genetic calcium channel blocker by targeted gem gene transfer in the heart. Circ. Res. 2004;95:398–405. doi: 10.1161/01.RES.0000138449.85324.c5. [DOI] [PubMed] [Google Scholar]
- 11.Yang T., Xu X., Colecraft H.M. Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J. Physiol. 2010;588:1665–1681. doi: 10.1113/jphysiol.2010.187203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang T., Puckerin A., Colecraft H.M. Distinct RGK GTPases differentially use α1- and auxiliary β-binding-dependent mechanisms to inhibit CaV1.2/CaV2.2 channels. PLoS ONE. 2012;7:e37079. doi: 10.1371/journal.pone.0037079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meza U., Beqollari D., Bannister R.A. Potent inhibition of L-type Ca2+ currents by a Rad variant associated with human cardiac hypertrophy. Biochem. Biophys. Res. Commun. 2013;439:270–274. doi: 10.1016/j.bbrc.2013.08.044. [DOI] [PubMed] [Google Scholar]
- 14.Yang T., Colecraft H.M. Regulation of voltage-dependent calcium channels by RGK proteins. Biochim. Biophys. Acta Biomembr. 2013;1828:1644–1654. doi: 10.1016/j.bbamem.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bannister R.A., Colecraft H.M., Beam K.G. Rem inhibits skeletal muscle EC coupling by reducing the number of functional L-type Ca2+ channels. Biophys. J. 2008;94:2631–2638. doi: 10.1529/biophysj.107.116467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Romberg C.F., Beqollari D., Bannister R.A. RGK protein-mediated impairment of slow depolarization-dependent Ca2+ entry into developing myotubes. Channels. 2014;8:3. doi: 10.4161/chan.27686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiFranco M., Capote J., Vergara J.L. Voltage-dependent dynamic FRET signals from the transverse tubules in mammalian skeletal muscle fibers. J. Gen. Physiol. 2007;130:581–600. doi: 10.1085/jgp.200709831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reynet C., Kahn C.R. Rad: a member of the Ras family overexpressed in muscle of type II diabetic humans. Science. 1993;262:1441–1444. doi: 10.1126/science.8248782. [DOI] [PubMed] [Google Scholar]
- 19.Finlin B.S., Andres D.A. Rem is a new member of the Rad- and Gem/Kir Ras-related GTP-binding protein family repressed by lipopolysaccharide stimulation. J. Biol. Chem. 1997;272:21982–21988. doi: 10.1074/jbc.272.35.21982. [DOI] [PubMed] [Google Scholar]
- 20.Paulik M.A., Hamacher L.L., Lenhard J.M. Identification of Rad’s effector-binding domain, intracellular localization, and analysis of expression in Pima Indians. J. Cell. Biochem. 1997;65:527–541. [PubMed] [Google Scholar]
- 21.Hawke T.J., Kanatous S.B., Garry D.J. Rad is temporally regulated within myogenic progenitor cells during skeletal muscle regeneration. Am. J. Physiol. Cell Physiol. 2006;290:C379–C387. doi: 10.1152/ajpcell.00270.2005. [DOI] [PubMed] [Google Scholar]
- 22.Halter B., Gonzalez de Aguilar J.L., Loeffler J.P. Oxidative stress in skeletal muscle stimulates early expression of Rad in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2010;48:915–923. doi: 10.1016/j.freeradbiomed.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Beam K.G., Knudson C.M. Calcium currents in embryonic and neonatal mammalian skeletal muscle. J. Gen. Physiol. 1988;91:781–798. doi: 10.1085/jgp.91.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z.M., Messi M.L., Delbono O. Patch-clamp recording of charge movement, Ca2+ current, and Ca2+ transients in adult skeletal muscle fibers. Biophys. J. 1999;77:2709–2716. doi: 10.1016/s0006-3495(99)77104-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prosser B.L., Hernández-Ochoa E.O., Schneider M.F. The Qγ component of intra-membrane charge movement is present in mammalian muscle fibers, but suppressed in the absence of S100A1. J. Physiol. 2009;587:4523–4541. doi: 10.1113/jphysiol.2009.177238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen H., Puhl H.L., 3rd, Ikeda S.R. Expression of Rem2, an RGK family small GTPase, reduces N-type calcium current without affecting channel surface density. J. Neurosci. 2005;25:9762–9772. doi: 10.1523/JNEUROSCI.3111-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Correll R.N., Pang C., Andres D.A. Plasma membrane targeting is essential for Rem-mediated Ca2+ channel inhibition. J. Biol. Chem. 2007;282:28431–28440. doi: 10.1074/jbc.M706176200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang T., Suhail Y., Colecraft H.M. Genetically encoded molecules for inducibly inactivating CaV channels. Nat. Chem. Biol. 2007;3:795–804. doi: 10.1038/nchembio.2007.42. [DOI] [PubMed] [Google Scholar]
- 29.Flynn R., Chen L., Zamponi G.W. Molecular determinants of Rem2 regulation of N-type calcium channels. Biochem. Biophys. Res. Commun. 2008;368:827–831. doi: 10.1016/j.bbrc.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 30.Béguin P., Mahalakshmi R.N., Hunziker W. 14-3-3 and calmodulin control subcellular distribution of Kir/Gem and its regulation of cell shape and calcium channel activity. J. Cell Sci. 2005;118:1923–1934. doi: 10.1242/jcs.02321. [DOI] [PubMed] [Google Scholar]
- 31.Béguin P., Mahalakshmi R.N., Hunziker W. Roles of 14-3-3 and calmodulin binding in subcellular localization and function of the small G-protein Rem2. Biochem. J. 2005;390:67–75. doi: 10.1042/BJ20050414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Béguin P., Mahalakshmi R.N., Hunziker W. Nuclear sequestration of β-subunits by Rad and Rem is controlled by 14-3-3 and calmodulin and reveals a novel mechanism for Ca2+ channel regulation. J. Mol. Biol. 2006;355:34–46. doi: 10.1016/j.jmb.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 33.Beam K.G., Knudson C.M., Powell J.A. A lethal mutation in mice eliminates the slow calcium current in skeletal muscle cells. Nature. 1986;320:168–170. doi: 10.1038/320168a0. [DOI] [PubMed] [Google Scholar]
- 34.Adams B.A., Tanabe T., Beam K.G. Intramembrane charge movement restored in dysgenic skeletal muscle by injection of dihydropyridine receptor cDNAs. Nature. 1990;346:569–572. doi: 10.1038/346569a0. [DOI] [PubMed] [Google Scholar]
- 35.Xu X., Marx S.O., Colecraft H.M. Molecular mechanisms, and selective pharmacological rescue, of Rem-inhibited CaV1.2 channels in heart. Circ. Res. 2010;107:620–630. doi: 10.1161/CIRCRESAHA.110.224717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crump S.M., Correll R.N., Satin J. L-type calcium channel α-subunit and protein kinase inhibitors modulate Rem-mediated regulation of current. Am. J. Physiol. Heart Circ. Physiol. 2006;291:H1959–H1971. doi: 10.1152/ajpheart.00956.2005. [DOI] [PubMed] [Google Scholar]
- 37.Yada H., Murata M., Fukuda K. Dominant negative suppression of Rad leads to QT prolongation and causes ventricular arrhythmias via modulation of L-type Ca2+ channels in the heart. Circ. Res. 2007;101:69–77. doi: 10.1161/CIRCRESAHA.106.146399. [DOI] [PubMed] [Google Scholar]
- 38.Wang G., Zhu X., Cheng H. Rad as a novel regulator of excitation-contraction coupling and β-adrenergic signaling in heart. Circ. Res. 2010;106:317–327. doi: 10.1161/CIRCRESAHA.109.208272. [DOI] [PubMed] [Google Scholar]
- 39.Jhun B.S., O-Uchi J., Jin Z.G. Adrenergic signaling controls RGK-dependent trafficking of cardiac voltage-gated L-type Ca2+ channels through PKD1. Circ. Res. 2012;110:59–70. doi: 10.1161/CIRCRESAHA.111.254672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magyar J., Kiper C.E., Satin J. Rem-GTPase regulates cardiac myocyte L-type calcium current. Channels (Austin) 2012;6:166–173. doi: 10.4161/chan.20192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.