

Abstract
One cornerstone of Chronic Kidney Disease (CKD) is fibrosis, as kidneys are susceptible due to their high vascularity and predisposition to ischemia. Presently, only therapies targeting the angiotensin receptor are used in clinical practice to retard the progression of CKD. Thus, there is a pressing need for new therapies designed to treat the damaged kidney. Several independent laboratories have identified a number of microRNAs that are dysregulated in human and animal models of CKD. We will explore the evidence suggesting that by blocking the activity of such dysregulated microRNAs, new therapeutics could be developed to treat the progression of CKD.
Keywords: kidney, fibrosis, PPAR, chronic kidney disease, chronic allograft disease, ROS
Introduction
Chronic kidney disease (CKD) is a growing epidemic across the globe, but its effects are especially noticed throughout industrialized nations. In the USA alone, CKD affects approximately 26 million people—a total of more than 8% of the population.1 Furthermore, the prevalence of CKD amongst adults in Beijing, China is about 13%,2 and about 20% of the Japanese adult population is predicted to have stage 3–5 CKD.3 One of the cornerstones of CKD is fibrosis, as the kidneys are particularly susceptible due to their high vascularity and predisposition to ischemia.4 CKD itself is defined as a decrease in the glomerular filtration rate, and signs of chronic kidney damage include the leakage of various plasma proteins into the urine. Pathologically, CKD is characterized by fibrosis of the glomeruli (glomerulosclerosis), interstitial fibrosis, injury with flattening (atrophy), loss of tubule epithelium, inflammation due to leukocyte recruitment, and loss of peritubular capillaries5 (Figure 1). Recent studies indicate that myofibroblasts, the main cells responsible for fibrosis deposition in the kidney, may play a crucial role in many of the features of CKD, such as inflammation, loss of capillaries, and fibrosis.6 Myofibroblasts therefore represent a new therapeutic target against CKD.
Figure 1. Characteristics of chronic kidney disease in glomeruli and interstitium of kidney cortex.
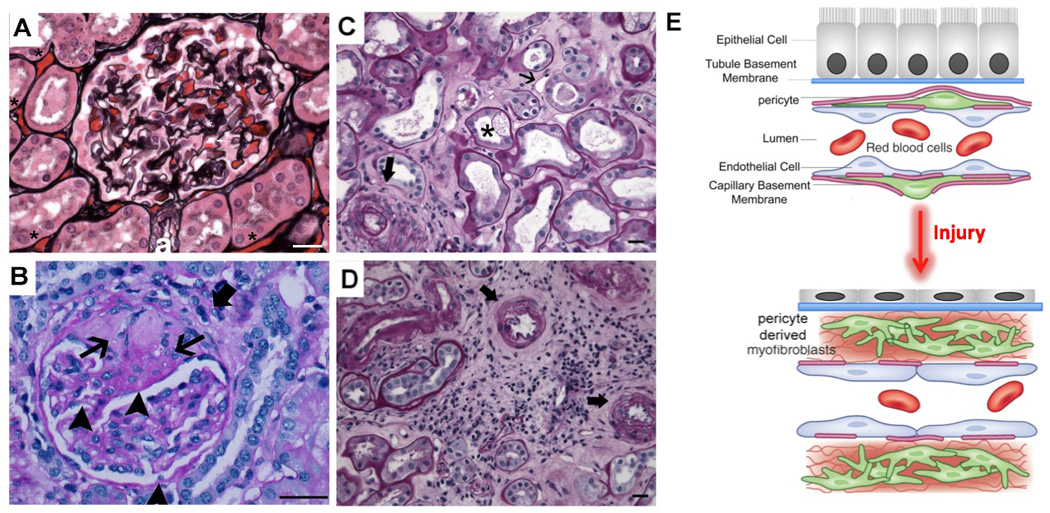
(A) Normal human glomerulus and surrounding tubules and peritubular capillaries (PTCs) filled with erythrocytes (* placed above examples of PTCs) stained with Silver methenamine combined with PAS (Jones) stain which highlights collagens. Arteriole (a) is shown. Note back-to-back tubules with cuboidal or columnar epithelium. (B) Sclerotic glomerulus showing wedge shaped sclerotic region showing dense pink material on PAS stained section (arrowhead) and rather acellular weaker pink stained material more peripherally (arrow) and obliteration of capillary loops. Sclerotic region is fused to Bowman’s capsule where there is local destruction of the basement membrane and periglomerular fibrosis (thick arrow). At the lower pole, a combination of increased cellularity and fibrosis in the mesangium, and basement membrane thickening in glomerular loops. (C) Jones stained image of cortex from diabetic nephropathy, showing injured tubules (tubule atrophy and tubule cell vacuolization, apoptotic cells, arrow), marked reduction in capillary density (* placed adjacent to examples of PTCs), expansion of the interstitial space with fibrotic material (fine black stain), and an increase in inflammatory cells. Note also thickening of the tubule basement membrane (black). (D) Trichrome stained image of kidney cortex from ischemic kidney disease showing marked expansion of interstitial fibrosis (cyan color), which has overtaken all of the tubules. The fibrosis is cellular showing inflammatory cells and myofibroblasts. The remaining tubules all show tubular atrophy with intraluminal debris. (E) Schema showing cellular mechanisms of CKD development in the kidney cortical interstitium.
Numerous diseases and conditions may affect renal function, leading to CKD, including diabetes mellitus, hypertension and cancer. Recipients of heart, liver and lung transplants are also predisposed to developing progressive CKD, and many drugs that are used to treat life threatening diseases are toxic to the kidney with long lasting consequences. Currently, there is a large unmet need for new therapies to counteract the progression of CKD and the equivalent process affecting kidney transplants, known as chronic allograft dysfunction (CAD). CKD and CAD often follow a relentless course, which progresses toward organ failure, even if the initiating factors have been adequately addressed. Furthermore, episodes of acute kidney damage that occur during a number of illnesses or as a consequence of medical treatment have been shown to accelerate the progression of CKD.7 Presently, only therapies targeting the angiotensin receptor (angiotensin receptor-1 blockers [ARB] or angiotensin converting enzyme [ACE] inhibitors) are used in clinical practice to retard the progression of CKD or CAD. Thus, there is a pressing need for new therapies designed to treat and protect the damaged kidney.
Several independent laboratories have identified a number of microRNAs (miRNA) that are dysregulated in human CKD with fibrosis and in animal models of CKD. MicroRNAs are a family of small, non-coding RNAs that control gene expression by inhibiting the translation of their complementary ‘target’ messenger RNAs (mRNA).8–10 They predominantly do so by facilitating the degradation of target mRNAs and also by inhibiting protein translation of target mRNAs (translational suppression).11 A single miRNA silences a number of functionally related genes, and the suppression of these genes gives miRNA a functional purpose. Dysregulated miRNA expression has, however, been identified in human diseases and is also readily observed in animal models.12 In vivo, modulation of dysregulated miRNAs can attenuate the manifestation of disease, suggesting that aberrant miRNA can contribute to disease pathogenesis.13,14 We will therefore explore the evidence suggesting that by blocking the activity of one or more of such dysregulated miRNA, new therapeutics could be developed to treat the progression of CKD and CAD.15
The Role of Fibrosis and Epithelial Cell Injury in Kidney Disease
Recent studies, primarily from animal models of kidney disease, have identified a separate lineage of cells of mesenchymal origin in the normal kidney referred to as ‘pericytes’ or ‘resident fibroblasts’. These cells represent >5% of normal kidney cells and perform critical homeostatic and regenerative functions, particularly with respect to microvascular homeostasis.16–18 In CKD, these pericytes and resident fibroblasts become activated and are then referred to as myofibroblasts (Figure 1). Myofibroblasts are the contractile cells that deposit fibrillar pathological matrix, known as fibrosis or scar tissue, in the kidney. Normally, pericytes nurse capillaries and support microvascular stability; however, when they become myofibroblasts, they no longer perform these functions. Activated pericytes (i.e. myofibroblasts) leave capillaries unstable and prone to ineffective angiogenesis, increasing their permeability and often leading to capillary demise, which is seen in the kidney as capillary rarefaction.19
Myofibroblasts in the glomerulus deposit fibrillar pathological matrix known as mesangial matrix expansion or mesangial nodules, and this is referred to as glomerulosclerosis when combined with loss of glomerular capillaries.19 Pathological fibrillar matrix (fibrosis) also accumulates in the virtual space between capillaries and tubules of the nephron or around capillaries of the glomerulus, thereby destroying local structures (Figure 1). In the glomerulus, fibrosis frequently accumulates initially in the mesangial area, along the capillary loop itself, or amongst proliferating cells that occupy the urinary space, often known as a ‘crescent’. The fibrotic material formed throughout these regions of the kidney encroaches on capillaries and prevents them from functioning. In addition, myofibroblast are contractile and can distort tissue architecture, as these cells are also inflammatory cells secreting innate immune cytokines, chemokines and oxygen radicals (Figure 1). Overall, fibrosis reduces nephron function and promotes tissue ischemia, distorting normal tissue architecture. Myofibroblasts, which cause fibrosis, are a source of inflammation and promote loss of capillaries. Myofibroblast are a major new target for therapeutics in kidney disease.19
Numerous studies have identified cell stress or cell injury in the tubule epithelial compartment, particularly the proximal tubule, as a stimulus for fibrosis.20 Increasingly, it is recognized that damaged or stressed epithelial cells exhibit a number of stereotyped responses: endoplasmic reticulum (ER) stress and the unfolded protein response; apoptosis and necrosis; activation of epithelial to mesenchymal transition genes; activation of transforming growth factor β (TGFβ); and cell-cycle arrest. Proximal tubule cells are particularly dependent on aerobic generation of high levels of ATP for survival and health. Therefore, factors that affect or compromise ATP generation have a profound impact on epithelial cell function. Not only do injured and stressed epithelial cells fail to perform normal functions, which are vital to the kidney, but they also generate a wide array of pro-fibrotic and inflammatory factors that can drive the manifestations of CKD from cell to cell signaling mechanisms (Figure 1).
Evidence of microRNA Dysregulation in Kidney Disease
Although there appears not to be kidney-specific microRNAs (miRNAs or miRs), a kidney signature has been reported by a number of recent studies using microarray approaches.21 In addition, a consistent pattern of up-regulated and down-regulated miRNAs has been described in response to acute and chronic kidney injuries.11,22 Some of these miRNA changes will reflect recruitment of inflammatory cells, but many changes reflect abnormal regulation of genes by miRNA, known as dysregulation. In transplanted human kidneys, miRNA profiles from kidney biopsies have been shown to distinguish patients with acute immunological rejection of the kidney transplant (allograft) from patients with a kidney transplant but no organ rejection. Acute rejection can be diagnosed with a high degree of accuracy by determining miRNA levels. Among the miRNA that were identified and indicative of organ rejection were 17 miRNAs—10 (let- 7c, miR-10a, miR-10b, miR-125a, miR-200a, miR-30a-3p, miR-30b, miR30c, miR30e- 3p, and miR-32) that were down-regulated in acute rejection biopsies compared to normal allograft biopsies, and 7 miRNAs (miR-142–5p, miR-142–3p, miR-155, miR- 223, miR-146b, miR-146a, and miR-342) that were up-regulated.23
In a diabetic nephropathy mouse model, Putta et al.24 showed that reduction of miR-192 retards renal fibrosis and improves proteinurea. Administration of modified RNA oligonucleotides that are complementary to the miR-192 sequence resulted in miR-192 degradation and attenuated histological evidence of glomerular expansion and renal interstitial fibrosis, as well as conferred improvement in renal function in the diabetic mice.24
Utilizing a mouse model of unilateral ureteral obstruction nephropathy, Qin et al.25 demonstrated that miR-29 negatively regulated fibrosis by targeting the process of collagen matrix synthesis rather than by inhibiting myofibroblast accumulation. MiR-29 is negatively regulated by TGFβ when it signals via the Smad3 dependent pathway. By using miRNA microarrays and real-time PCR, the investigators found that miR-29a, -b, -c family members were substantially reduced in the fibrotic kidney of UUO wild type mice but significantly increased in Smad3−/− mice in which renal fibrosis was reduced. These findings implicate miR-29 in TGFβ dependent fibrosis. From a therapeutic perspective, the identification of miRNAs that are elevated in disease and contribute to pathogenesis by silencing genes is critical for the innovation of new therapies.25
MicroRNAs in the Circulation and Urine as Markers to Identify and Predict Kidney Disease
It has recently been shown that miRNAs are found in the circulation in exosomes or stably bound to the assembly protein Argonaut. In addition to the blood stream, miRNAs have been detected in urine and other secreted fluids in a stable form.26–28 An increasing number of investigations suggest that several circulating miRNAs are biomarkers for cancer growth and organ injuries, such as cardiac ischemia. Therefore, it is probable that miRNAs will be released into urine or blood during kidney disease. Recently, miR-21 was shown to be upregulated in both the medulla and cortex of a rat model after gentamicin-induced nephrotoxicity.29 The researchers went further to investigate urine samples from patients with acute kidney injury (AKI) in the intensive care unit and compared those samples to healthy subjects. They found a significant 1.2 fold increase of miR-21 in the urine of AKI patients compared to healthy patients. Although there does not appear to be a miRNA restricted to the kidney in disease or development, future studies should be focused on the identification of patterns of miRNA that are released into the urine or blood by damaged kidneys.
MicroRNA-21 in kidney disease
Transgenically over-expressed microRNA-21 (miR-21) in lymphocytes appears to function as an oncogenic miRNA (i.e. an oncomir).13 In these lymphocytic cancer cells, miR-21 prevents apoptotic cell death; however, miR-21 is not specific to cancer cells as it is widely expressed in many tissues, including the normal kidney. Conversely, up-regulation of miR-21 has been indicated in a range of kidney diseases, and is one of the most highly expressed miRNAs in kidney disease.11 Recent studies reported that miR-21 levels were increased in cardiac fibroblasts of failing hearts and it was shown that miR-21 results in over-activation of the P42/P44 mitogen activated protein (MAP) kinase signaling pathway in cardiac fibroblasts, but not in cardiomyocytes.10 Silencing of miR-21 by a specific modified RNA oligonucleotide complementary to the miR-21 sequence, which was conjugated to cholesterol and known as ‘antagomir’, reduced cardiac P42/P44 MAP kinase activity and interstitial fibrosis and attenuated cardiac dysfunction in models of cardiac failure in mice. Although follow-up studies failed to replicate these findings,30 these cardiac studies suggested that miR-21 may play a role in amplifying the fibrogenic process and is therefore a potential target in other organs, such as the kidney.9
Chau et al. produced a mouse where the locus for miR-21 was successfully mutated.11 These mice were normal at 8 months of age in a sterile animal housing facility, exhibited normal fertility, and the kidneys developed normally. However, in several models of CKD, the miR21−/− kidneys suffered less tubule injury/atrophy, less fibrosis, less capillary destruction, and reduced P42/P44 MAP kinase pathway activation in response to the same degree of injury (Figure 2).11 The investigators developed and synthesized modified oligonucleotides based around ribose nucleotides that are complementary to miR-21. These freely enter cells in the kidney when injected subcutaneously, bind to intracellular miR-21 and stimulate its degradation, effectively silencing miR-21.11 Administration of these oligonucleotides (anti-miR21) to mice recapitulated the results of miR21−/− in kidney disease, and additionally reversed kidney disease. In a microarray analysis of the miR21−/− mouse kidneys, it was found that in non-diseased kidneys, there were no genes that were normally silenced by miR-21, even though it is expressed at quite high levels. However, in response to kidney injury, a characteristic pattern of genes, with the complementary sequence in their 3’UTRs to miR-21, were silenced in the miR21+/+ kidneys compared to the miR21−/− kidneys.11 These observations suggested that, unlike many other miRNAs, miR-21 is sequestered in an intracellular compartment and released into the cytoplasm in response to cell stress where it becomes active. The genes (more than 80) that were silenced in diseased kidneys by miR-21 were surprising. Rather than inflammatory, innate immunity or fibrotic/matrix turnover genes, the genes were all involved in cell metabolic functions. In particular, there were genes that play crucial roles in lipid metabolism, fatty acid oxidation, and redox regulation in the mitochondria.11
Figure 2. Kidney disease in mice that lack miR-21 compared to WT mice following kidney injury.
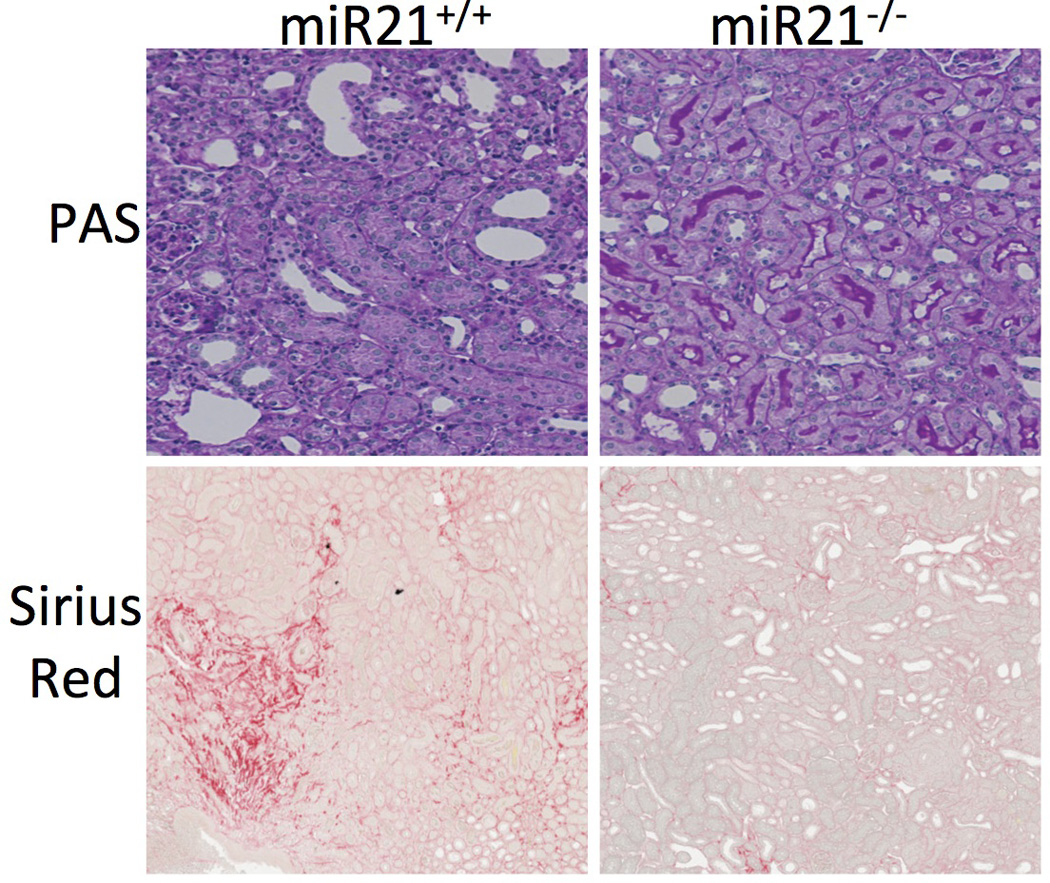
Sirius red stained low power images showing red stained interstitial fibrosis following kidney injury, and medium power images showing PAS stain of kidney cortex after injury. Note reduced fibrosis in kidneys lacking miR-21. Also note that kidney epithelial cells are more injured in WT showing increased flattening and loss of typical purple brush border, whereas these features are more preserved in miR21−/− kidneys.
The peroxisomal and mitochondrial fatty acid oxidation metabolic pathway, regulated by the transcription factor Peroxisome Proliferator Activated Receptor-α (PPARα), was identified as a major target for miR-21; in fact 9 distinct enzymes induced by PPARα in this catalytic pathway were all specifically silenced by miR-21 in the kidney (Figure 3).11 MiR-21 engaged this pathway in kidney epithelial cells, particularly proximal tubules, but also in myofibroblasts. Furthermore, over-expression of PPARα protected kidneys from the development of fibrosis. Finally, it was shown that while Ppara−/− mice exhibited increased fibrogenesis and epithelial injury, oligonucleotides that block miR-21 action were no longer able to inhibit epithelial injury and fibrogenesis, implicating the PPARα pathway as a major target of miR-21 in kidney disease.11 Previously, PPARα was identified as a protective transcription factor in acute kidney injury.31,32 PPARα stimulates the proliferation of peroxisomes, and these organelles play a critical role in the oxidation of fatty acids, also providing detoxification and metabolism of oxygen radicals, including H2O2. Detoxification of accumulated fatty acids in injured cells and metabolism of excess oxygen radicals are important factors that promote cell survival and function. In addition, peroxisomes are a major site of long chain fatty acid metabolism by the α- and β-oxidation pathways. Epithelial cells rely heavily on fatty acids for ATP generation. While peroxisomal β-oxidation enzymes reduce very long chain fatty acids, mitochondrial β-oxidation enzymes oxidize fatty acids with less than 22 carbon atoms, resulting in ATP generation.31 In proximal tubules, nuclear receptor PPARα plays a pivotal role in regulating peroxisomal and mitochondrial fatty acid oxidation in kidney tissue.32 Moreover, many of the enzymes critical in this pathway (Acyl-CoA oxidase 1; Acyl-CoA Dehydrogenases; and Carnitine Palmitoyl Transferase) are also silenced by miR-21.11
Figure 3. Schema showing importnat gene products and pathways silenced by miR-21 in animal models of kidney disease.
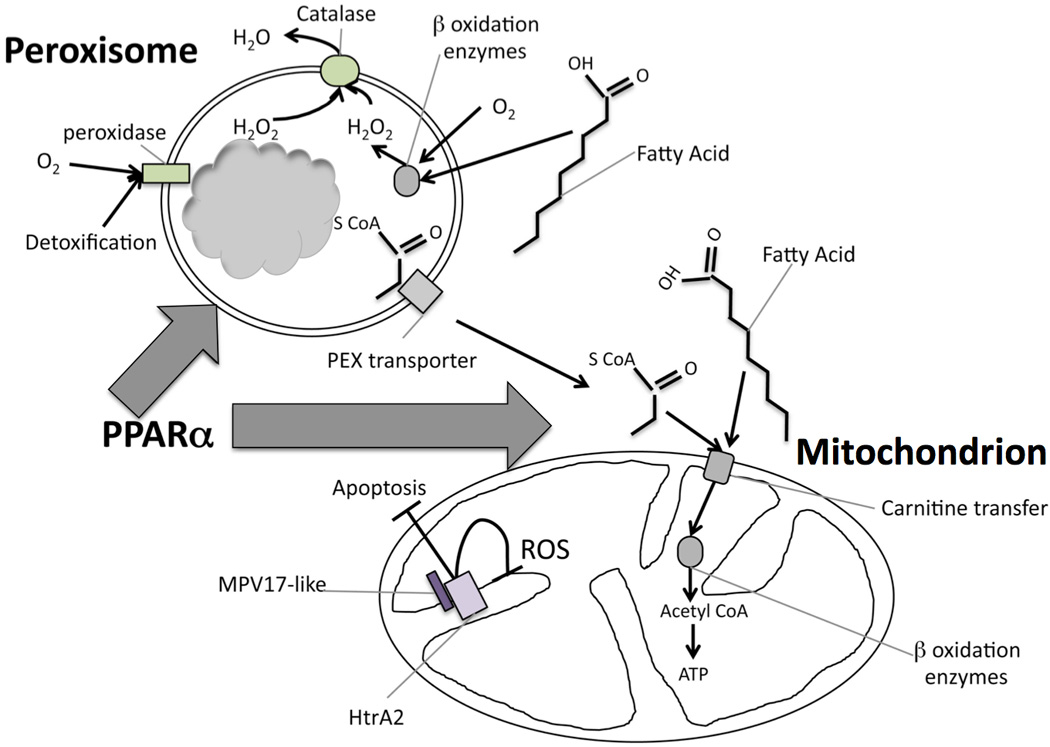
MiR-21 silences the transcriptional regulator PPARα and many of the downstream enzymes in fatty acid metabolism that are also regulated by PPARα, including transporters and enzymes (shown in grey) of the β-oxidation metabolic pathway of fatty acids that occurs in peroxisomes and mitochondria. The consequence of miR-21 activity is to reduce metabolism of fatty acids. In addition, miR-21 increases reactive oxygen species and toxin formation/accumulation. First, by suppressing peroxisome formation and activity, the metabolism of H2O2 is retarded by miR-21, and second, miR-21 silences genes that inhibit ROS generation in mitochondria, including MPV17-like which exerts its inhibitory function by binding to the mitochondrial protease HrtA2. In this configuration HrtA2 is also anti-apoptotic.
In addition to the fatty acid metabolic pathway, Chau et al. identified enzymes and co-factors involved in the intracellular redox state and inhibition of reactive oxygen species (ROS) generation as miR-21 targets, particularly in epithelial cells. Consistent with miR-21 stimulating ROS generation in kidney epithelium, they showed that in diseased kidneys from miR21−/− mice there was reduced evidence of ROS generation, in interstitial as well as epithelial cells.11 One of the proteins silenced by miR-21 was Mpv17-like (Figure 3). This, obscurely named protein was identified in kidney epithelial cell mitochondrial membranes and has recently been reported to interact via its PDZ domain with a serine protease enzyme, HTRA2, which prevents mitochondrial ROS generation and prevents mitochondrial triggered apoptosis.33 Therefore, it is feasible that miR-21 also stimulates ROS generation in epithelial cells in response to cell stress. Impaired fatty acid metabolism and enhanced mitochondrial ROS generation in CKD development and progression are all implicated in these studies.11 It is therefore interesting that excessive production of ROS by mitochondria has been strongly implicated in CKD progression in a number of independent studies and in development of kidney abnormalities that occur with normal aging.34,35 The gene Mpv17, an orthologue of Mpv17-like, was identified more than 20 years ago as an epithelial and neuronal restricted protein located in the mitochondrial inner membrane and implicated in metabolism of ROS. Mice lacking Mpv17 spontaneously develop kidney disease with glomerulosclerosis and interstitial diseases similar to CKD. 36–38
In addition to these links between mitochondrial ROS generation, kidney disease progression, and miR-21, the families of PPAR transcription factors including PPARγ and PPARα have been drug targets for a number of years using a class of agents known as glitazones as ligands for PPARγ and fibrates as ligands for PPARα.32 Glitazones have been used to treat diabetes mellitus by enhancing insulin sensitivity, but have been documented in animals to exhibit anti-inflammatory and anti-fibrotic effects in kidney disease.39 Moreover, PPARγ has been suggested as a transcription factor that inhibits myofibroblast activation in a number of tissues including lung, liver and skin.40 Fibrates were used in patients with diabetes and it was demonstrated that end points of cardiovascular disease events and nonfatal myocardial infarction were significantly reduced by fibrates. Similarly, fenofibrates significantly reduced the microvascular complications of type 2 diabetes, including nephropathy.39 These clinical studies suggest that stimulation of peroxisome functions and fatty acid metabolism, potentially by anti- miR21, are desirable strategies in kidney disease (Figure 3).
Numerous investigators have identified that miR-21 is upregulated in models of kidney disease. This upregulation is also observed in human CKD, kidney transplants and in native kidney disease.11,21 Moreover, independent groups have shown that inhibition of miR-21 has therapeutic benefits.22 These independent findings add weight to the significance of miR-21 as a candidate target for kidney disease.
MicroRNAs as a potential therapeutic target in kidney disease
Since microRNAs are located in cytoplasm of cells and because they control cell functions, they are obvious candidates for therapy. In addition, because of sequence complementarity, drugs can be designed to specifically target a single miRNA, potentially avoiding conventional small molecule drug side effects. Recent advances in the development of oligonucleotides that can bind to mRNA and silence the translation of those mRNA (known as RNA silencing), heralded the development of small (22nt) oligonucleotides that are stable in the circulation, can freely enter cells and, because of their sequence complementarity, bind specifically causing their targets to be silenced.11 Such anti-microRNA oligonucleotides are already in clinical trials as therapeutics in cancer. The animal studies reported by Chau et al. showed that anti-miR-21 oligonucleotides accumulate in the kidney and effectively block miR-21 functions. Although miR-21 is normally expressed widely, it appears that it is not active in healthy cells, but becomes active only in stressed or injured cells. The fact that miR21−/− mice are healthy attests to a dormant role for miR-21 in cell physiology. It is quite likely therefore that anti-miR21 oligonucleotides will only block miR-21 actions in areas of tissue injury or inflammation.11 Finally, the current anti-miR-21 oligonucleotides have been administered to animals for several months without any toxicity. An additional question that arises from these studies is what is the functional role for miR-21 without disease, since it appears to be detrimental only during disease. Currently, miR-21 has no clear physiological role during disease-free conditions.
Conclusion
Recent studies have identified dysregulated miRNA in animal models and human tissue samples of kidney disease. Evidence from knockout mice and miRNA-silencing oligonucleotides in rodents indicates that miR-21 is an important pathological player in chronic kidney disease. The silencing of metabolic pathways by miR-21 dictates an important function in fatty acid metabolism and in the removal of reactive oxygen species in peroxisomes and mitochondria, both critical process for reducing fibrosis and disease. It is thus likely that recent advances in oligonucleotide technology will lead to a potential new type of therapeutic that specifically targets and degrades miRNAs.
Acknowledgements
Duffield Laboratory is supported by NIH grants (DK84077, DK87389, DK93493) University of Washington, Institute for Stem Cell & Regenerative Medicine, Genzyme Research in Progress Grant, the Nephcure Foundation and Regulus Therapeutics. The Portilla laboratory is supported by NIH grant DK075976, and a VA Merit Award.
Footnotes
Conflict of Interest
JSD serves on the Scientific Advisory Board of Regulus therapeutics and has a Research Grant sponsored by Regulus Therapeutics.
References
- 1.Snyder JJ, Foley RN, Collins AJ. Prevalence of CKD in the United States: a sensitivity analysis using the National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am. J.Kidney Dis. 2009;53:218–228. doi: 10.1053/j.ajkd.2008.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang L, Zhang P, Wang F, Zuo L, Zhou Y, Shi Y, et al. Prevalence and factors associated with CKD: a population study from Beijing. Am. J. Kidney Dis. 2008;51:373–384. doi: 10.1053/j.ajkd.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Imai E, Horio M, Iseki K, Yamagata K, Watanabe T, Hara S, et al. Prevalence of chronic kidney disease (CKD) in the Japanese general population predicted by the MDRD equation modified by a Japanese coefficient. Clin. Exp. Nephrol. 2007;11:156–163. doi: 10.1007/s10157-007-0463-x. [DOI] [PubMed] [Google Scholar]
- 4.Johnson RJ, Herrera-Acosta J, Schreiner GF, Rodriguez-Iturbe B. Subtle acquired renal injury as a mechanism of salt-sensitive hypertension. N. Engl. J. Med. 2002;346:913–923. doi: 10.1056/NEJMra011078. [DOI] [PubMed] [Google Scholar]
- 5.Ishii Y, Sawada T, Kubota K, Fuchinoue S, Teraoka S, Shimizu A. Injury and progressive loss of peritubular capillaries in the development of chronic allograft nephropathy. Kidney Int. 2005;67:321–332. doi: 10.1111/j.1523-1755.2005.00085.x. [DOI] [PubMed] [Google Scholar]
- 6.Schrimpf C, Duffield JS. Mechanisms of fibrosis: the role of the pericyte. Curr. Opin. Nephrol. Hypertens. 2011;20:297–305. doi: 10.1097/MNH.0b013e328344c3d4. [DOI] [PubMed] [Google Scholar]
- 7.Ishani A, Xue JL, Himmelfarb J, Eggers PW, Kimmel PL, Molitoris BA, et al. Acute kidney injury increases risk of ESRD among elderly. J. Am. Soc. Nephrol. 2009;20:223–228. doi: 10.1681/ASN.2007080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee R, Feinbaum R, Ambros V. A short history of a short RNA. Cell. 2004 doi: 10.1016/s0092-8674(04)00035-2. p. S89-92-1pfollowingS96. [DOI] [PubMed] [Google Scholar]
- 9.Dugas DV, Bartel B. MicroRNA regulation of gene expression in plants. Curr. Opin. Plant Biol. 2004;7:512–520. doi: 10.1016/j.pbi.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 10.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 11.Chau BN, Xin C, Hartner J, Ren S, Castano AP, Linn G, et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med. 2012;4:121ra18. doi: 10.1126/scitranslmed.3003205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melo SA, Esteller M. Dysregulation of microRNAs in cancer: playing with fire. FEBS Lett. 2011;585:2087–2299. doi: 10.1016/j.febslet.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010;467:86–90. doi: 10.1038/nature09284. [DOI] [PubMed] [Google Scholar]
- 14.Gao P, Tchernyshyov I, Chang T-C, Lee Y-S, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, et al. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am. J. Pathol. 2007;170:1831–1840. doi: 10.2353/ajpath.2007.061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Humphreys BD, Lin S-L, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin S-L, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asada N, Takase M, Nakamura J, Oguchi A, Asada M, Suzuki N, et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J. Clin. Invest. 2011;121:3981–3990. doi: 10.1172/JCI57301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang F-C, Yu-Hsiang C, Chen Y-T, Lin S-L. Novel insights into pericyte-myofibroblast transition and therapeutic targets in renal fibrosis. Journal of the Formosan Medical Association. 2012;XX:1–10. doi: 10.1016/j.jfma.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 20.Friedman S. Therapy for Fibrotic Disease: Nearing the Start Line. Science Tans. Med; 2012 doi: 10.1126/scitranslmed.3004700. in press. [DOI] [PubMed] [Google Scholar]
- 21.Godwin JG, Ge X, Stephan K, Jurisch A, Tullius SG, Iacomini J. Identification of a microRNA signature of renal ischemia reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 2010;107:14339–14344. doi: 10.1073/pnas.0912701107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zarjou A, Yang S, Abraham E, Agarwal A, Liu G. Identification of a microRNA signature in renal fibrosis: role of miR-21. Am. J. Physiol. Renal Physiol. 2011;301:F793–F801. doi: 10.1152/ajprenal.00273.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anglicheau D, Sharma VK, Ding R, Hummel A, Snopkowski C, Dadhania D, et al. MicroRNA expression profiles predictive of human renal allograft status. Proc. Natl. Acad. Sci. U.S.A. 2009;106:5330–5335. doi: 10.1073/pnas.0813121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Putta S, Lanting L, Sun G, Lawson G, Kato M, Natarajan R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J. Am. Soc. Nephrol. 2012;23:458–469. doi: 10.1681/ASN.2011050485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qin W, Chung ACK, Huang XR, Meng X-M, Hui DSC, Yu C-M, et al. TGF-β/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 2011;22:1462–1474. doi: 10.1681/ASN.2010121308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volinia S, Visone R, Galasso M, Rossi E, Croce CM. Identification of microRNA activity by Targets' Reverse EXpression. Bioinformatics. 2010;26:91–97. doi: 10.1093/bioinformatics/btp598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. U.S.A. 2008;105:10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zampetaki A, Willeit P, Drozdov I, Kiechl S, Mayr M. Profiling of circulating microRNAs: from single biomarkers to re-wired networks. Cardiovasc. Res. 2012;93:555–562. doi: 10.1093/cvr/cvr266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saikumar J, Hoffmann D, Kim T-M, Gonzalez VR, Zhang Q, Goering PL, et al. Expression, Circulation, and Excretion Profile of MicroRNA-21-155, and-18a Following Acute Kidney Injury. Toxicol. Sci. 2012;129:256–267. doi: 10.1093/toxsci/kfs210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patrick DM, Montgomery RL, Qi X, Obad S, Kauppinen S, Hill JA, et al. Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J. Clin. Invest. 2010;120:3912–3916. doi: 10.1172/JCI43604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li S, Nagothu KK, Desai V, Lee T, Branham W, Moland C, et al. Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-alpha in mice confers protection during acute kidney injury. Kidney Int. 2009;76:1049–1062. doi: 10.1038/ki.2009.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li S, Basnakian A, Bhatt R, Megyesi J, Gokden N, Shah SV, et al. PPAR-alpha ligand ameliorates acute renal failure by reducing cisplatin-induced increased expression of renal endonuclease G. Am. J. Physiol. Renal Physiol. 2004;287:F990–F998. doi: 10.1152/ajprenal.00206.2004. [DOI] [PubMed] [Google Scholar]
- 33.Krick S, Shi S, Ju W, Faul C, Tsai S-Y, Mundel P, et al. Mpv17l protects against mitochondrial oxidative stress and apoptosis by activation of Omi/HtrA2 protease. Proc. Natl. Acad. Sci. U.S.A. 2008;105:14106–14111. doi: 10.1073/pnas.0801146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kashihara N, Haruna Y, Kondeti VK, Kanwar YS. Oxidative stress in diabetic nephropathy. Curr. Med. Chem. 2010;17:4256–4269. doi: 10.2174/092986710793348581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Modlinger PS, Wilcox CS, Aslam S. Nitric oxide, oxidative stress, and progression of chronic renal failure. Semin. Nephrol. 2004;24:354–365. doi: 10.1016/j.semnephrol.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 36.O'Bryan T, Weiher H, Rennke HG, Kren S, Hostetter TH. Course of renal injury in the Mpv17-deficient transgenic mouse. J. Am. Soc. Nephrol. 2000;11:1067–1074. doi: 10.1681/ASN.V1161067. [DOI] [PubMed] [Google Scholar]
- 37.Clozel M, Hess P, Fischli W, Löffler BM, Zwacka RM, Reuter A, et al. Age-dependent hypertension in Mpv17-deficient mice, a transgenic model of glomerulosclerosis and inner ear disease. Exp. Gerontol. 1999;34:1007–1015. doi: 10.1016/s0531-5565(99)00074-1. [DOI] [PubMed] [Google Scholar]
- 38.Schenkel J, Zwacka RM, Rutenberg C, Reuter A, Waldherr R, Weiher H. Functional rescue of the glomerulosclerosis phenotype in Mpv17 mice by transgenesis with the human Mpv17 homologue. Kidney Int. 1995;48:80–84. doi: 10.1038/ki.1995.270. [DOI] [PubMed] [Google Scholar]
- 39.Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–1861. doi: 10.1016/S0140-6736(05)67667-2. [DOI] [PubMed] [Google Scholar]
- 40.Burgess HA, Daugherty LE, Thatcher TH, Lakatos HF, Ray DM, Redonnet M, et al. PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2005;288:L1146–L1153. doi: 10.1152/ajplung.00383.2004. [DOI] [PubMed] [Google Scholar]