

Abstract
NMR spectroscopy is a principal tool in metabolomic studies and can, in theory, yield atom-level information critical for understanding biological systems. Nevertheless, NMR investigations on biological tissues generally have to contend with field inhomogeneities originating from variations in macroscopic magnetic susceptibility; these field inhomogeneities broaden spectral lines and thereby obscure metabolite signals. The congestion in one-dimensional NMR spectra of biological tissues often leads to ambiguities in metabolite identification and quantification. We propose an NMR approach based on intermolecular double-quantum coherences to recover high-resolution two-dimensional (2D) J-resolved spectra from inhomogeneous magnetic fields, such as those created by susceptibility variations in intact biological tissues. The proposed method makes it possible to acquire high-resolution 2D J-resolved spectra on intact biological samples without recourse to time-consuming shimming procedures or the use of specialized hardware, such as magic-angle-spinning probes. Separation of chemical shifts and J couplings along two distinct dimensions is achieved, which reduces spectral crowding and increases metabolite specificity. Moreover, the apparent J coupling constants observed are magnified by a factor of 3, facilitating the accurate measurement of small J couplings, which is useful in metabolic analyses. Dramatically improved spectral resolution is demonstrated in our applications of the technique on pig brain tissues. The resulting spectra contain a wealth of chemical shift and J-coupling information that is invaluable for metabolite analyses. A spatially localized experiment applied on an intact fish (Crossocheilus siamensis) reveals the promise of the proposed method in in vivo metabolite studies. Moreover, the proposed method makes few demands on spectrometer hardware and therefore constitutes a convenient and effective manner for metabonomics study of biological systems.
Introduction
NMR spectroscopy is a powerful tool for examining molecular structures and compositions, revealing a cornucopia of useful information in the guise of chemical shifts, J couplings, and multiplet patterns. This is especially evident in in vivo applications, where spectroscopic approaches can provide useful metabolic information complementary to the insight delivered by MRI for biological systems. Due to its efficacy, NMR spectroscopy has wide-ranging applications in a variety of fields (1–3). In metabonomic applications, NMR analysis enables an unbiased identification and quantification of metabolic markers of diseases, toxic insult, genetic manipulation, environmental stress, etc (4–6). One-dimensional (1D) 1H NMR is a commonly employed analytical method in this field and has the advantage of fast spectral acquisition. However, 1D NMR measurements of biological samples containing complex metabolites generally yields complicated and highly crowded spectra with overlapping signals, which significantly hinders metabolite identification and quantification. The two-dimensional J-resolved (2D JRES) experiment, represented as (π/2) − t1/2 − (π) − t1/2 + t2, solves this problem by achieving a complete separation of chemical shifts and J couplings along two distinct dimensions (7–9). Utilization of the 2D JRES technique has allowed metabolite identification in human biofluids, such as urine (10), cerebral spinal fluid (11), and blood plasma (12). However, the applicability of 2D JRES NMR is in general limited to the study of homogeneous systems, such as biofluids or tissue extracts. Commonly observed variations of macroscopic magnetic susceptibility in biological tissues impose this limitation on the conventional 2D JRES technique. Magnetic susceptibility variations produce field inhomogeneities that broaden lines along the F2 (chemical shift) dimension of 2D J-resolved spectra, concealing information necessary for metabolite identification. Although effects of field inhomogeneities can be removed in the F1 (J-coupling) dimension (8), spectral overlap in the F2 dimension may make extracting accurate J-coupling information challenging.
Field shimming is often the first option to alleviate field inhomogeneities. However, field inhomogeneities caused by variations of macroscopic magnetic susceptibility in biological samples are hardly removed by conventional field shimming methods. High-resolution 2D J-resolved spectra of biological samples can also be obtained by employing the magic-angle-spinning (MAS) technique (13,14), which involves spinning the sample around an axis oriented at the so-called magic angle, or 54.7°, relative to the static magnetic field. Although this method is able to average magnetic-field inhomogeneities arising from magnetic susceptibility variations throughout the sample volume, it requires specialized hardware and careful attention to detail in the experimental setup. The ultraslow MAS (e.g., 1.5 Hz spinning rate) has been applied to in vivo or intact biological tissues (15,16), however, removing spinning sidebands from the resulting spectra is challenging and spectral resolution is still insufficient. In view of the resolution challenges faced by experimentalists in the field of NMR metabonomics on biological systems, a great demand for high-resolution methodologies easily adapted to the commonly available spectrometer hardware features has arisen. Several modified sequences have been proposed to improve the performance of 2D JRES, such as spin-selective multiple-quantum J-resolved spectroscopy for measurements of small J couplings (17,18) and 2D J-point-resolved spectroscopy for spatially localized applications (19). However, for these techniques, it remains difficult to acquire high-resolution 2D J-resolved information from biological systems.
It has been shown that intermolecular multiple-quantum coherences (iMQCs), originating from distant dipolar interactions among spins in different molecules, can be used to improve the resolution of NMR spectra obtained in inhomogeneous fields (20–23). Both experimental and simulated results show that the iMQC signal is immune to field inhomogeneities from complex variations of macroscopic magnetic susceptibility in biological samples (24,25). We present a pulse sequence, named iDQCJRES, designed based on intermolecular double-quantum coherence (iDQC) to deliver high-resolution 2D J-resolved spectra in inhomogeneous fields. This sequence is well suited to studies of biological tissues with intense water signal and intrinsic macroscopic susceptibility variations, even spatially localized spectra for in vivo applications. The iDQCJRES approach can be easily implemented on standard NMR spectrometers without special hardware requirements. Three-dimensional (3D) acquisition is needed, but the indirect detection periods are optimized for improved acquisition efficiency. A high-resolution 2D J-resolved spectrum free from inhomogeneous line broadening can be generated using a simple data shearing and 2D projection. Experiments are performed on a chemical solution to show the detailed implementation of iDQCJRES. Demonstrations of the method applied to pig brain tissues and an intact fish are used to test the performance of iDQCJRES.
Theory
A pulse diagram of the iDQCJRES sequence is shown in Fig. 1. We consider a solution consisting of I (solvent) and S (solute) components, where I is a single spin-1/2 system, and S is an AX spin-1/2 system that includes Sk and Sl spins coupled by a Jkl scalar interaction. The evolution of two-spin order terms resulting in the desired iDQC signals can be understood intuitively, using the raising and lowering operator formalism as
| (1) |
where DISIzSz represents distant dipolar interactions for iDQCs between solvent and solute spins. In the iDQCJRES sequence, a pair of linear coherence selection gradients (CSGs) with an area ratio of 1:−2 is applied along the z direction to retain only the desired coherence transfer pathway. Two indirect detection periods, t1 and t2, are utilized and each is further divided into two equal parts to form an iDQC delay acquisition scheme and a spin-echo scheme, respectively, for the desired signal evolution. Under this signal evolution, only half the frequency range of field inhomogeneity plus J-coupling splitting needs to be covered with the t1 period, and only the frequency range of J-coupling splitting needs to be covered with the t2 period, resulting in greatly improved acquisition efficiency. Since the water suppression is indispensable for NMR measurements on biological samples, a water suppression (WS) module using the excitation sculpting (26) is added to the iDQCJRES sequence. A Localization module (27) is optionally inserted for spatially volume-localized 2D J-resolved spectroscopy.
Figure 1.
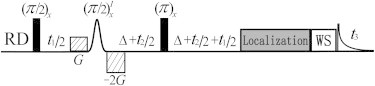
Pulse diagram of the iDQCJRES sequence for high-resolution 2D J-resolved spectra in inhomogeneous fields. WS is the module for solvent suppression, and Localization is the optional module for spatial localization. Related parameters are defined in text.
In this work, the iMQC treatments (28,29) are employed to deduce theoretical expressions for signals resulting from the iDQCJRES sequence. Assume that the solvent spin I is abundant, that ωm is the frequency offset of the m (m = I, Sk, Sl) spins in the rotating frame and in the absence of field inhomogeneity, and that ΔBm(r) is the inhomogeneous deviation of the magnetic field at the location of a particular m spin. The frequency offset, Ωm(r), of spin m at position r is given by
| (2) |
where γ is the gyromagnetic ratio. Equation 2 suggests that magnetic field inhomogeneity causes a shift of angular frequency from ωm. For simplicity, the effects of radiation damping, diffusion, relaxation, and intermolecular nuclear Overhauser effects are ignored. The behavior of the Sk spin is similar to that of the Sl spin; hence, without loss of generality, we consider only the behavior of the Sk spin. The density operator, , after the action of the iDQCJRES sequence can be written as
| (3) |
where is the Boltzmann factor, Skj represents the jth Sk spin operator, and NS is the total number of Sk spin. The dipolar demagnetizing time is defined as , in which is the equilibrium magnetization per unit volume of the I spin, and is the vacuum magnetic permeability. Then the observed signal for Sk can be shown to be
| (4) |
where is the equilibrium magnetization per unit volume of the S spin. Equation 4 provides quantitative expressions of the 3D iDQCJRES signal between solute spin, Sk, and solvent spin, I. It shows that the 3D signal is doublet and is located at and . The three coordinates of the 3D signal are labeled Ω1, Ω2, and Ω3, respectively. Note that the frequency offset of solvent with J coupling is contained in the F1 dimension, only J coupling information is retained in the F2 dimension, and the frequency offset of solute with J coupling is shown in the F3 dimension. If the spectrometer reference frequency is set to the resonant frequency of I spin, i.e., , the signal will be observed at and . Therefore, it is apparent that only half the frequency range of field inhomogeneity ΔBI(r) plus J coupling needs to be covered in the F1 dimension, and only the frequency range of J coupling needs to be covered in the F2 dimension. This allows spectral widths in the F1 and F2 dimensions to be significantly decreased, reducing experimental time and decreasing the size of the data matrix that needs to be stored. Solute and solvent spins on two molecules coupled by intermolecular dipolar interactions are physically close to each other, and therefore the magnetic field over the distance between the two spins should not vary significantly, i.e., ΔBI(r) is very nearly equal to . Although it would seem, prima facie, given the inhomogeneous broadening of both F1 and F3 dimensions, that a high-resolution 2D J-resolved spectrum cannot be extracted from 3D data obtained in this manner, a 3D shearing of the F1-F3 plane along the F3 axis will eliminate inhomogeneous line broadening along the F3 dimension, thereby facilitating the construction of a high-resolution 2D spectrum. As a result of this shearing transform, in the sheared spectrum. Thus, in the sheared 3D spectrum, the signals are finally observed at and . It is obvious that the F2 and F3 dimensions, which form the 2D J-resolved spectrum, are free from field inhomogeneities in the sheared 3D iDQCJRES spectrum. A projection of the 3D spectrum onto the F2-F3 plane produces the desired high-resolution 2D J-resolved spectrum. After the accumulated projection, analytical expressions for the signal from the Sk spin in the F2-F3 plane can be written as
| (5) |
where p is a factor related to accumulated projection. Equation 5 shows that the signal in the resulting 2D spectrum is located at and , similar to the signal observed in a conventional 2D J-resolved spectrum, located at and . The J-coupling constant in the F3 dimension is magnified threefold, which facilitates the accurate measurement of small J-coupling constants. Similar to conventional J-resolved spectroscopy, a clockwise rotation of 45° along F2 = 0 for the 2D iDQCJRES spectrum can be performed to separate chemical shifts and J couplings, resulting in and . Hence, the same spectral features observed in a conventional 2D J-resolved spectrum obtained in homogeneous fields are also observed in a 2D iDQCJRES spectrum acquired in inhomogeneous fields, but with a triple magnification of J-coupling splitting in the iDQCJRES spectrum.
Although the theoretical analyses presented above are based on a simple model sample, they should also hold for complicated biological systems, in which the abundant water is analogous to the I spin, the metabolites behave similar to the S spins, and ΔBm(r) takes the role of the field inhomogeneity originating from variations of macroscopic magnetic susceptibility. In a similar way, for a biological sample, the iDQCJRES experiment relies on the formation of iDQCs between the water and metabolite spins. As was the case before, the coupled pairs of the water and metabolite spins are physically proximate and should experience almost the same magnetic field. Therefore, field inhomogeneities will be removed along the F3 dimension in the sheared 3D iDQCJRES spectrum, and high-resolution 2D J-resolved spectral information for metabolites can be obtained.
Materials and Methods
Hardware
All experiments were performed at 298 K using a Varian NMR System 500 MHz spectrometer (Varian, Palo Alto, CA) equipped with a 5 mm 1H {15N-31P} XYZ indirect detection probe with 3D gradient coils.
Experiments on a chemical solution
A solution of butyl methacrylate (C8H14O2) in dimethylsulfoxide (DMSO) (C2H6SO) with a molar ratio of 1:8 was used to demonstrate implementation details of the iDQCJRES sequence. The magnetic field was intentionally deshimmed by randomly altering currents in shimming coils to produce broadened peaks with a line width of 200 Hz. The 2D J-resolved spectra were acquired in such an inhomogeneous field using iDQCJRES and conventional 2D JRES sequences. In addition, a conventional 2D J-resolved spectrum was acquired in a well-shimmed field as a reference. For conventional 2D JRES experiments in inhomogeneous and well-shimmed fields, the pulse repetition time was 1.5 s, the number of averages was 4, and points were acquired with spectral widths of 50 Hz × 4000 Hz (F1 × F2) in 5 min. For the iDQCJRES experiment in the inhomogeneous field, the width of a hard RF pulse was 10 μs, the solvent selective pulse was in Gaussian shape with a pulse-width of 6.0 ms, and the CSGs with strength G = 10 G/cm and duration δ = 1.2 ms were applied. The W5 binomial π pulse (30) was used as a solvent-exclusive π pulse in the WS module, and parameters for crusher gradient pulses in this module were G1 = 7 G/cm, G2 = 18 G/cm, and . The optional Localization module was unused, and no phase cycling was applied in this iDQCJRES experiment. The pulse repetition time was 1.0 s, the echo time (2Δ) was 100 ms, and 35 × 40 × 1056 points were acquired with spectral widths of 50 Hz × 140 Hz × 4000 Hz (F1 × F2 × F3) in 23.3 min. The iDQCJRES 3D data were processed using our custom-written program on MATLAB 7.8 (The MathWorks, Natick, MA). The sensitivity, which is defined as (31), was calculated for each 2D J-resolved spectrum. The resonance at 5.96 ppm and noise signals in the region between 4.7 and 5.2 ppm were used for this calculation.
Global-volume experiments on pig brain tissues
To test the ability of the iDQCJRES sequence on biological tissues with intense water signal and intrinsic macroscopic susceptibility variations, we carried out global-volume experiments on pig brain tissues fitted in a 5-mm NMR tube. Experiments were performed without field locking and shimming while the probe was well tuned to preserve high sensitivity. In the iDQCJRES experiment, the widths of hard radiofrequency (RF) pulse and the solvent-selective Gaussian pulse were 11 μs and 6.0 ms, respectively. Parameters of the CSGs were G = 10 G/cm and δ = 1.2 ms. The WS module was applied to suppress intense water signal, and parameters for crusher gradient pulses were G1 = 7 G/cm, G2 = 18 G/cm, and . A two-step phase cycling was applied: phases for the first RF pulse, the second RF pulse, and the receiver were (x, y), (x, x), and (x, −x), respectively. The pulse repetition time was 1.0 s, the echo time (2Δ) was 60 ms, and 30 × 12 × 600 points were acquired with spectral widths of 50 Hz × 100 Hz × 4000 Hz (F1 × F2 × F3) in 12 min. A conventional 2D water-presaturated JRES experiment was performed for comparison. For this experiment, the pulse repetition time was 2.0 s, the number of averages was 2, and points were acquired with spectral widths of 50 Hz × 4000 Hz (F1 × F2) in 2.7 min. To confirm results acquired from the iDQCJRES sequence, we carried out a 1D water-presaturated spin echo MAS experiment using a 1H Nano NMR probe. The sample of pig brain tissues was spun along the magic-angle direction at a rate of 2 kHz. In this 1D experiment, a T2-edited Carr-Purcell-Meiboom-Gill segment with a total echo time of 360 ms was incorporated into the pulse sequence to selectively record the signals of small metabolites in the brain tissue. The pulse repetition time was 2.0 s, the number of averages was 64, and 1100 points were acquired with a spectral width of 4000 Hz in 128 s.
Spatially localized experiments on an intact fish
To show the applicability of iDQCJRES sequence on real biological samples, we performed a postmortem study on an intact fish (Crossocheilus siamensis) of size suitable for a 5-mm NMR tube. Both iDQCJRES and existing 2D JPRESS sequences were used to detect metabolites in the fish body, with the spatially localized voxel containing the spinal cord. Before spectral measurements, spin-echo images of sagittal and axial planes were acquired with repetition time (TR)/echo time (TE) = 2500/25 ms and imaging matrix = 256 × 512 in 10.7 min. For the Localization module in the iDQCJRES experiment, three sinc-shaped selective pulses with corresponding slice-selective gradients along three orthogonal directions were applied to select the desired region of the fish. The width of sinc-shaped pulses was 2.0 ms, and parameters for three slice-selective gradients were Gx = 1.32 G/cm, Gy = 1.32 G/cm, Gz = 0.41 G/cm, and , resulting in a 4 × 4 × 13 mm3 voxel. The duration of the Localization module was 8.0 ms. A four-step phase cycling was applied: phases for the first RF pulse, the second RF pulse, and the receiver were (x, y, x, y), (x, x, −x, −x), and (x, −x, −x, x), respectively. Other parameters set for iDQCJRES were the width of the hard RF pulse, 12 μs, and the solvent-selective Gaussian pulse, 7.0 ms. Parameters of the CSGs and the WS module were the same as in the pig brain tissue experiment; the pulse repetition time was 1.0 s; the echo time (2Δ) was 40 ms; and 40 × 12 × 500 points were acquired with spectral widths of 80 Hz × 150 Hz × 5000 Hz (F1 × F2 × F3) in 32 min. For the JPRESS experiment, the voxel was the same as the iDQCJRES experiment, and the variable power and optimized relaxation delays (VAPOR) module was used for water suppression. The TR/TE value was 2000/50 ms, the average number was 4, and 40 × 600 points were acquired with spectral widths of 80 Hz × 5000 Hz in 5.3 min.
Results and Discussion
Chemical solution
Experimental results of butyl methacrylate in DMSO are presented in Figs. 2 and 3. The 3D shearing process applied to iDQCJRES data is described in Fig. 2. The original 3D iDQCJRES spectrum is achieved by a direct 3D fast Fourier transform (Fig. 2 a). It can be seen that this 3D spectrum suffers from inhomogeneous line broadening along both F1 and F3 dimensions but not along the F2 dimension, resulting in the streaks observed parallel to the F1-F3 plane and perpendicular to the F2 dimension. After the F1-F3 plane is sheared along the F3 axis, all streaks stretch along the F1 dimension and are perpendicular to the F2-F3 plane (Fig. 2 b). Consequently, the sheared 3D spectrum is free from field inhomogeneities along the F2 and F3 dimensions. A high-resolution 2D J-resolved spectrum can then be constructed by projecting the sheared 3D spectrum onto the F2-F3 plane (Fig. 2 b, gray-shaded plane).
Figure 2.
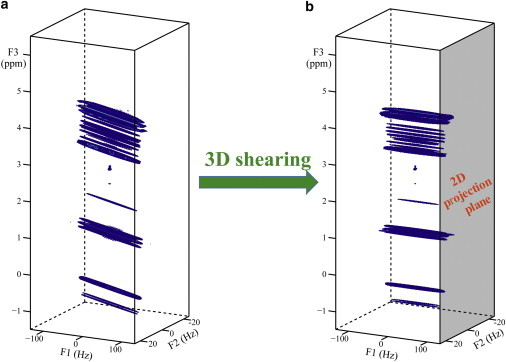
Shearing process for 3D iDQCJRES data of butyl methacrylate in DMSO with 200 Hz field inhomogeneity. (a) Original 3D spectrum before shearing. The streaks are parallel to the F1-F3 plane and perpendicular to the F2 dimension. (b) Sheared 3D spectrum. The streaks stretch along the F1 dimension and are perpendicular to the F2-F3 plane, so a high-resolution 2D J-resolved spectrum can be constructed through 2D projection onto the F2-F3 plane (gray-shaded plane). To see this figure in color, go online.
Figure 3.
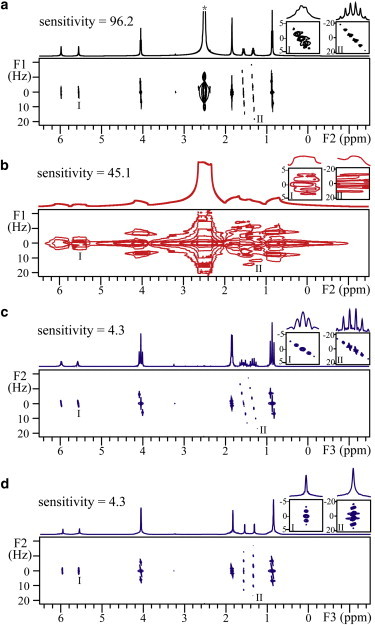
2D J-resolved spectra of butyl methacrylate in DMSO. Two regions marked I and II, containing peaks at 5.56 ppm and 1.32 ppm, respectively, are expanded for all 2D spectra. (a) Conventional J-resolved spectrum in a well-shimmed field and its projection along the F2 axis. The truncated peak marked with an asterisk is the solvent peak. (b) Conventional J-resolved spectrum acquired in an inhomogeneous field with a line width of 200 Hz. (c) iDQCJRES 2D projection spectrum along the F3 axis in the same inhomogeneous field as in b. (d) iDQCJRES 2D projection spectrum after a clockwise rotation of the image in c by 45° along F2 = 0. To see this figure in color, go online.
The 2D J-resolved spectra acquired using iDQCJRES and conventional JRES sequences are shown in Fig. 3. The conventional J-resolved spectrum acquired in a well-shimmed magnetic field is presented in Fig. 3 a. The standard J-resolved information is clearly presented and seven coupled peaks from the solute are shown. Detail of the two regions marked I and II containing signal peaks at 5.56 ppm and 1.32 ppm, respectively, are shown in the insets above the spectrum. The peak at 5.56 ppm is a quintuplet with a J-coupling constant of 1.77 Hz and the peak at 1.32 ppm is a sextet with a J-coupling constant of 7.57 Hz. A clockwise rotation of 45° along F1 = 0 can be performed to separate chemical shifts and J couplings along two different axes. When a conventional 2D JRES sequence is applied in inhomogeneous fields, high-resolution spectral information is lost and the signal peaks stretch along the F2 axis, resulting in the overlap of neighboring peaks (Fig. 3 b). Although the field inhomogeneity can be refocused by spin echo in the F1 dimension, the overlap of neighboring signals makes it difficult to read out exact J-coupling information in the F1 dimension (see the expanded regions shown in Fig. 3 b, insets I and II). Under the same inhomogeneous field, a high-resolution 2D J-resolved spectrum can be obtained using the iDQCJRES sequence (Fig. 3 c), which results from a 2D projection of the sheared 3D spectrum onto the F2-F3 plane (the shearing process is described in Fig. 2 b). Compared to the conventional J-resolved spectrum shown in Fig. 3 b, the line width is reduced from 200 Hz to 4 Hz along the F3 dimension, indicating that the influence of field inhomogeneity is eliminated in the 2D iDQCJRES spectrum. The solvent signal is fully suppressed, and J-coupling constants are magnified threefold along the F3 dimension (see the 1D projection). The true J-coupling constants are observed in the F2 dimension and threefold-magnified J-coupling constants are available in the F3 dimension. For example, the signal peak at 5.56 ppm in expanded region I yields a J-coupling constant of 1.79 Hz along the F2 dimension and 5.41 Hz along the F3 dimension. Except for the J-coupling magnification, the spectra shown in Fig. 3, a and c, are qualitatively identical. Similar to conventional JRES, a clockwise rotation of 45° along F2 = 0 can be performed on the 2D iDQCJRES spectrum to separate J couplings and chemical shifts along two distinct axes. The resulting 2D spectrum is shown in Fig. 3 d. A 1D decoupled spectrum can be obtained from its projection along the F3 dimension. All results above coincide with theoretical deductions.
Since the iDQCJRES sequence requires selective excitation of the solvent resonance, the maximum field inhomogeneity allowed for a high-resolution iDQCJRES spectrum is related to the chemical-shift difference between the solvent peak and the nearest-neighbor solute peak. When the field inhomogeneity reaches to such a degree that the broadened solvent peak severely overlaps with the broadened solute peaks, unintentional excitation of solute resonances will emerge and degrade spectral quality by introducing undesired crosspeaks arising from distant dipolar fields generated by solute spins, and the resulting spectrum will not be amenable to straightforward interpretation (32,33). In addition, it can be noted that the sensitivity of the iDQCJRES spectrum is only ∼9.5% of that of the conventional 2D J-resolved spectrum in the same inhomogeneous field due to the intrinsic low SNR of iMQCs. Hence, all experimental parameters should be optimized to obtain the best sensitivity.
Pig brain tissue
The inhomogeneous line broadening caused by intrinsic macroscopic susceptibility variations and intense water signal represent the two main obstacles to acquiring high-resolution NMR spectra on biological samples. Herein, measurements on pig brain tissues are performed to demonstrate the ability of the iDQCJRES method to overcome these obstacles. Experimental results are presented in Fig. 4. The line width of the water resonance near 4.8 ppm is ∼80 Hz in the conventional 1D NMR spectrum (Fig. 4 a), and metabolites in the brain tissues are hardly observable. The 1D water-presaturated spin-echo spectrum of the brain tissue sample acquired using a Nano probe is shown in Fig. 4 b. Except for two resonances at 1.74 ppm and 2.92 ppm from 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt (DSS), most of the metabolites in the pig brain tissues are assigned according to the literature (34,35). The line width in the 1D MAS spectrum is 3.5 Hz when displayed in phase-sensitive mode (Fig. 4 b). Most of the metabolite J couplings can be read out in this measurement. Therefore, we use a 1D MAS spectrum as a standard for comparison when acquiring NMR spectra of the brain tissues. Since chemical shift and J coupling information is contained in the same spectral dimension, the 1D MAS spectrum is slightly crowded. Fig. 4 c shows the conventional 2D water-presaturated J-resolved spectrum after a clockwise rotation of 45° and its 1D projection along the F2 dimension. Chemical shifts and J couplings are separated in this 2D spectrum. However, valuable J coupling and chemical shift information cannot be recovered for the metabolite analyses, since the spectral resolution is degraded by macroscopic susceptibility variations in the sample. A high-resolution 2D iDQCJRES spectrum is shown in Fig. 4 d. Using the same data processing procedure demonstrated in Fig. 2, the final 2D iDQCJRES spectrum with its 1D J-decoupled projection along the F3 axis is shown in Fig. 4 d. In contrast with the conventional J-resolved spectrum in Fig. 4 c, the 1D J-decoupled spectrum from the 2D iDQCJRES spectrum in Fig. 4 d yields much better spectral resolution, and signal resonances are well resolved in a manner satisfactory for metabolite analysis. For example, the line width of creatine (Cr) at 3.01 ppm is 15 Hz (Fig. 4 d). Metabolites are assigned according to the literature (34,35). Most metabolites detected in the MAS spin-echo spectrum (Fig. 4 b) can be clearly observed in the iDQCJRES spectrum (Fig. 4 d). Spectral overlap is avoided in the F3 dimension and metabolite J couplings are clearly observed in the F2 dimension. For example, lactate (Lac) shows two peaks. One is a quadruplet at 4.08 ppm, and the other is a doublet at 1.31 ppm. Both peaks exhibit J-coupling constants of 7.1 Hz. Even the weak signal of aspartate (Asp) at 2.79 ppm can be identified as a double doublet with J-coupling constants of 4.5 Hz and 17.6 Hz. In a similar way, the metabolites N-acetyl-DL-aspartic acid (NAA), glutamine (Glu), γ-amino butyric acid (GABA), scyllo-inositol (s-Ins), and myo-inositol (m-Ins) can clearly be seen.
Figure 4.
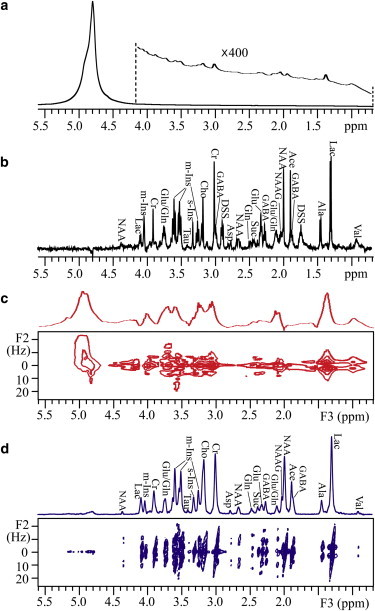
NMR results of pig brain tissues fitted in a 5-mm NMR tube. (a) Conventional 1D spectrum and the expanded region for metabolites. (b) 1D water-presaturated MAS spectrum acquired using a Nano probe. (c) Conventional 2D water-presaturated J-resolved spectrum after a clockwise rotation of 45° and its projection along the F2 axis. (d) Processed 2D iDQCJRES spectrum and its 1D J-decoupled projection along the F3 axis. To see this figure in color, go online.
To facilitate comparison of results obtained using iDQCJRES, MAS, and conventional JRES methods, we list in Table 1 the 1H chemical shifts, multiplet patterns, and J-coupling constants extracted from the spectra shown in Fig. 4, b–d. As shown in Table 1, 26 peaks in the 2D iDQCJRES spectrum could be assigned to 16 metabolites, 27 peaks in the 1D MAS spectrum could be assigned to 16 metabolites, yet in the conventional 2D J-resolved spectrum, only seven signal peaks could be assigned to seven metabolites. Except for the peak of GABA at 3.03 ppm, all peaks observed in the iDQCJRES spectrum exhibit chemical shifts, J-coupling constants, and multiplet patterns similar to those seen in the 1D MAS spectrum. These results convincingly demonstrate the superiority of the iDQCJRES method over the conventional 2D J-resolved method for NMR metabonomic investigations of biological tissues. Although there is a slight difference in J-coupling constants, this difference is <0.5 Hz and is well within tolerable limits. Due to the spectral resolution limitations, the accuracy of the J-coupling constant measurement in the 1D MAS spectrum is 3.7 Hz, i.e., J coupling less than this value is immeasurable. The accuracy of the J-coupling measurements in the iDQCJRES and conventional J-resolved spectra are 3.9 Hz and 5.1 Hz, respectively. The similarity of results obtained by iDQCJRES and MAS measurements is obvious. However, note that due to the intrinsic short relaxation times of metabolites in the tissue, the line width in the iDQCJRES decoupled projection appears larger than that observed in the 1D MAS spectrum. Therefore, weak signals may be obscured by the shoulders of nearby strong resonances in the iDQCJRES spectrum. For example, the peak of GABA at 3.03 ppm is totally covered by the Cr peak at 3.01 ppm. However, the iDQCJRES method enjoys an advantage in practical applications. It delivers high-resolution spectra without requiring the use of specialized equipment (i.e., Nano probes) or requiring that the sample is spun rapidly. This makes the iDQCJRES method well suited to in vivo NMR applications, whereas MAS based methods are not.
Table 1.
Comparisons of 1H chemical shift assignments, multiplet patterns, and J coupling constants of metabolites in pig brain tissues using iDQCJRES, MAS, and conventional JRES methods
| Metabolite | Group |
1H Chemical shifts (ppm) |
Multiplet patterns |
J-coupling constants (Hz) |
|---|---|---|---|---|
| iDQCJRES/MAS/JRES | iDQCJRES/MAS/JRES | iDQCJRES/MAS/JRES | ||
| Acetate (Ace) | -CH3 | 1.89/1.90/NO | s/s/NO | -/-/NO |
| N-Acetyl aspartate (NAA) | -CH3 | 2.00/2.00/2.05 | s/s/s | −/−/- |
| -CH-CH2 | 2.67/2.67/NO | dd/dd/NO | 3.9&15.6/4.1&15.8/ NO | |
| -CH-CH2 | 4.36/4.37/NO | m/m/NO | 3.9/3.7/ NO | |
| N-Acetyl aspartate glutamate (NAAG) | -CH3 | 2.04/2.05/NO | s/s/NO | −/−/NO |
| Alanine (Ala) | -CH3 | 1.46/1.46/NO | d/d/NO | 7.4/7.2/NO |
| γ-Aminobutyric acid (GABA) | -CH2-CH2 | 1.88/1.89/NO | q/NO/NO | 7.3/ NO/NO |
| -CH2-CH2 | 2.27/2.28/NO | t/t/NO | 7.4/7.3/NO | |
| -CH2 | NO/3.03/NO | NO/t/NO | NO/7.0/NO | |
| Aspartate (Asp) | -CH | 2.79/2.80/NO | dd/dd/NO | 4.5&17.6/4.3&17.7/ NO |
| Choline (Cho) | -CH3 | 3.18/3.19/3.22 | s/s/s | −/−/- |
| Creatine (Cr) | -CH3 | 3.01/3.02/3.05 | s/s/s | −/−/- |
| -CH2 | 3.90/3.91/NO | s/s/NO | −/−/NO | |
| Glutamate/Glutamine (Glu/Gln) | -CH2 | 2.09/2.10/NO | m/m/NO | 8.0/7.2/NO |
| -CH2 | 2.34/2.34/NO | m/m/NO | 7.4/7.5/NO | |
| -CH2 | 2.46/2.47/NO | m/m/NO | 7.4/7.2/NO | |
| -CH | 3.75/3.76/NO | dd/dd/NO | 6.1&9.9/6.8&9.7/ NO | |
| Myo-inositol (m-Ins) | -CH | 3.27/3.27/NO | t/t/NO | 9.3/9.2/NO |
| -CH | 3.50/3.52/NO | t/t/NO | 10.2/10.4/NO | |
| -CH | 3.60/3.61/NO | t/t/NO | 9.7/9.8/NO | |
| -CH | 4.05/4.07/4.01 | t/s/s | 4.7/−/− | |
| Scyllo-inositol (s-Ins) | -CH | 3.32/3.34/NO | s/s/NO | −/−/NO |
| Lactate (Lac) | -CH-CH3 | 1.31/1.30/1.35 | d/d/d | 7.1/7.1/7.1 |
| -CH-CH3 | 4.08/4.10/NO | q/q/NO | 7.1/7.1/NO | |
| Succinate (Suc) | -CH2 | 2.38/2.41/NO | s/s/NO | −/−/NO |
| Taurine (Tau) | -CH2 | 3.40/3.42/NO | t/t/NO | 6.8/6.3/NO |
| Valine (Val) | -CH3 | 0.92/0.94/0.97 | d/m/d | 7.3/6.9/7.5 |
The left, center, and right values in each entry correspond to results of iDQCJRES, MAS, and conventional JRES, respectively. Multiplet patterns are defined as singlets (s), doublets (d), triplets (t), quartets (q), double doublets (dd), and multiplets (m). FA, fatty acids; NO, not observable.
Fish
Localized 2D J-resolved spectra and postmortem images of an intact fish (Crossocheilus siamensis) are shown in Fig. 5. Fig. 5 a shows the sagittal and axial spin-echo images with TR/TE = 2500/25 ms. Both iDQCJRES and conventional JPRESS sequences are performed with spatial localization in three dimensions. The localized voxel containing the fish spinal cord, with a size of 4 × 4 × 13 mm3, is marked by dashed boxes in the images. Fig. 5 b shows a conventional 1D NMR spectrum of the fish and illustrates variations of the static magnetic field over this region of space. The line width of the water resonance near 4.8 ppm is 120 Hz in this 1D spectrum. Because of intense water signal and inhomogeneous line broadening, signals from metabolites of the fish tissue are obscured in the 1D spectrum. In the 2D J-resolved spectrum acquired utilizing the conventional JPRESS method (Fig. 5 c), the water signal is suppressed, but inhomogeneous line broadening is still present. Assigning metabolites correctly and measuring J-coupling constants accurately are nevertheless still considerably challenging tasks with this methodology. Several main peaks are observed and assigned to All fatty acids (FA) except n-3 (0.84 ppm), All FA except 20:5 and 22:6 n-3 (1.23 ppm), Acetate (Ace, 1.90 ppm), and Betaine (Bet, 3.18 ppm). However, chemical shift and J-coupling information remain inaccessible for most of the metabolites. The iDQCJRES method provides an alternative method for the J-resolved detection on biological tissues. From the 2D iDQCJRES spectra acquired on the fish (Fig. 5 d), it can be seen that spectral resolution is greatly enhanced; for example, the line width of the Bet peak at 3.19 ppm is 20 Hz. Many more peaks could be assigned from the spectrum shown in Fig. 5 d than could be assigned from the 2D JPRESS spectrum shown in Fig. 5 c. Chemical shift information is contained in the J-decoupled dimension (F3 axis) and J coupling constants are measurable along the J-coupling dimension (F2 axis). According to the literature (34,35), all signal peaks observed in iDQCJRES and JPRESS spectra are assigned, and their J coupling constants are also measured. A comparison of these observed metabolite data, acquired from iDQCJRES and JPRESS methods, is presented in Table 2 . The comparison includes 1H chemical-shift assignments, multiplet patterns, and J-coupling constants of metabolites. As shown in Table 2, 25 peaks in the 2D iDQCJRES spectrum could be assigned to 17 metabolites and these include three peaks assigned to fatty acids contained within larger macromolecular structures. Only six peaks could be assigned to six metabolites in the conventional 2D JPRESS. The advantages of the 2D iDQCJRES method over the conventional 2D JPRESS method in the 2D J-resolved detection of real biological samples are self-evident.
Figure 5.
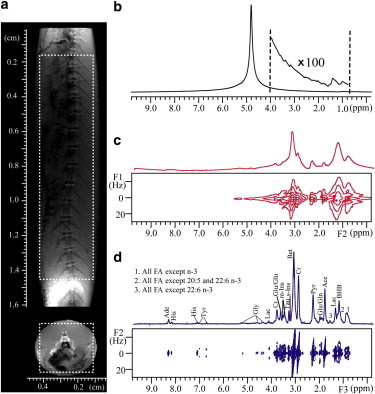
Postmortem studies of an intact fish (Crossocheilus siamensis). (a) Sagittal and axial spin-echo images with a marked voxel (4 × 4 × 13 mm3). (b) Conventional 1D spectrum and the expanded region for metabolites. (c) 2D JPRESS spectrum; (d) 2D iDQCJRES spectrum with assigned metabolites. To see this figure in color, go online.
Table 2.
Comparisons of 1H chemical shift assignments, multiplet patterns, and J coupling constants of metabolites in an intact fish using iDQCJRES and conventional JPRESS methods
| Metabolite | Group |
1H Chemical shifts (ppm) |
Multiplet patterns |
J-coupling constants (Hz) |
|---|---|---|---|---|
| iDQCJRES/JPRESSa | iDQCJRES/JPRESS | iDQCJRES/JPRESS | ||
| All FA except n-3 | -CH3 | 0.88/0.84 | d/q | 6.8/4.6 |
| All FA except 20:5 and 22:6 | -(CH2)n | 1.21/1.23 | s/s | −/− |
| All FA except 22:6 n-3 | -CH2 | 1.58/NO | s/NO | -/NO |
| Glyceryl (Gly) | -CH2 | 4.37/NO | s/NO | -/NO |
| -CH2 | 4.56/NO | s/NO | -/NO | |
| -CH2 | 5.21/NO | s/NO | -/NO | |
| Acetate (Ace) | -CH3 | 1.88/1.90 | s/s | −/− |
| Adenosine (Ade) | -CH | 8.24/NO | s/NO | -/NO |
| β-Hydroxybutyrate (BHB) | -CH3 | 2.18/NO | s/NO | -/NO |
| Betaine (Bet) | -CH3 | 3.19/3.18 | s/s | −/− |
| Creatine (Cr) | -CH3 | 2.91/2.88 | s/s | −/− |
| -CH2 | 3.82/NO | s/NO | -/NO | |
| Glutamate/Glutamine (Glu/Gln) | -CH2 | 2.01/NO | m/NO | 7.7/NO |
| -CH | 3.62/NO | m/NO | 7.1/NO | |
| Hisditine (His) | -CH | 7.08/NO | s/NO | -/NO |
| -CH | 8.15/NO | s/NO | -/NO | |
| Myo-inositol (m-Ins) | -CH | 3.46/NO | dd/NO | 10.1&5.4/NO |
| -CH | 3.55/NO | dd/NO | 9.8&5.5/NO | |
| Scyllo-inositol (s-Ins) | -CH | 3.23/NO | s/NO | -/NO |
| Lactate (Lac) | -CH-CH3 | 1.34/NO | d/NO | 7.1/NO |
| -CH-CH3 | 4.09/NO | q/NO | 7.3/NO | |
| Taurine (Tau) | -CH2 | 3.31/NO | t/NO | 6.7/ NO |
| Tyrosine (Tyr) | -CH | 6.72/NO | d//NO | 8.8/NO |
| -CH | 7.03/NO | d//NO | 8.6/NO | |
| Pyruvate (Pyr) | -CH3 | 2.32/2.31 | s/s | −/− |
The left and right values in each entry correspond to results of iDQCJRES and conventional JPRESS, respectively. Multiplet patterns are coded as in Table 1. FA, fatty acids; NO, not observable.
Conclusions
In this work, a pulse sequence, named iDQCJRES, is proposed to acquire high-resolution 2D J-resolved spectra in inhomogeneous magnetic fields. With its immunity to field inhomogeneities arising from variations in macroscopic magnetic susceptibility, the iDQCJRES sequence is well suited to applications on biological tissues, even spatially localized spectra for in vivo applications. The iDQCJRES approach is not constrained by the hardware requirements of MAS methods and respresents a useful and complementary avenue for recovering high-resolution 2D J-resolved spectra of biological tissues without recourse to time-consuming shimming procedures. In addition, the iDQCJRES sequence is applicable on most modern NMR spectrometers. Experiments performed on a chemical solution in a deliberately deshimmed magnetic field are first used to show the detailed implementation of iDQCJRES, outlining the data processing and experimental steps taken to achieve a high-resolution spectrum. Spectral features, such as resolution enhancement obtained, magnification of J-coupling constants observed, and effective solvent suppression, are characterized. The performance of iDQCJRES on biological systems is then verified using pig brain tissues. The information contained in iDQCJRES and MAS spectra are shown to be nearly identical. Because the iDQCJRES does not require sample spinning, it is suited to NMR studies of intact biological tissues and, notably, is expected to prove especially useful in in vivo applications. Finally, we demonstrate the spatial benefits accrued by applying the iDQCJRES method, as opposed to the conventional JPRESS method, to obtain localized 2D J-resolved spectra on an intact fish. Theoretical deductions and experimental observations illuminate the strengths of the iDQCJRES sequence for obtaining high-resolution 2D J-resolved spectra in inhomogeneous fields and show that it is potentially appropriate for in vivo metabonomic studies.
Acknowledgments
We thank Pieter Smith for helping to revise the manuscript.
This work was partially supported by the National Natural Science Foundation of China under grants 11205129,11174239, and 11275161, and the Prior Research Field Fund for the Doctoral Program of Higher Education of China under grant 20120121130003.
References
- 1.Tzeng S.R., Kalodimos C.G. Protein activity regulation by conformational entropy. Nature. 2012;488:236–240. doi: 10.1038/nature11271. [DOI] [PubMed] [Google Scholar]
- 2.Nath N., Lokesh, Suryaprakash N. Measurement and applications of long-range heteronuclear scalar couplings: recent experimental and theoretical developments. ChemPhysChem. 2012;13:645–660. doi: 10.1002/cphc.201100748. [DOI] [PubMed] [Google Scholar]
- 3.Wu E.L., Engström O., Im W. Molecular dynamics and NMR spectroscopy studies of E. coli lipopolysaccharide structure and dynamics. Biophys. J. 2013;105:1444–1455. doi: 10.1016/j.bpj.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akira K., Hichiya H., Mitome H. Metabonomic study on the biochemical response of spontaneously hypertensive rats to chronic taurine supplementation using (1)H NMR spectroscopic urinalysis. J. Pharm. Biomed. Anal. 2013;85:155–161. doi: 10.1016/j.jpba.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 5.Raison C. NMR profiling of urinary metabolites could improve hepatocellular carcinoma diagnosis. Expert Rev. Mol. Diagn. 2013;13:418. [Google Scholar]
- 6.Gupta A., Mahdi A.A., Sankhwar S.N. Efficacy of Withania somnifera on seminal plasma metabolites of infertile males: a proton NMR study at 800 MHz. J. Ethnopharmacol. 2013;149:208–214. doi: 10.1016/j.jep.2013.06.024. [DOI] [PubMed] [Google Scholar]
- 7.Bruch R.C., Bruch M.D. Two-dimensional J-resolved proton NMR spectroscopy of oligomannosidic glycopeptides. J. Biol. Chem. 1982;257:3409–3413. [PubMed] [Google Scholar]
- 8.Ludwig C., Viant M.R. Two-dimensional J-resolved NMR spectroscopy: review of a key methodology in the metabolomics toolbox. Phytochem. Anal. 2010;21:22–32. doi: 10.1002/pca.1186. [DOI] [PubMed] [Google Scholar]
- 9.Viant M.R. Improved methods for the acquisition and interpretation of NMR metabolomic data. Biochem. Biophys. Res. Commun. 2003;310:943–948. doi: 10.1016/j.bbrc.2003.09.092. [DOI] [PubMed] [Google Scholar]
- 10.Foxall P.J.D., Parkinson J.A., Nicholson J.K. Analysis of biological fluids using 600 MHz proton NMR spectroscopy: application of homonuclear two-dimensional J-resolved spectroscopy to urine and blood plasma for spectral simplification and assignment. J. Pharm. Biomed. Anal. 1993;11:21–31. doi: 10.1016/0731-7085(93)80145-q. [DOI] [PubMed] [Google Scholar]
- 11.Lutz N.W., Maillet S., Cozzone P.J. Further assignment of resonances in 1H NMR spectra of cerebrospinal fluid (CSF) FEBS Lett. 1998;425:345–351. doi: 10.1016/s0014-5793(98)00262-2. [DOI] [PubMed] [Google Scholar]
- 12.Nicholson J.K., Foxall P.J.D., Lindon J.C. 750 MHz 1H and 1H-13C NMR spectroscopy of human blood plasma. Anal. Chem. 1995;67:793–811. doi: 10.1021/ac00101a004. [DOI] [PubMed] [Google Scholar]
- 13.Righi V., Durante C., Schenetti L. Discrimination of healthy and neoplastic human colon tissues by ex vivo HR-MAS NMR spectroscopy and chemometric analyses. J. Proteome Res. 2009;8:1859–1869. doi: 10.1021/pr801094b. [DOI] [PubMed] [Google Scholar]
- 14.Misra D., Gupta V., Roy R. Proton HR-MAS NMR spectroscopic characterization of metabolites in various human organ tissues: pancreas, brain and liver from trauma cases. Physiol. Chem. Phys. Med. NMR. 2008;40:67–88. [PubMed] [Google Scholar]
- 15.Wind R.A., Hu J.Z., Rommereim D.N. High-resolution 1H NMR spectroscopy in a live mouse subjected to 1.5 Hz magic angle spinning. Magn. Reson. Med. 2003;50:1113–1119. doi: 10.1002/mrm.10650. [DOI] [PubMed] [Google Scholar]
- 16.Wind R.A., Hu J.Z., Majors P.D. Localized in vivo isotropic-anisotropic correlation 1H NMR spectroscopy using ultraslow magic angle spinning. Magn. Reson. Med. 2006;55:41–49. doi: 10.1002/mrm.20740. [DOI] [PubMed] [Google Scholar]
- 17.Hebbar S., Prabhu U.R., Suryaprakash N. Selective double quantum resolved correlation experiment for the complete separation of entire proton NMR spectra of enantiomers. J. Magn. Reson. 2012;215:23–26. doi: 10.1016/j.jmr.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 18.Baishya B., Reddy G.N.M., Suryaprakash N. Simplifying the complex 1H NMR spectra of fluorine-substituted benzamides by spin system filtering and spin-state selection: multiple-quantum-single-quantum correlation. J. Phys. Chem. A. 2008;112:10526–10532. doi: 10.1021/jp8055174. [DOI] [PubMed] [Google Scholar]
- 19.Thomas M.A., Lange T., Boesiger P. Two-dimensional MR spectroscopy of healthy and cancerous prostates in vivo. MAGMA. 2008;21:443–458. doi: 10.1007/s10334-008-0121-7. [DOI] [PubMed] [Google Scholar]
- 20.Chen Z., Chen Z.W., Zhong J.H. High-resolution NMR spectra in inhomogeneous fields via IDEAL (intermolecular dipolar-interaction enhanced all lines) method. J. Am. Chem. Soc. 2004;126:446–447. doi: 10.1021/ja036491f. [DOI] [PubMed] [Google Scholar]
- 21.Vathyam S., Lee S., Warren W.S. Homogeneous NMR spectra in inhomogeneous fields. Science. 1996;272:92–96. doi: 10.1126/science.272.5258.92. [DOI] [PubMed] [Google Scholar]
- 22.Hoerr V., Purea A., Faber C. NMR separation of intra- and extracellular compounds based on intermolecular coherences. Biophys. J. 2010;99:2336–2343. doi: 10.1016/j.bpj.2010.06.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang B., Liu H., Mao X. Double quantum CRAZED NMR signal in inhomogeneous fields. Chem. Phys. 2008;351:33–36. [Google Scholar]
- 24.Garrett-Roe S., Warren W.S. Numerical studies of intermolecular multiple quantum coherences: high-resolution NMR in inhomogeneous fields and contrast enhancement in MRI. J. Magn. Reson. 2000;146:1–13. doi: 10.1006/jmre.2000.2096. [DOI] [PubMed] [Google Scholar]
- 25.Faber C., Pracht E., Haase A. Resolution enhancement in in vivo NMR spectroscopy: detection of intermolecular zero-quantum coherences. J. Magn. Reson. 2003;161:265–274. doi: 10.1016/s1090-7807(03)00006-5. [DOI] [PubMed] [Google Scholar]
- 26.Hwang T.L., Shaka A.J. Water suppression that works—excitation sculpting using arbitrary wave-forms and pulsed-field gradient. J. Magn. Reson. A. 1995;112:275–279. [Google Scholar]
- 27.Balla D.Z., Faber C. Localized intermolecular zero-quantum coherence spectroscopy in vivo. Concept Magn. Reson. A. 2008;32A:117–133. doi: 10.1002/mrm.21007. [DOI] [PubMed] [Google Scholar]
- 28.Jeener J. Equivalence between the “classical” and the “Warren” approaches for the effects of long range dipolar couplings in liquid nuclear magnetic resonance. J. Chem. Phys. 2000;112:5091–5094. [Google Scholar]
- 29.Ahn S., Warren W.S., Lee S. Quantum treatment of intermolecular multiple-quantum coherences with intramolecular J coupling in solution NMR. J. Magn. Reson. 1997;128:114–129. doi: 10.1006/jmre.1997.1226. [DOI] [PubMed] [Google Scholar]
- 30.Liu M.L., Mao X.A., Lindon J.C. Improved WATERGATE pulse sequences for solvent suppression in NMR spectroscopy. J. Magn. Reson. 1998;132:125–129. [Google Scholar]
- 31.Ernst R.R., Bodenhausen G., Wokaun A. Clarendon Press; Oxford: 1987. Principles of Nuclear Magnetic Resonance in One and Two Dimensions; pp. 148–152. [Google Scholar]
- 32.Zhang W., Cai C.B., Chen Z. Intermolecular double-quantum coherence NMR spectroscopy in moderate inhomogeneous fields. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2009;74:1138–1144. doi: 10.1016/j.saa.2009.09.023. [DOI] [PubMed] [Google Scholar]
- 33.Balla D.Z., Faber C. Intermolecular zero-quantum coherence NMR spectroscopy in the presence of local dipole fields. J. Chem. Phys. 2008;128:154522. doi: 10.1063/1.2904564. [DOI] [PubMed] [Google Scholar]
- 34.Govindaraju V., Young K., Maudsley A.A. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed. 2000;13:129–153. doi: 10.1002/1099-1492(200005)13:3<129::aid-nbm619>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 35.de Graaf R.A. Vivo NMR Spectroscopy: Principles and Techniques. 2nd ed. John Wiley & Sons; Oxford, United Kingdom: 2007. pp. 62–96. [Google Scholar]