

Abstract
Previous studies have shown that muscle atrophy is associated with mitochondrial dysfunction and an increased rate of mitochondrial reactive oxygen species production. We recently demonstrated that fatty acid hydroperoxides (FA-OOH) are significantly elevated in mitochondria isolated from atrophied muscles. The purpose of the current study is to determine whether FA-OOH can alter skeletal muscle mitochondrial function. We found that FA-OOH (at low micromolar concentrations) induces mitochondrial dysfunction assessed by decrease in the rate of ATP production, oxygen consumption and activity of respiratory chain complexes I and III. Using methods to distinguish superoxide release towards the matrix and inter-membrane space, we demonstrate that FA-OOH significantly elevates oxidative stress in the mitochondrial matrix (and not the inter-membrane space) with complex I as the major site of superoxide production (most likely from a site upstream of the ubiquinone binding site but downstream from the flavin binding site-the iron sulfur clusters). Our results are the first to indicate that FA-OOH’s are important modulators of mitochondrial function and oxidative stress in skeletal muscle mitochondria and may play an important role in muscle atrophies that are associated with increased generation of FA-OOH’s, e.g., denervation-induced muscle atrophy.
Keywords: Oxidative stress, superoxide, fatty acid hydroperoxides, hydrogen peroxide, mitochondria
INTRODUCTION
Studies from our group and others suggest that mitochondrial oxidative stress and mitochondrial dysfunction may be key mechanisms involved in the loss of muscle mass during aging and in neurodegenerative diseases [1-4]. Enzymatic activity of respiratory chain complexes, the respiratory control ratio (RCR, state 3/state 4) and the rate of mitochondrial ATP production are each significantly decreased in mitochondria isolated from skeletal muscle in old mice, data that indicate age-related mitochondrial dysfunction [4-6]. In addition, the rate of mitochondrial reactive oxygen species (ROS) production is significantly elevated by mitochondria isolated from skeletal muscle in aging rats and mice [2, 7]. Although the studies mentioned above indicate a strong association between ROS production, mitochondrial dysfunction and muscle atrophy, little is known about the pathways or mediators that are responsible.
Previous studies using isolated mitochondria suggest that fatty acids and fatty acid hydroperoxides (FA-OOH’s) are important modulators of mitochondrial function and ROS production [8-12]. It has been proposed that fatty acids exert these effects by 1) acting as protonophoric uncouplers, 2) by interfering with specific components of the electron transport chain, and 3) by inhibiting adenine nucleotide translocase [8-10, 13, 14]. Under normal conditions, tissues contain small amounts of fatty acids bound to proteins and membranes [15]. However, in pathological conditions including ischemia, fatty acids and their hydroperoxides are known to accumulate in target tissues [16-18]. Our recent study showed that FA-OOH’s are significantly elevated in mitochondria isolated from atrophic skeletal muscles [19]. We further showed that attenuation of muscle atrophy in calorie-restricted and glutathione peroxidase 4 transgenic mice is associated with decrease in FA-OOH’s in skeletal muscle mitochondria [19]. Based on these preliminary studies, we hypothesized that FA-OOH are important modulators of mitochondrial function and ROS production in skeletal muscle mitochondria, which may have a causative role in muscle atrophy.
Therefore, the purpose of this study was 1) to determine whether FA-OOH’s alter mitochondrial function (rate of ATP production, RCR and enzymatic activity of respiratory chain complexes) in skeletal muscle mitochondria and 2) to determine the effect of FA-OOH on the topology and sites of superoxide production, using methodologies that can distinguish between superoxide released towards the matrix and towards the intermembrane space. We demonstrate for the first time that low micromolar concentrations of FA-OOH decreases the rate of mitochondrial ATP production, RCR and the enzymatic activity of respiratory chain complexes I and III in skeletal muscle mitochondria. Additionally, using methodologies that distinguish between superoxide generation towards the matrix and intermembrane space, we demonstrate that in skeletal muscle mitochondria, FA-OOH (but not FA-OH) significantly increases the rate of mitochondrial ROS production directed towards the matrix (and not the intermembrane space) with complex I as the major site of ROS production.
EXPERIMENTAL PROCEDURES
Reagents
All reagents were obtained from Sigma (St. Louis, MO). Amplex Red was obtained from Molecular Probes (Eugene, OR). 15-hydroxyeicosatetraenoic acid (hereafter referred to as FA-OH) and 15-hydroxyperoxyeicosatetraenoic acid (hereafter referred to as FA-OOH) were from Cayman Chemicals (Ann Arbor, MI). Reagents were dissolved in either incubation buffer or ethanol as appropriate.
Experimental animals
All experiments were performed using 6- to 8-month-old wild-type C57BL/6J mice. The mice were maintained under specific pathogen-free conditions and housed 3-4/cage on a 12:12 (light:dark) cycle at 22 ± 2°C and 50 ± 10% relative humidity. Mice were euthanized using a CO2 chamber followed by cervical dislocation. Lower hind limb skeletal muscle was then collected. All procedures were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at San Antonio and the Audie L. Murphy Veterans Hospital.
Isolation of skeletal muscle mitochondria
Mitochondria were purified from lower hind limb skeletal muscle according to Chappell and Perry, as described previously [2, 19]. Briefly, hind limb skeletal muscle was excised, weighed, bathed in 150 mM KCl and placed in Chappell-Perry buffer with the protease nagarse. The minced skeletal muscle was homogenized, and the homogenate was centrifuged for 10 min at 600 × g; the supernatant was then passed through cheesecloth and centrifuged at 14,000 × g for 10 min. The mitochondrial pellet was washed in modified Chappell-Perry buffer with 0.5% bovine serum albumin (BSA) and centrifuged at 7,000 × g for 10 min. The pellet was further washed twice in modified Chappell-Perry buffer without BSA, centrifuging each time at 3,500 × g for 10 min. Isolated mitochondria were used immediately. Protein concentration was measured with the Bradford method.
Experimental design
To test our hypothesis that fatty acid hydroperoxides are important modulators of mitochondrial function and ROS production, we performed the following experiments. To determine mitochondrial function, we measured 1) oxygen consumption (state 3/state 4 respiration) 2) the rate of mitochondrial ATP production and 3) enzymatic activity of respiratory chain complexes I-IV in isolated skeletal muscle mitochondria treated with FA-OOH (or the corresponding FA-OH). To determine the role of FA-OOH on mitochondrial ROS production, we measured the following variables - 1) Amplex Red fluorescence to determine the rate of mitochondrial H2O2 release 2) aconitase activity to measure superoxide production directed towards the matrix and 3) electron paramagnetic resonance to measure extra-mitochondrial superoxide production. In addition, we also determined mitochondrial membrane potential by fluorescence quenching of safranin O dye. All these measures were performed in the presence of substrates/inhibitors specific for the respiratory chain complexes.
Mitochondrial respiration
The rate of mitochondrial oxygen consumption was measured using a Clark electrode (Oxytherm, Oxygen electrode system from PP System, Hansatech Instruments Ltd.) as originally described by Estabrook [20]. Mitochondrial preparation (500 μg/ml) was suspended in respiratory buffer consisting of 125 mM KCl, 10 mM HEPES, 5 mM MgCl2, 2 mM K2HPO4, pH 7.44; with 0.3% BSA. Glutamate/malate (G/M, 5 mM) was added as the respiratory substrate. FA-OH or FA-OOH were added at the final concentration of 0.75 μM. State 3 respiration was induced with the addition of 0.3 mM ADP.
Mitochondrial ATP production
Aliquots of the mitochondrial suspension were used for the measurement of the rate of mitochondrial ATP production with use of a luciferin–luciferase ATP-monitoring reagent (Roche, Indianapolis, IN). The reaction mixture (in mM: 124 KCl, 5 MgCl2, 2 K2HPO4, and 10 HEPES at pH 7.44) included luciferin–luciferase, respiratory substrate (5 mM G/M) or 5 mM succinate plus 10 μM rotenone (S/R), and 30μg of mitochondrial protein (in 200 μl total volume). FA-OH and FA-OOH were added the final concentration of 0.75 μM. Reaction was initiated by the addition of ADP (final concentration 75 μM). The rate of ATP production was expressed as nmoles ATP/min/mg protein. The assay was calibrated with the addition of an ATP standard.
Measurement of electron transport complex activity
Mitochondria isolated from skeletal muscle were treated with either FA-OH or FA-OOH (final concentration 0.75 μM) for 15 min followed by treatment with 10% (w/v) lauryl maltoside in extraction buffer containing 0.75 M aminocaproic acid, 0.05 M Bis–Tris. NADH-ubiquinone oxidoreductase (complex I) activity was assayed [21] by following the rotenone-sensitive rate of NADH oxidation at 340 nm using decylubiquinone as electron acceptor in the presence of 2,6-dichlorophenolindophenol (DCPIP). The reaction mixture contained 100 μM NADH, 2 μM antimycin A, malonate (5 μM) and 2 mM KCN. The reaction was initiated by adding 50 μM decylubiquinone and 20 μg of mitochondrial protein. Succinate-ubiquinone oxidoreductase (complex II) activity was measured by following the malonate-sensitive succinate-dependent reduction of DCPIP at 600 nm [22]. The reaction mixture contained 200 μM rotenone, 2 μM antimycin A, 2 mM KCN, 50 μM DCPIP, and 20 mM succinate. The reaction was initiated by adding 50 μM decylubiquinone and 20 μg of mitochondrial protein. Ubiquinol-cytochrome c oxidoreductase (complex III) activity was determined by measuring antimycin A-sensitive cytochrome c reduction upon addition of 100 μM reduced decylubiquinone at 550 nm [23]. The reaction medium for complex III activity contained 2 mM KCN, 200 μM rotenone, 2 μM antimycin A, malonate (5 μM), 100 μM ferricytochrome c and 5 μg of mitochondrial protein. Cytochrome c oxidase (complex IV) activity was determined by measuring the KCN-sensitive rate of ferrocytochrome c oxidation at 550 nm [24]. The reaction mixture was supplemented with 40 μM ferrocytocrome c, 2 μM antimycin A, 200 μM rotenone, malonate (5 μM) and 5 μg of mitochondrial protein.
Measurement of the membrane potential with Safranin O
Membrane potential was monitored by fluorescence quenching of the positively charged dye Safranin O, as described by Votyakova and Reynolds [25]. Safranin O fluorescence was followed at a λex of 485 nm, a λem of 590 nm using a Fluroskan-FL Ascent Type 374 multi-well plate reader (Labsystems, Finland). 30 μg of mitochondrial protein in 200 μL of reaction buffer (125 mM KCl, 10 mM HEPES, 5 mM MgCl2 and 2 mM K2HPO4 (pH 7.44) in 96-well black plates were pre-incubated with FA-OH or FA-OOH (final concentration 0.75 μM) for 10 min followed by the addition of safranin O (final concentration 5 μM). G/M and S/R were added at concentrations similar to that used for measuring the rate of ATP production. Safranin O fluorescence quenching was determined in the presence of respiratory chain substrates and inhibitors. The relative decrease in safranin O fluorescence was taken as measure of mitochondrial membrane potential and the results are expressed as change in fluorescence.
Mitochondrial H2O2 release
The rate of mitochondrial H2O2 production was measured by the amplex red (AR, Molecular Probes, Eugene, OR) horseradish peroxidase (HRP) method [26]. HRP (2 Units/mL) catalyzes the H2O2-dependent oxidation of the non-fluorescent AR (80 μM) to the fluorescent resorufin [2]. Amplex red buffer containing 125 mM KCl, 10 mM HEPES, 5 mM MgCl2, 2 mM K2HPO4, pH 7.44 (ROS buffer) was prepared with ± 30 Units/mL of CuZnSOD. Skeletal muscle mitochondria (30 μg, in amplex red buffer) were pre-incubated with increasing doses of FA-OH or FA-OOH (0.375 μM - 2.25 μM) for 10 mins before the addition of respiratory chain substrates/inhibitors (at concentrations similar to that used for measuring the rate of ATP production). All the assays were performed in black 96-well plates at 37°C and fluorescence was followed at an excitation wavelength of 545 nm and an emission wavelength of 590 nm using a Fluoroskan Ascent type 374 multi-well plate reader (Labsystems, Helsinki, Finland). The slope of the increase in fluorescence was converted to the rate of H2O2 production with use of a H2O2 standard curve. Additional experiments were performed in the presence of catalase to determine the specificity of assay for H2O2.
Electron paramagnetic spectroscopy (EPR)
Extra-mitochondrial superoxide release was measured by EPR using the spin trap, 5-Diisopropoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide (DIPPMPO, Alexis Biochemicals). DIPPMPO forms an adduct with superoxide, resulting in the generation of DIPPMPO-OOH, which decays to the DIPPMPO-OH adduct by the action of glutathione peroxidases. EPR measurements were performed using an X-band MS200 spectrometer (Magnetech, Berlin, Germany). Mitochondria (20 μg) were pre-incubated with 0.75 uM FA-OH or FA-OOH for 10 min followed by incubation with substrates (24 mM G/M or succinate), inhibitor (2.4 μM rotenone) and DIPPMPO (50 mM) in 125 mM KCl, 10 mM MOPS, 2 mM DTPA, 5 mM MgCl2, 2 mM K2HPO4, pH 7.44 for 10 min at 37°C. Catalase (10 U/mL) was added because it prevented the appearance of small additional and unidentified spectral peaks after extended incubation. Forty μL of sample was transferred to a 50 μL capillary tube, and EPR spectra were measured at room temperature with the following settings: receiver gain, 5×105; microwave power, 20 mW; microwave frequency, 9.55 GHz; modulation amplitude, 2G; scan time, 40 s; and scan width, 100 G, with an accumulation of 10 scans [19]. Experiments were performed in the presence or absence of exogenously added CuZnSOD to confirm the presence of superoxide released from the mitochondria.
Aconitase activity
Aconitase activity was assayed (in detergent-dispersed samples) by measuring the reduction of NADP+ in the presence of citrate and isocitrate dehydrogenase (IDH, Sigma, St. Louis, MO), the conversion of citrate to isocitrate by aconitase being the rate-limiting step [27]. We used a fluorometric method (excitation at 355 nm and emission at 460 nm) to quantify the reduction of NADP+. Mitochondria (20 μg) were resuspended in buffer containing 125 mM KCl, 10 mM HEPES, 5 mM MgCl2, 2 mM K2HPO4, pH 7.44, aliquoted (40 μL) into a 96-well plate and incubated at 30°C up to 10 min with either FA-OH or FA-OOH. Substrates (5 mM succinate or 5 mM G/M) and inhibitor (10 μM rotenone) were added thereafter. Aconitase activity measurements were begun by the addition of 160 μL of 50 mM Tris, 0.6 mM MnCl2, 60 mM citrate, 0.2% Triton X-100, 100 μM NADP+, and 1 Unit of IDH. Fluorometric measurements were then started immediately (Fluoroskan-FL Ascent type 374 microplate reader). The “blank”, used to measure aconitase-independent NADP+ reduction, consisted of the same buffer except with IDH omitted. The slope of the increase in NADPH fluorescence is proportional to aconitase activity [28]. Results were expressed as percentage relative to control (untreated mitochondria).
Statistics
Data are expressed as mean ± SEM. Statistical analysis was performed using Prism 3.0 software (GraphPad Software, San Diego, CA). Significance was examined by either Student’s t-test (when comparing two averages) or by one-way ANOVA with Newman Keul’s multiple comparison test. A p-value of p < 0.05 was considered statistically significant.
RESULTS
FA-OOH decreases skeletal muscle mitochondrial function
To determine the effect of FA-OOH (and the corresponding FA-OH) on mitochondrial function, we measured the rate of mitochondrial oxygen consumption, the rate of mitochondrial ATP production and enzymatic activity of electron transport respiratory complexes I-IV. The rate of mitochondrial oxygen consumption was measured in mitochondria isolated from skeletal muscle respiring on the complex I substrate, G/M, in the presence (State 3) and absence (State 4) of ADP. FA-OOH (0.75 μM) significantly decreased RCR (State 3/State 4, ~ 20%) but there was no effect of FA-OH at the same concentration (Figure 1A). This decrease in RCR is attributed to decrease in State 3 respiration as there was no change in State 4 respiration (data not shown).
Figure 1. FA-OOH decreases skeletal muscle mitochondrial function.
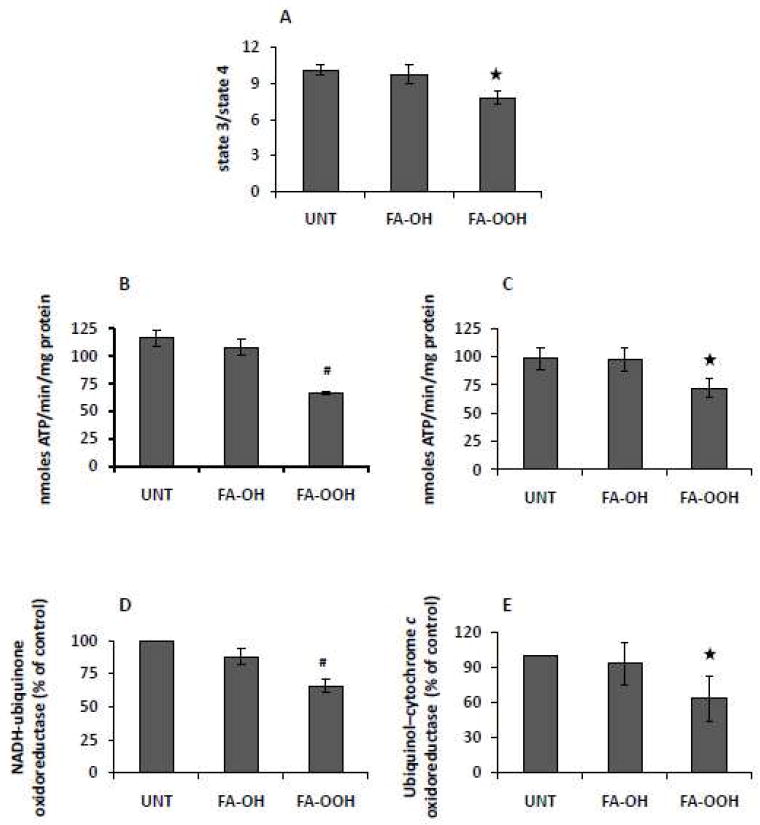
Oxygen consumption as a measure of mitochondrial function was determined in actively phosphorylating (State 3) and resting (State 4) mitochondria respiring on 5 mM glutamate/malate (G/M). The respiratory control ratio (RCR) was determined as the ratio of State 3 to State 4 (A). The rate of mitochondrial ATP production was measured using a luciferase based assay as described under ‘Experimental Procedures’ in mitochondria respiring on (B) G/M (5 mM) and (C) succinate (5 mM) + rotenone (0.5 μM) (S/R). Activities of NADH-ubiquinone oxidoreductase (complex I, D) and Ubiquinol–cytochrome c oxidoreductase (complex III, E) were measured as described under ‘Experimental Procedures’. FA-OH and FA-OOH were added at a final concentration of 0.75 μM. Results shown represent means ± S.E.M. for 5-6 individual mitochondrial preparations. Statistical significance was assessed by one-way ANOVA with Newman Keul’s multiple comparison test (#p < 0.01, *p < 0.05 vs untreated mitochondria).
We next measured the effect of FA-OH and FA-OOH on the rate of mitochondrial ATP production by mitochondria isolated from skeletal muscle respiring on either G/M or S/R. FA-OOH (0.75 μM) significantly decreased the rate of mitochondrial ATP production in mitochondria respiring on G/M (~ 40%, Figure 1B) and S/R (~ 25%, Figure 1C), whereas FA-OH had no effect at a similar concentration.
We also measured the enzymatic activity of respiratory chain complexes I-IV as index of mitochondrial function. While FA-OOH (0.75 μM) significantly decreased the activity of complexes I (~ 40%; Figure 1D) and III (~ 25%; Figure 1E) there was no effect on complexes II or IV (data not shown), whereas FA-OH had no effect on the activity of complex I-IV activity at a similar concentration.
FA-OOH increases the rate of skeletal muscle mitochondrial ROS production
In this study, we used three different methods to detect ROS production in isolated mitochondria, namely EPR, amplex red fluorescence and aconitase activity. Each of these methods detects mitochondrial ROS production in a different manner and in combination, allows for the comparison of superoxide release directed towards the matrix versus superoxide release directed towards the intermembrane space [27, 29-31]. The amplex red probe detects superoxide indirectly after its conversion to H2O2 in the presence of matrix manganese SOD (MnSOD) and both inter-membrane space and exogenously added CuZnSOD. The size of horseradish peroxidase (HRP, 40 kDa) used in the amplex red assay prohibits its entry into the mitochondria. Therefore, use of the amplex red system detects H2O2 that has been released from the mitochondria [26]. H2O2 diffuses out of the mitochondria and in the presence of horseradish peroxidase, converts the non-fluorescent amplex red to the fluorescent resorufin.
The amplex red assay was used to measure the rate of mitochondrial H2O2 production by mitochondria isolated from skeletal muscle in the presence of substrates specific for complex I (G/M) and II (S/R), and, either FA-OOH or FA-OH. While the G/M-supported rate of mitochondrial H2O2 production is derived through forward electron transfer (FET), the succinate-supported rate of mitochondrial H2O2 production is mainly derived through reverse electron transfer (RET) from ubiquinol to complex I [32]. FA-OOH increased the rate of mitochondrial H2O2 production by mitochondria respiring on G/M in a dose-dependent manner, whereas FA-OH had no effect at the same concentration (Figure 2A). FA-OOH (but not FA-OH) also increased the rate of mitochondrial H2O2 production by mitochondria respiring on succinate in a dose-dependent manner, when RET was inhibited with rotenone (Figure 2B). In contrast, the lowest concentrations of both FA-OH and FA-OOH (0.375 μM) significantly inhibited (> 50%) the rate of mitochondrial H2O2 production when mitochondria respired on succinate alone (Figure 2C).
Figure 2. FA-OOH increases the rate of mitochondrial ROS production during forward electron transfer.
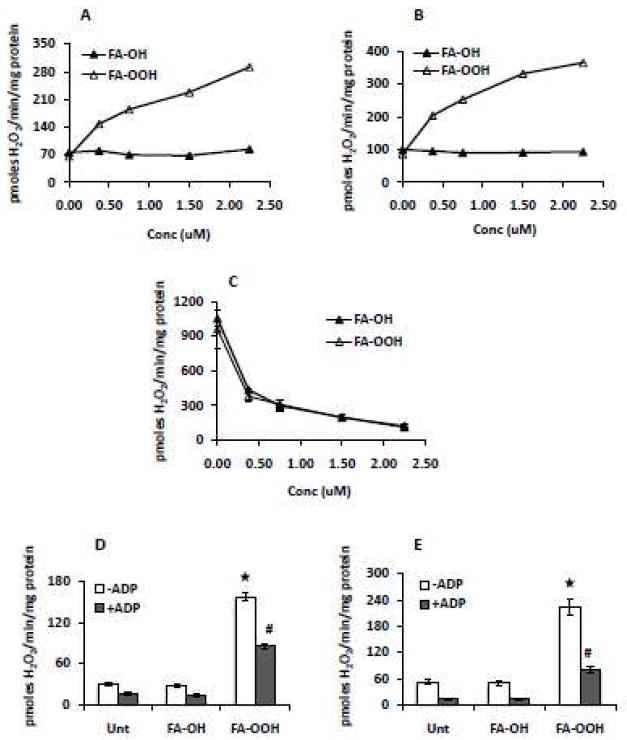
Amplex red was used to measure H2O2 production in the presence of respiratory substrates specific for complex I and complex II and complex I inhibitor, rotenone - (A) glutamate/malate (G/M), 5 mM; (B) succinate (5 mM) + rotenone (0.5 μM) (S/R) and (C) succinate, 5 mM. FA-OH or FA-OOH was added at the concentrations indicated. The amplex red assay was performed in the absence/presence of 2 mM ADP to determine the effect of FA-OH or FA-OOH (0.75 μM) on ROS production in resting vs phosphorylating mitochondria in the presence of (D) G/M (5 mM) and (E) succinate (5 mM) + rotenone (0.5 μM). The results represent means ± S.E.M. for 6-7 individual mitochondrial preparations. Statistical significance was assessed by one-way ANOVA with Newman Keul’s multiple comparison test (*p < 0.0001 vs. untreated mitochondria (State 4), #p < 0.001 vs. untreated mitochondria (State 3).
We also compared the effect of FA-OH and FA-OOH on the rate of mitochondrial H2O2 production in resting (State 4) versus phosphorylating (State 3) mitochondria. FA-OOH (0.75 μM) increased the rate of mitochondrial H2O2 production in the presence of substrates specific for complex I and II (G/M, left panel, ~ 6 fold, Figure 2D) and S/R (right panel; ~ 5-6 fold, Figure 2E) under both State 4 and State 3 conditions, relative to untreated mitochondria.
FA-OH and FA-OOH both decrease mitochondrial membrane potential during RET
Masini et al. (1994) reported that fatty acid hydroperoxides decrease mitochondrial membrane potential in liver mitochondria (during succinate oxidation) only when mitochondrial glutathione content is depleted and not during control conditions [33, 34]. Fatty acid hydroxides, on the other hand, did not affect mitochondrial membrane potential in either of the experimental conditions [34]. The effect of FA-OOH on mitochondrial membrane potential in skeletal muscle mitochondria has not been previously investigated. We measured the effect of FA-OH and FA-OOH on mitochondrial membrane potential in skeletal muscle mitochondria respiring on complex I or II linked substrates. As shown in Table 1, membrane potential was unaffected by FA-OH or FA-OOH (0.75 μM) during forward electron transfer (G/M and S/R). Interestingly, during succinate oxidation, both FA-OH and FA-OOH decreased the membrane potential by ~ 40%, relative to untreated mitochondria.
Table 1.
FA-OH and FA-OOH decrease mitochondrial membrane potential during reverse electron transfer.
| Subs/Inhibit | Untreated | FA-OH | FA-OOH |
|---|---|---|---|
| G/M | 738.83±44.33 | 704.65±39.55 | 711.48±37.08 |
| S/R | 721.88±45.27 | 702.68±44.63 | 692.90±45.93 |
| S | 507.73±38.08 | 326.23±45.27* | 309.83±25.34* |
Mitochondrial membrane potential was measured using Safranin O (5 μM) in the presence of substrates specific for complex I or II (glutamate/malate (G/M), 5 mM; succinate (S), 5 mM) and inhibitor (rotenone (R), 0.5 μM). FA-OH or FA-OOH was added at a final concentration of 0.75 μM per assay. Fluorescence was measured at excitation/emission wavelengths set at 485 nm/590 nm. Results are expressed as change in fluorescence. The results shown represent means ± S.E.M. for 4 individual mitochondrial preparations. Statistical significance was assessed by one-way ANOVA with Newman Keul’s multiple comparison test
(p < 0.0001 vs. untreated mitochondria).
Subs/Inhibit = substrates/inhibitors.
FA-OOH does not increase extra-mitochondrial superoxide release in the presence of substrates specific for complex I or II
EPR with use of the spin-trap DIPPMPO detects superoxide released from the mitochondria [35, 36]. Superoxide and DIPPMPO form the DIPPMPO/·OOH adduct, which rapidly decays to DIPPMPO/·OH and can be quantitatively determined using EPR. Superoxide generated in this manner largely derives from the Q cycle of complex III [37]. Although DIPPMPO is membrane permeable [35], the rate constants for the spontaneous dismutation of superoxide (~106) or, for the reaction of superoxide with MnSOD and CuZnSOD (~109), far exceeds the rate constant for the DIPPMPO-superoxide reaction (~10). Thus, DIPPMPO will be outcompeted by MnSOD and CuZnSOD in the mitochondrial matrix and intermembrane space, respectively, and will detect only the superoxide that has been released from the mitochondria. We measured the effect of FA-OH and FA-OOH on the mitochondrial release of superoxide in the presence of respiratory substrates specific for complex I or II. As shown in Table 2, FA-OH or FA-OOH (0.75 μM) did not increase the DIPPMPO/·OH signal when compared with mitochondria respiring on substrates specific for complex I or II. The possibility that the DIPPMPO/·OH signal could originate from hydroxyl radical and not from the spontaneous decay of the DIPPMPO/·OOH adduct was tested using exogenously added, membrane impermeable CuZnSOD [31]. Addition of CuZnSOD abolished the DIPPMPO/·OH signal (data not shown), demonstrating the specificity of the assay for measuring extra-mitochondrially released superoxide.
Table 2.
Extra-mitochondrial superoxide release by isolated skeletal muscle mitochondria respiring on complex I and II-linked substrates (measured by electron paramagnetic resonance).
| Subs/Inhibit | Untreated | FA-OH | FA-OOH |
|---|---|---|---|
| G/M | 423.44±25.7 | 394.38±22.7 | 454.06±27.5 |
| S/R | 498.75±57.0 | 473.75±42.8 | 425.00±55.8 |
| S | 536.67±27.6 | 501.88±21.6 | 503.44±10.3 |
Skeletal muscle mitochondria (20 μg) were incubated with substrates (24 mM glutamate/malate (G/M) or succinate (S)), inhibitor (2.4 μM rotenone (R)) and DIPPMPO (50 mM) for 10 min at 37°C. Fatty acid hydroxide (FA-OH), or fatty acid hydroperoxide (FA-OOH) were added at the final concentration of 0.75 μM. Units are in relative intensity/20 μg protein. Values are in mean ± S.E.M. Statistical significance was assessed by one-way ANOVA with Newman Keul’s multiple comparison test (n = 6). Subs/Inhibit = substrates/inhibitors.
We next measured the rate of mitochondrial H2O2 production by the amplex red assay in the presence or absence of exogenously added CuZnSOD to indirectly detect mitochondrial superoxide release. Since exogenously added CuZnSOD is membrane impermeable, the difference in the rate of mitochondrial H2O2 production in the absence and presence of exogenous CuZnSOD indicates extra-mitochondrial superoxide release arising from the cytoplasmic side of the inner mitochondrial membrane [38]. FA-OOH (but not FA-OH) increased the rate of mitochondrial ROS production (~ 3-4 fold) using amplex red buffer without CuZnSOD in the presence of substrates specific for complex I (G/M, Figure 3A) and II (S/R, Figure 3B). However, the addition of CuZnSOD did not further increase ROS production in FA-OOH- treated mitochondria, further indicating that FA-OOH does not increase the extra-mitochondrial release of superoxide.
Figure 3. FA-OOH does not affect extra-mitochondrial superoxide release during forward electron transfer.
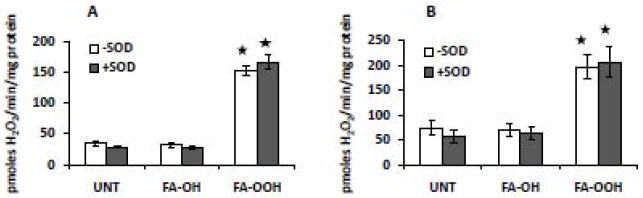
Amplex red was used to measure H2O2 production in the presence of substrates specific for complex I or II and complex I inhibitor, rotenone - (A) glutamate/malate, 5 mM and (B) succinate, 5 mM + rotenone, 0.5 μM. The amplex red buffer was prepared with ± 30 Units/mL of CuZnSOD. FA-OH or FA-OOH was added at a final concentration of 0.75 μM. Statistical significance was assessed by one-way ANOVA with Newman Keul’s multiple comparison test (* p < 0.0001 vs. untreated mitochondria). The results shown represent means ± S.E.M. for 5-6 individual mitochondrial preparations.
FA-OOH inhibits aconitase activity in mitochondria during forward electron transfer
We measured aconitase activity as an indicator of superoxide generation directed toward the mitochondrial matrix [39, 40]. Aconitase is a Krebs cycle enzyme present within the mitochondrial matrix and catalyzes the reversible isomerization of citrate to isocitrate. Aconitase has been shown to be sensitive to inactivation by superoxide. At the active site of aconitase is a cubane [4Fe–4S]2+ cluster, in which only three of the four irons are ligated to cysteine residues. The fourth iron is exposed to the aqueous media of the mitochondrial matrix and is open to attack from superoxide. Superoxide causes a one-electron oxidation of the iron-sulfur cluster, releasing the exposed iron in the ferrous state and inactivating the enzyme [28, 41]. In skeletal muscle, aconitase is present exclusively in the mitochondrial matrix so the measure of the activity of this enzyme is a sensitive measure of superoxide content inside the matrix. FA-OOH (0.75 μM) significantly decreased aconitase activity ((G/M, ~ 15% Figure 4A) and (S/R, ~ 40% Figure 4B)) compared to mitochondria respiring on G/M and S/R alone. The addition of FA-OH had no effect on aconitase activity under the same experimental conditions. Aconitase activity was strongly inhibited in the presence of succinate (~ 80%, mostly due to superoxide production by RET originating from complex I) when compared with aconitase activity in untreated mitochondria. The decrease in aconitase activity in mitochondria respiring on succinate alone was significantly inhibited in the presence of both FA-OH and FA-OOH (Figure 4C).
Figure 4. FA-OOH inhibits aconitase activity during forward electron transfer.
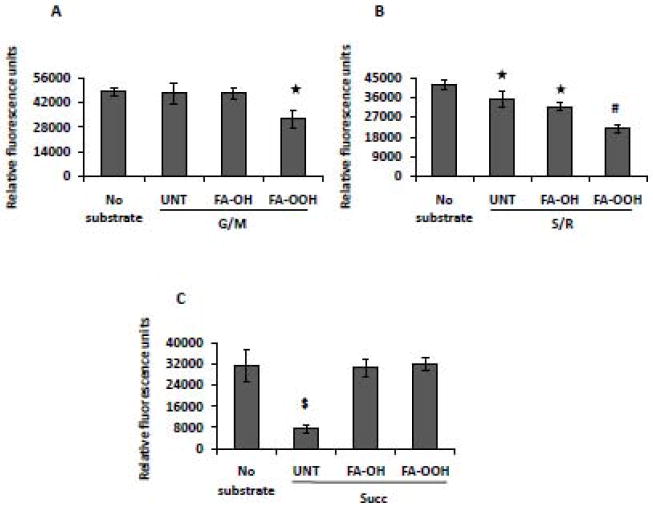
Muscle mitochondria (0.5 mg of protein/ml; final concentration 0.1 mg/ml) were incubated with EGTA-free incubation buffer (with or without isocitrate dehydrogenase), and aconitase activity was measured via fluorescence at 355 nm (excitation) and 460 nm (emission). The difference in the fluorescence reading in the presence/absence of isocitrate dehydrogenase was taken as a measure of aconitase activity. FA-OH or FA-OOH was added at a final concentration of 0.75 μM per assay. The assays were performed with respiratory substrates specific for complex I or complex II and complex I inhibitor, rotenone - (A) glutamate/malate (G/M, 5 mM); (B) succinate (S, 5 mM) + rotenone (R, 0.5 uM) and (C) succinate (Succ, 5 mM). Statistical significance was assessed by one-way ANOVA with Newman Keul’s multiple comparison test (* p < 0.05; #p < 0.001; $ p < 0.0001 vs. untreated mitochondria). The results shown represent means ± S.E.M. for 8-10 individual mitochondrial preparations.
FA-OOH increases mitochondrial H2O2 production and inhibits aconitase activity during rotenone inhibition of complex I
To determine potential binding sites and sources of superoxide production in the presence of oxidized fatty acids, we tested the effect of FA-OH and FA-OOH on superoxide originating from complex I in mitochondria respiring on G/M and inhibited by rotenone. Under these experimental conditions, mitochondrial ROS production is directed exclusively towards the matrix [27]. FA-OOH (0.75 μM) dramatically increased the rate of mitochondrial H2O2 production (~3 fold, Figure 5A) and significantly inhibited aconitase activity (~ 30%; Figure 5B), whereas FA-OH had no effect. These data suggest that FA-OOH stimulates superoxide production from a site upstream of the rotenone binding site of complex I.
Figure 5. FA-OOH increases the rate of mitochondrial ROS production and inhibits aconitase activity in rotenone inhibited mitochondria.
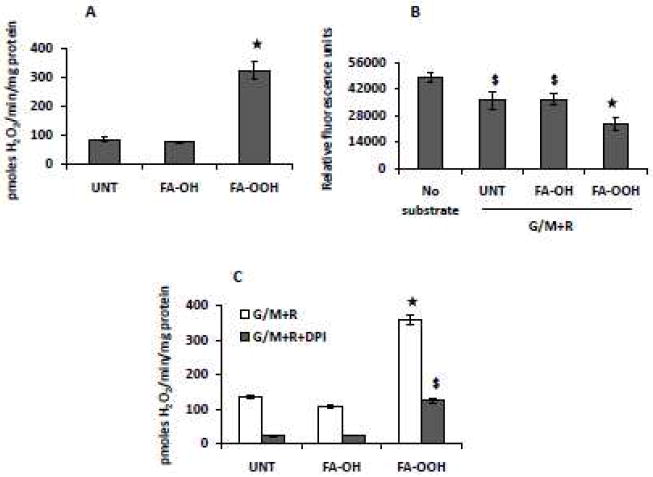
(A) Amplex red assay to measure H2O2 production and (B) aconitase assay were performed in the presence of 5 mM glutamate/malate (G/M) + 0.5 μM rotenone (R). FA-OH or FA-OOH was added at a final concentration of 0.75 μM. The effect of DPI (final concentration 25 μM) on the rate of mitochondrial ROS production was measured using amplex red in the presence of 5 mM G/M + 0.5 μM R. Statistical significance was assessed by one-way ANOVA with Newman Keul’s multiple comparison test (* p < 0.0001; $p < 0.001 vs. untreated mitochondria The results shown represent means ± S.E.M. for 7-8 (amplex red assay) and 8-10 (aconitase activity) individual mitochondrial preparations.
The FMN binding site of complex I has been shown to produce superoxide [42-44]. To address whether the FMN binding site of complex I was responsible for superoxide production in the presence of FA-OOH, we added diphenyleneiodonium (DPI), an inhibitor shown to be specific for FMN [45]. Addition of 25 μM DPI to mitochondria respiring on G/M and in the presence of rotenone inhibited the rate of mitochondrial H2O2 production by ~65% both in the presence and absence of FA-OOH (Figure 5C). If the DPI binding site was responsible for the increased rate of mitochondrial H2O2 production in the presence of FA-OOH, we would expect to see a greater decrease in the rate of mitochondrial H2O2 production in FA-OOH-treated mitochondria, relative to untreated mitochondria. From these data, we propose that FA-OOH does not stimulate superoxide production from the DPI binding site of complex I but from a site downstream of the flavin site, most likely the iron-sulfur clusters.
Vitamin E does not inhibit FA-OOH induced increase in the rate of mitochondrial ROS production
Finally, we wanted to determine if oxidized fatty acids increase the rate of mitochondrial ROS production by the propagation of lipid peroxidation reaction using the amplex red assay in the presence of vitamin E (vit E). Skeletal muscle mitochondria were incubated with FA-OH and FA-OOH (0.75 μM) for 15 mins. Thereafter, substrates specific for complex I (G/M, Figure 6A) and complex II (S/R, Figure 6B) were added followed by the addition of vit E (25 μM) after 5 mins and the rate of mitochondrial ROS production was measured. This concentration of vit E has been shown to decrease ROS production in mitochondria isolated from hepatocytes [46]. FA-OOH induced increase in the rate of mitochondrial H2O2 production was not affected by the presence of vit E in the reaction mixture suggesting that the effect of FA-OOH on mitochondrial ROS production is unlikely to be related to the propagation of lipid peroxidation reaction.
Figure 6. The addition of vitamin E does not inhibit FA-OOH induced increase in the rate of mitochondrial ROS production.
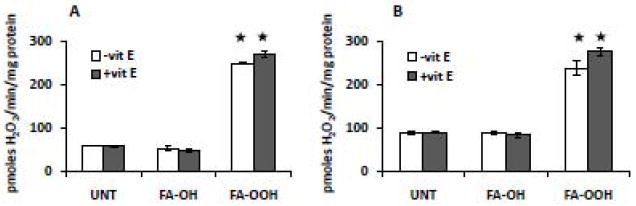
Skeletal muscle mitochondria were incubated with FA-OH or FA-OOH (final concentration 0.75 μM) for 10 mins followed by the addition of 25 μM vitamin E (vit E). The rate of mitochondria H2O2 production was measured using amplex red in the presence of substrates specific for complex I or II and complex I inhibitor, rotenone - (A) glutamate/malate, 5 mM and (B) succinate, 5 mM + rotenone, 0.5 μM. Statistical significance was assessed by one-way ANOVA with Newman Keul’s multiple comparison test (* p < 0.0001 vs. untreated mitochondria). The results shown represent means ± S.E.M. for 3-4 individual mitochondrial preparations.
DISCUSSION
Previous studies by our group and others have shown that muscle atrophy is associated with an increase in mitochondrial oxidative stress and dysfunction [2, 7, 47, 48]. Our recent study showed that mitochondria isolated from atrophied muscles generate significant levels of lipid hydroperoxides [19]. However, no studies thus far have investigated the role of fatty acid hydroperoxides in the modulation of mitochondrial oxidative stress and dysfunction in skeletal muscle mitochondria. Therefore, the aims of the present study were 1) to determine whether FA-OOH alters skeletal muscle mitochondrial function and 2) to determine whether FA-OOH increases mitochondrial oxidative stress. We show that FA-OOH (but not the corresponding FA-OH) at low micromolar concentration decreases the rate of mitochondrial ATP production, RCR and enzymatic activity of respiratory chain complexes I and III in skeletal muscle mitochondria. We further show that 1) FA-OOH (but not the corresponding FA-OH) significantly increases the rate of mitochondrial ROS production during forward electron transfer; 2) FA-OOH-induced superoxide production is directed mainly towards the matrix suggesting an increase in oxidative stress in the mitochondrial matrix and 3) FA-OOH induced increase in mitochondrial oxidative stress originates mainly from complex I.
FA-OOH induces mitochondrial dysfunction in skeletal muscle mitochondria
The fatty acid hydroperoxide, linoleate hydroperoxide has been previously shown to increase state 4 respiration and inhibit oxidative phosphorylation in rat heart mitochondria [12]. Imagawa et al. (1982) later showed that the autoxidation product of methyl linoleate, methyl 9-hydroperoxy-12, 13-epoxy-10-octadecenoate, decreases state 3 respiration in heart and liver mitochondria respiring on the complex I-linked substrate, G/M but not in mitochondria respiring on the complex II-linked substrate, succinate [11]. Other studies in heart, brain and liver mitochondria also indicate that fatty acids and fatty acid hydroperoxides are important modulators of mitochondrial respiration and ATP production [8, 11, 12, 49, 50]. However, the effect of fatty acid hydroperoxides on mitochondrial function in skeletal muscle mitochondria has not been studied. In agreement with previous studies, our results demonstrate that FA-OOH decreases RCR and ATP production in skeletal muscle mitochondria indicating a decline in mitochondrial function.
FA-OOH induces superoxide production from site(s) upstream of the rotenone binding site on complex I during NADH oxidation
Complex I is one of the major sites of superoxide production from the ETC. The ubiquinone reduction site, the iron-sulfur clusters and the flavin binding site have each been proposed as plausible sites of superoxide release from complex I [44, 51-54]. In mitochondria respiring on NADH-linked substrates, the rate of mitochondrial H2O2 production is low, because the redox centers on complex I are relatively oxidized. However, inhibition of the ubiquinone acceptor site of complex I with rotenone leads to the reduction of redox centers located upstream and is associated with significant increase in the rate of mitochondrial H2O2 production [27, 55-57]. In this study, FA-OOH increased the rate of mitochondrial ROS production in mitochondria respiring on NADH-linked substrate, which increased further during rotenone inhibition. Previous studies have suggested that long-chain fatty acids increase the rate of complex I derived ROS production by a rotenone-like action [8, 9]. However, based on our finding, we propose that the FA-OOH induced increase in the rate of mitochondrial ROS production from complex I (during FET) is likely due to its interaction with a site different from the rotenone binding site. Addition of DPI, an inhibitor specific for the flavin binding site of complex I decreased the rate of mitochondrial ROS production to a similar extent in the presence or absence of FA-OOH, in mitochondria respiring on G/M (in the presence of rotenone). These data suggest that FA-OOH probably does not stimulate superoxide production from the flavin binding site of complex I. Rotenone acting at complex I has been hypothesized to induce a conformational change in the enzyme, allowing for potential electron leakage from the N2-Iron-Sulfur cluster to oxygen [58]. It is possible that FA-OOH stimulates superoxide production from this site in mitochondria respiring on G/M, in the presence of rotenone. The hypothesis that FA-OOH stimulates superoxide production from complex I at a site upstream of the rotenone binding site is further supported by our data involving aconitase activity. We found that FA-OOH decreased aconitase activity in mitochondria respiring on G/M, in the presence of rotenone. This data suggests that FA-OOH stimulates superoxide production from a site different from the rotenone binding site on complex I. Our future experiments will investigate the possibility of the N2-Iron-Sulfur cluster as a plausible site of FA-OOH-mediated superoxide release by complex I by determining the effect of stigmatellin, which has been proposed to bind to this site when complex I is inhibited with rotenone [58].
FA-OOH induces matrix directed superoxide production from complex III during succinate oxidation (in the presence of rotenone)
Mitochondrial ROS production in the presence of succinate (when RET is inhibited with rotenone) is generally low, with values produced that are similar to mitochondria respiring on NADH-linked substrates [37, 59]. Under these experimental conditions (S/R), ROS production is thought to be entirely confined to complex III of the ETC [60, 61] as superoxide is released towards both sides of the mitochondrial inner membrane [27]. Superoxide generated at the Qo site is released towards the intermembrane space, whereas superoxide generated at the Qi site is directed towards the matrix [27]. In this study, FA-OOH (but not FA-OH) increased the rate of mitochondrial ROS production in mitochondria respiring on S/R. The addition of CuZnSOD to mitochondria under the same experimental conditions failed to increase the rate of mitochondrial H2O2 production. Furthermore, FA-OOH did not increase the EPR-derived superoxide signal in mitochondria respiring on S/R. These findings suggest that FA-OOH does not stimulate mitochondrial superoxide release from the Qo site of complex III. As mentioned earlier, complex III has been shown to release superoxide on both sides of the inner mitochondrial membrane [27, 31]. FA-OOH increased the rate of mitochondrial ROS production and decreased aconitase activity in mitochondria respiring on S/R. These data suggest that FA-OOH stimulates superoxide production from complex III but that is only directed exclusively towards the mitochondrial matrix.
Mitochondrial superoxide production during succinate oxidation suggests a rotenone-like effect of FA-OH and FA-OOH
Mitochondria respiring on the complex II substrate succinate (in the absence of rotenone) exhibit high rates of mitochondrial ROS production [44, 62]. The addition of rotenone decreases the high rate of mitochondrial ROS production during succinate oxidation, suggesting that the majority of this ROS is derived by RET from ubiquinol to complex I [53, 63]. The inhibition of aconitase activity (~80%) in mitochondria respiring on succinate in the present study supports this finding. EPR-based superoxide measurements showing no difference in extra-mitochondrial superoxide release in mitochondria respiring on succinate or S/R further suggest that majority of the ROS production during succinate oxidation is derived from complex I. The rate of mitochondrial ROS production during succinate oxidation was strongly inhibited by both FA-OH and FA-OOH, decreasing ~50% even with the lowest concentration tested. This finding was further supported by the observation that both FA-OH and FA-OOH inhibited the decline in aconitase activity during succinate oxidation. The high rate of mitochondrial ROS production during RET has been shown to be extremely sensitive to changes in membrane potential [64] with decrease in membrane potential associated with decline in the rate of mitochondrial ROS production [65]. In the present study, both FA-OH and FA-OOH decreased the mitochondrial membrane potential during succinate oxidation. Thus, the inhibition of the high rate of mitochondrial ROS production during RET by both FA-OH and FA-OOH may be attributed to depolarization of the inner mitochondrial membrane, or a rotenone-like effect for these fatty acid metabolites, under these experimental conditions [8]. Interestingly, previous studies in liver and heart mitochondria have shown that fatty acid hydroperoxides (but not fatty acid hydroxides) decrease membrane potential but only during conditions that inhibit mitochondrial glutathione content [33, 34].
FA-OOH induced increase in mitochondrial ROS production is associated with decrease in the activity of respiratory chain complexes I and III
The inhibition of ETC complexes has been proposed as one of the likely mechanisms for the effect of fatty acids on mitochondrial dysfunction and ROS production [8, 50]. In particular, complexes I and III have been shown to be partially inactivated in the presence of unsaturated fatty acids [8, 13, 50, 66]. In agreement with earlier findings, we found that FA-OOH partially inactivates respiratory chain complexes I and III which, in part, may explain its effect on mitochondrial dysfunction and ROS production. It is likely that the hydroperoxide group on FA-OOH’s interact with specific proteins on respiratory chain complexes I and III, thereby altering their function and leading to decline in activity of complexes I and III.
Decrease in mitochondrial content of reduced glutathione (GSH) could be one of the likely mechanisms by which FA-OOH may increase mitochondrial H2O2 release. Matrix GSH provides the reducing equivalents necessary for glutathione peroxidase to convert mitochondrial H2O2 to H2O plus O2. Depletion of GSH is associated with an increase in mitochondrial H2O2 release [44, 67]. Moreover, it has been shown that the branched chain fatty acid; phytanic acid decreases the ratio of reduced to oxidized glutathione and increases mitochondrial H2O2 release in isolated brain mitochondria [8]. We found that depletion of glutathione with 1-chloro-2,4-dinitrobenzene increased mitochondrial H2O2 release which increased further in the presence of FA-OOH (data not shown). This observation suggests that FA-OOH’s probably do not increase mitochondrial H2O2 release by this mechanism. We plan to examine this in detail in future experiments using skeletal muscle mitochondria isolated from glutathione peroxidase 1 knockout mice.
In conclusion, we describe an important finding that at low micromolar concentrations, FA-OOH (but not FA-OH) induces skeletal muscle mitochondrial dysfunction. Moreover, using methods that can distinguish between superoxide released towards the matrix or towards the intermembrane space, we demonstrate that FA-OOH significantly increases oxidative stress in the mitochondrial matrix, with complex I as the major site of superoxide production (most likely from a site upstream of the ubiquinone binding site but downstream from the flavin binding site). These findings are of particular interest in conditions that exhibit an increased generation of fatty acid hydroperoxides by skeletal muscle mitochondria, e.g., denervation-induced muscle atrophy.
Acknowledgments
This work was supported by the National Institute of Aging [grant AG20591 (H.V.R.)], and Muscular Dystrophy Association [grant # MDA 10047 (H.V.R.)]. We would like to greatly acknowledge the editorial support provided by Ms. Corinne Price.
LIST OF ABBREVIATIONS
The abbreviations used are:
- FA-OH
fatty acid hydroxide
- FA-OOH
fatty acid hydroperoxide
- ETC
electron transport chain
- RCR
respiratory control ratio
- RET
reverse electron transfer
- FET
forward electron transfer
- EPR
electron paramagnetic resonance
- ROS
reactive oxygen species
- DIPPMPO
5-diisopropoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Muller FL, Song W, Liu Y, Chaudhuri A, Pieke-Dahl S, Strong R, Huang TT, Epstein CJ, Roberts LJ, 2nd, Csete M, Faulkner JA, Van Remmen H. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic Biol Med. 2006;40:1993–2004. doi: 10.1016/j.freeradbiomed.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 2.Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A, Van Remmen H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1159–1168. doi: 10.1152/ajpregu.00767.2006. [DOI] [PubMed] [Google Scholar]
- 3.Fulle S, Protasi F, Di Tano G, Pietrangelo T, Beltramin A, Boncompagni S, Vecchiet L, Fano G. The contribution of reactive oxygen species to sarcopenia and muscle ageing. Exp Gerontol. 2004;39:17–24. doi: 10.1016/j.exger.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Mansouri A, Muller FL, Liu Y, Ng R, Faulkner J, Hamilton M, Richardson A, Huang TT, Epstein CJ, Van Remmen H. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mech Ageing Dev. 2006;127:298–306. doi: 10.1016/j.mad.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Desai VG, Weindruch R, Hart RW, Feuers RJ. Influences of age and dietary restriction on gastrocnemius electron transport system activities in mice. Arch Biochem Biophys. 1996;333:145–151. doi: 10.1006/abbi.1996.0375. [DOI] [PubMed] [Google Scholar]
- 6.Drew B, Phaneuf S, Dirks A, Selman C, Gredilla R, Lezza A, Barja G, Leeuwenburgh C. Effects of aging and caloric restriction on mitochondrial energy production in gastrocnemius muscle and heart. Am J Physiol Regul Integr Comp Physiol. 2003;284:R474–480. doi: 10.1152/ajpregu.00455.2002. [DOI] [PubMed] [Google Scholar]
- 7.Adhihetty PJ, O’Leary MF, Chabi B, Wicks KL, Hood DA. Effect of denervation on mitochondrially mediated apoptosis in skeletal muscle. J Appl Physiol. 2007;102:1143–1151. doi: 10.1152/japplphysiol.00768.2006. [DOI] [PubMed] [Google Scholar]
- 8.Schonfeld P, Reiser G. Rotenone-like action of the branched-chain phytanic acid induces oxidative stress in mitochondria. J Biol Chem. 2006;281:7136–7142. doi: 10.1074/jbc.M513198200. [DOI] [PubMed] [Google Scholar]
- 9.Schonfeld P, Wojtczak L. Fatty acids decrease mitochondrial generation of reactive oxygen species at the reverse electron transport but increase it at the forward transport. Biochim Biophys Acta. 2007;1767:1032–1040. doi: 10.1016/j.bbabio.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Martinez B, Perez-Castillo A, Santos A. The mitochondrial respiratory complex I is a target for 15-deoxy-delta12,14-prostaglandin J2 action. J Lipid Res. 2005;46:736–743. doi: 10.1194/jlr.M400392-JLR200. [DOI] [PubMed] [Google Scholar]
- 11.Imagawa T, Kasai S, Matsui K, Nakamura T. Methyl hydroperoxy-epoxy-octadecenoate as an autoxidation product of methyl linoleate: a new inhibitor-uncoupler of mitochondrial respiration. J Biochem. 1982;92:1109–1121. doi: 10.1093/oxfordjournals.jbchem.a134027. [DOI] [PubMed] [Google Scholar]
- 12.Shiotani A, Watanabe T, Matsuoka I, Nakamura T. Comparative studies on the effects of linoleate and methyl linoleate and their hydroperoxides on the respiration and reactivities of rat heart mitochondria. J Biochem. 1980;88:677–683. doi: 10.1093/oxfordjournals.jbchem.a133019. [DOI] [PubMed] [Google Scholar]
- 13.Takeuchi Y, Morii H, Tamura M, Hayaishi O, Watanabe Y. A possible mechanism of mitochondrial dysfunction during cerebral ischemia: inhibition of mitochondrial respiration activity by arachidonic acid. Arch Biochem Biophys. 1991;289:33–38. doi: 10.1016/0003-9861(91)90438-o. [DOI] [PubMed] [Google Scholar]
- 14.Di Paola M, Lorusso M. Interaction of free fatty acids with mitochondria: coupling, uncoupling and permeability transition. Biochim Biophys Acta. 2006;1757:1330–1337. doi: 10.1016/j.bbabio.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 15.Wojtczak L, Schonfeld P. Effect of fatty acids on energy coupling processes in mitochondria. Biochim Biophys Acta. 1993;1183:41–57. doi: 10.1016/0005-2728(93)90004-y. [DOI] [PubMed] [Google Scholar]
- 16.Lesnefsky EJ, Gudz TI, Moghaddas S, Migita CT, Ikeda-Saito M, Turkaly PJ, Hoppel CL. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J Mol Cell Cardiol. 2001;33:37–47. doi: 10.1006/jmcc.2000.1273. [DOI] [PubMed] [Google Scholar]
- 17.Yoshida S, Inoh S, Asano T, Sano K, Shimasaki H, Ueta N. Brain free fatty acids, edema, and mortality in gerbils subjected to transient, bilateral ischemia, and effect of barbiturate anesthesia. J Neurochem. 1983;40:1278–1286. doi: 10.1111/j.1471-4159.1983.tb13567.x. [DOI] [PubMed] [Google Scholar]
- 18.Boudina S, Abel ED. Mitochondrial uncoupling: a key contributor to reduced cardiac efficiency in diabetes. Physiology (Bethesda) 2006;21:250–258. doi: 10.1152/physiol.00008.2006. [DOI] [PubMed] [Google Scholar]
- 19.Bhattacharya A, Muller FL, Liu Y, Sabia M, Liang H, Song W, Jang YC, Ran Q, Van Remmen H. Denervation induces cytosolic phospholipase A2-mediated fatty acid hydroperoxide generation by muscle mitochondria. J Biol Chem. 2009;284:46–55. doi: 10.1074/jbc.M806311200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Estabrook RW. Mitochondrial respiratory control and the polarographic measurement of ADP:O ratios. Methods Enzymol. 1974;10:41–47. [Google Scholar]
- 21.Birch-Machin MA, Turnbull DM. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. [DOI] [PubMed] [Google Scholar]
- 22.Boveris A, Cadenas E. Mitochondrial production of superoxide anions and its relationship to the antimycin insensitive respiration. FEBS Lett. 1975;54:311–314. doi: 10.1016/0014-5793(75)80928-8. [DOI] [PubMed] [Google Scholar]
- 23.Krahenbuhl S, Talos C, Wiesmann U, Hoppel CL. Development and evaluation of a spectrophotometric assay for complex III in isolated mitochondria, tissues and fibroblasts from rats and humans. Clin Chim Acta. 1994;230:177–187. doi: 10.1016/0009-8981(94)90270-4. [DOI] [PubMed] [Google Scholar]
- 24.Santos DL, Moreno AJ, Leino RL, Froberg MK, Wallace KB. Carvedilol protects against doxorubicin-induced mitochondrial cardiomyopathy. Toxicol Appl Pharmacol. 2002;185:218–227. doi: 10.1006/taap.2002.9532. [DOI] [PubMed] [Google Scholar]
- 25.Votyakova TV, Reynolds IJ. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem. 2001;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- 26.Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem. 1997;253:162–168. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- 27.Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 28.Gardner PR. Aconitase: sensitive target and measure of superoxide. Methods Enzymol. 2002;349:9–23. doi: 10.1016/s0076-6879(02)49317-2. [DOI] [PubMed] [Google Scholar]
- 29.Miwa S, Brand MD. The topology of superoxide production by complex III and glycerol 3-phosphate dehydrogenase in Drosophila mitochondria. Biochim Biophys Acta. 2005;1709:214–219. doi: 10.1016/j.bbabio.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 30.O’Malley Y, Fink BD, Ross NC, Prisinzano TE, Sivitz WI. Reactive oxygen and targeted antioxidant administration in endothelial cell mitochondria. J Biol Chem. 2006;281:39766–39775. doi: 10.1074/jbc.M608268200. [DOI] [PubMed] [Google Scholar]
- 31.Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem. 2003;278:5557–5563. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- 32.Hinkle PC, Butow RA, Racker E, Chance B. Partial resolution of the enzymes catalyzing oxidative phosphorylation. XV. Reverse electron transfer in the flavin-cytochrome beta region of the respiratory chain of beef heart submitochondrial particles. J Biol Chem. 1967;242:5169–5173. [PubMed] [Google Scholar]
- 33.Masini A, Ceccarelli D, Gallesi D, Giovannini F, Trenti T. Lipid hydroperoxide induced mitochondrial dysfunction following acute ethanol intoxication in rats. The critical role for mitochondrial reduced glutathione. Biochem Pharmacol. 1994;47:217–224. doi: 10.1016/0006-2952(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 34.Masini A, Ceccarelli D, Trenti T, Gallesi D, Muscatello U. Mitochondrial inner membrane permeability changes induced by octadecadienoic acid hydroperoxide. Role of mitochondrial GSH pool. Biochim Biophys Acta. 1992;1101:84–89. doi: 10.1016/0167-4838(92)90471-o. [DOI] [PubMed] [Google Scholar]
- 35.Chalier F, T P. 5-(Diisopropoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide, DIPPMPO, a crystalline analog of the nitrone DEPMPO: synthesis and spin trapping properties. Chem Soc Perkin Trans. 2002;2:2110–2117. [Google Scholar]
- 36.Lustgarten MS, Jang YC, Liu Y, Muller FL, Qi W, Steinhelper M, Brooks SV, Larkin L, Shimizu T, Shirasawa T, McManus LM, Bhattacharya A, Richardson A, Van Remmen H. Conditional knockout of Mn-SOD targeted to type IIB skeletal muscle fibers increases oxidative stress and is sufficient to alter aerobic exercise capacity. Am J Physiol Cell Physiol. 2009;297:C1520–1532. doi: 10.1152/ajpcell.00372.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 38.Lambert AJ, Brand MD. Reactive oxygen species production by mitochondria. Methods Mol Biol. 2009;554:165–181. doi: 10.1007/978-1-59745-521-3_11. [DOI] [PubMed] [Google Scholar]
- 39.Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, Miziorko H, Epstein CJ, Wallace DC. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci U S A. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gardner PR, Raineri I, Epstein LB, White CW. Superoxide radical and iron modulate aconitase activity in mammalian cells. J Biol Chem. 1995;270:13399–13405. doi: 10.1074/jbc.270.22.13399. [DOI] [PubMed] [Google Scholar]
- 41.Gardner PR, Nguyen DD, White CW. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc Natl Acad Sci U S A. 1994;91:12248–12252. doi: 10.1073/pnas.91.25.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kudin AP, Debska-Vielhaber G, Kunz WS. Characterization of superoxide production sites in isolated rat brain and skeletal muscle mitochondria. Biomed Pharmacother. 2005;59:163–168. doi: 10.1016/j.biopha.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 43.Vinogradov AD, Grivennikova VG. Generation of superoxide-radical by the NADH:ubiquinone oxidoreductase of heart mitochondria. Biochemistry (Mosc) 2005;70:120–127. doi: 10.1007/s10541-005-0090-7. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 45.Ino T, Nishioka T, Miyoshi H. Characterization of inhibitor binding sites of mitochondrial complex I using fluorescent inhibitor. Biochim Biophys Acta. 2003;1605:15–20. doi: 10.1016/s0005-2728(03)00060-4. [DOI] [PubMed] [Google Scholar]
- 46.Zhang JG, Nicholls-Grzemski FA, Tirmenstein MA, Fariss MW. Vitamin E succinate protects hepatocytes against the toxic effect of reactive oxygen species generated at mitochondrial complexes I and III by alkylating agents. Chem Biol Interact. 2001;138:267–284. doi: 10.1016/s0009-2797(01)00278-2. [DOI] [PubMed] [Google Scholar]
- 47.Chabi B, Ljubicic V, Menzies KJ, Huang JH, Saleem A, Hood DA. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell. 2008;7:2–12. doi: 10.1111/j.1474-9726.2007.00347.x. [DOI] [PubMed] [Google Scholar]
- 48.Max SR. Disuse atrophy of skeletal muscle: loss of functional activity of mitochondria. Biochem Biophys Res Commun. 1972;46:1394–1398. doi: 10.1016/s0006-291x(72)80130-x. [DOI] [PubMed] [Google Scholar]
- 49.Hillered L, Chan PH. Effects of arachidonic acid on respiratory activities in isolated brain mitochondria. J Neurosci Res. 1988;19:94–100. doi: 10.1002/jnr.490190113. [DOI] [PubMed] [Google Scholar]
- 50.Schonfeld P, Wojtczak L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic Biol Med. 2008;45:231–241. doi: 10.1016/j.freeradbiomed.2008.04.029. [DOI] [PubMed] [Google Scholar]
- 51.Galkin A, Brandt U. Superoxide radical formation by pure complex I (NADH:ubiquinone oxidoreductase) from Yarrowia lipolytica. J Biol Chem. 2005;280:30129–30135. doi: 10.1074/jbc.M504709200. [DOI] [PubMed] [Google Scholar]
- 52.Kussmaul L, Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci U S A. 2006;103:7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I) J Biol Chem. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 54.Genova ML, Ventura B, Giuliano G, Bovina C, Formiggini G, Parenti Castelli G, Lenaz G. The site of production of superoxide radical in mitochondrial Complex I is not a bound ubisemiquinone but presumably iron-sulfur cluster N2. FEBS Lett. 2001;505:364–368. doi: 10.1016/s0014-5793(01)02850-2. [DOI] [PubMed] [Google Scholar]
- 55.Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr. 1999;31:347–366. doi: 10.1023/a:1005427919188. [DOI] [PubMed] [Google Scholar]
- 56.Hansford RG, Hogue BA, Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J Bioenerg Biomembr. 1997;29:89–95. doi: 10.1023/a:1022420007908. [DOI] [PubMed] [Google Scholar]
- 57.Lambert AJ, Buckingham JA, Boysen HM, Brand MD. Diphenyleneiodonium acutely inhibits reactive oxygen species production by mitochondrial complex I during reverse, but not forward electron transport. Biochim Biophys Acta. 2008;1777:397–403. doi: 10.1016/j.bbabio.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 58.Fato R, Bergamini C, Bortolus M, Maniero AL, Leoni S, Ohnishi T, Lenaz G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim Biophys Acta. 2009;1787:384–392. doi: 10.1016/j.bbabio.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J. 2001;353:411–416. doi: 10.1042/0264-6021:3530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 61.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 2008;294:C460–466. doi: 10.1152/ajpcell.00211.2007. [DOI] [PubMed] [Google Scholar]
- 62.Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ohnishi ST, Ohnishi T, Muranaka S, Fujita H, Kimura H, Uemura K, Yoshida K, Utsumi K. A possible site of superoxide generation in the complex I segment of rat heart mitochondria. J Bioenerg Biomembr. 2005;37:1–15. doi: 10.1007/s10863-005-4117-y. [DOI] [PubMed] [Google Scholar]
- 64.Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 65.Starkov AA, Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem. 2003;86:1101–1107. doi: 10.1046/j.1471-4159.2003.01908.x. [DOI] [PubMed] [Google Scholar]
- 66.Cocco T, Di Paola M, Papa S, Lorusso M. Arachidonic acid interaction with the mitochondrial electron transport chain promotes reactive oxygen species generation. Free Radic Biol Med. 1999;27:51–59. doi: 10.1016/s0891-5849(99)00034-9. [DOI] [PubMed] [Google Scholar]
- 67.Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernandez-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem. 1997;272:11369–11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]