

Abstract
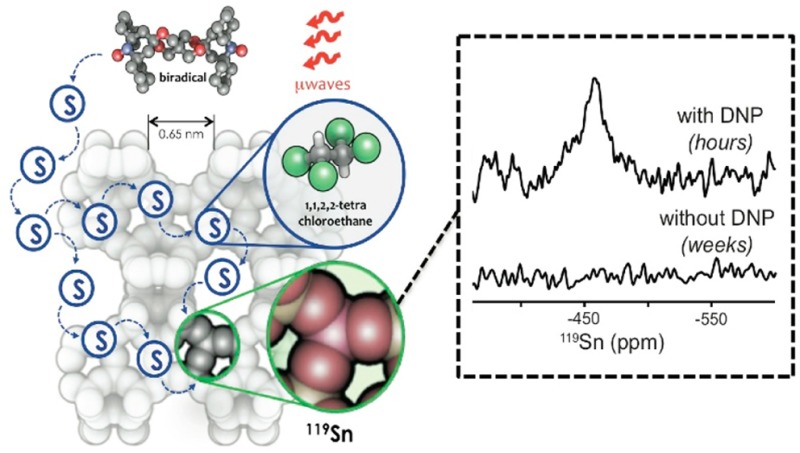
The catalytic activity of tin-containing zeolites, such as Sn-Beta, is critically dependent on the successful incorporation of the tin metal center into the zeolite framework. However, synchrotron-based techniques or solid-state nuclear magnetic resonance (ssNMR) of samples enriched with 119Sn isotopes are the only reliable methods to verify framework incorporation. This work demonstrates, for the first time, the use of dynamic nuclear polarization (DNP) NMR for characterizing zeolites containing ∼2 wt % of natural abundance Sn without the need for 119Sn isotopic enrichment. The biradicals TOTAPOL, bTbK, bCTbK, and SPIROPOL functioned effectively as polarizing sources, and the solvent enabled proper transfer of spin polarization from the radical’s unpaired electrons to the target nuclei. Using bCTbK led to an enhancement (ε) of 75, allowing the characterization of natural-abundance 119Sn-Beta with excellent signal-to-noise ratios in <24 h. Without DNP, no 119Sn resonances were detected after 10 days of continuous analysis.
Pure-silica zeolites containing a small amount of tetravalent heteroatoms with open coordination sites (e.g., Sn, Zr, or Ti) have emerged as highly active, water-tolerant solid Lewis acids for many important reactions.1 Tin-containing zeolites have been shown to promote the intermolecular Meerwein–Ponndorf–Verley (MPV) reaction between alcohols and ketones as well as the Baeyer–Villiger oxidation of cyclic ketones.2 Sn-Beta transforms hexoses, pentoses, and trioses through intramolecular hydride and carbon-atom shifts in both organic and aqueous media.3
The catalytic activity and water tolerance of Sn-Beta critically depend on three factors: the successful incorporation of the metal center into the zeolite framework, the presence of a defect-free pore structure that promotes a hydrophobic environment, and the hydrolysis of one Sn–O–Si bond to form an “open site” with a neighboring silanol group. These site requirements are essential to catalyze many industrially relevant reactions.4 Synthesizing the zeolite in fluoride media affords defect-free pores and minimizes extra framework species but also limits the maximum amount of heteroatom incorporation and slows down crystallization when compared to other methods.2b
For this reason, Sn-Beta is usually synthesized with Si/Sn ratios >100 (i.e., <1.9 wt % Sn) and crystallization times >20 days. Recently, other methods, including solid-state ion exchange, seeding, and grafting, have emerged as faster synthesis routes, but framework incorporation has been inferred mostly from indirect reactivity data or from qualitative infrared (IR) or diffuse reflectance ultraviolet (DR-UV) spectra.5
Although framework Sn incorporation can be quantitatively verified with 119Sn (I = 1/2, natural abundance = 8.6%) magic angle spinning (MAS) NMR, the coupled effects of low natural abundance of the 119Sn isotope, low intrinsic NMR sensitivity, and low Sn loadings in the sample make NMR analysis impractical without 119Sn isotopic enrichment.6 Unfortunately, the high cost of isotopic enrichment drastically hinders high-throughput screening, routine analysis, or analysis of low-yield syntheses with NMR. DNP addresses these challenges by transferring the larger polarization of electron spins, such as those found in stable exogenous radical compounds, to nuclear spins through irradiation with high-frequency microwaves. The target nuclei then become dynamically polarized, and their NMR signals are enhanced by orders of magnitude. DNP has been a valuable tool for studying local and medium range structure in challenging biological and inorganic materials,7 including SnO2 nanoparticles,8 by drastically reducing acquisition times.
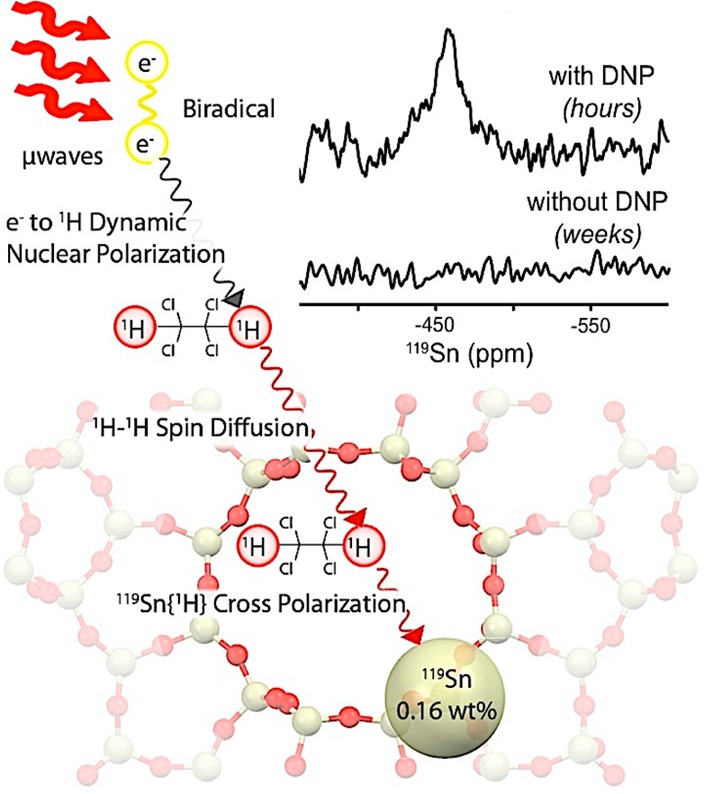
Here, we demonstrate DNP NMR characterization of zeolites containing ∼2 wt % of natural abundance 119Sn. Our approach, using indirect polarization, is exemplified in Scheme 1. First, high-power microwaves irradiate the sample treated with an exogenous biradical and a glassing agent (i.e., 1,1,2,2-tetrachloroethane, [TCE]). Next, the electron polarization is transferred from the radical to the protons in the solvent through electron–nuclear dipolar couplings. Freezing the sample at cryogenic temperatures (100 K) allows for 1H–1H spin diffusion to occur efficiently and enables the relay of polarization to the solvent molecules present inside the zeolite pores. Lastly, by using appropriate contact times (ranging from 1 to 8 ms), 1H polarization can be effectively transferred to the Sn sites using a cross-polarization (CP) step. Enhancements of ε > 35 (ε† > 100, accounting for the gain in Boltzmann levels when acquiring the data at 100 vs 300 K) are achieved with nitroxide-based biradicals, thus enabling the characterization of nonenriched Sn-Beta zeolites with superb signal-to-noise ratios (S/N) in <24 h.
Scheme 1. Hyperpolarization of Sn-Beta Zeolite Using Dynamic Nuclear Polarization.
The DNP method was first optimized using enriched 119Sn-Beta samples (Si/Sn = 102, 82% 119Sn enrichment). Figure 1 shows the 119Sn DNP NMR resonances for a hydrated and dehydrated 119Sn-Beta calcined sample using bis-cyclohexyl-TEMPO-bisketal (bCTbK) as the radical and TCE as the solvent. In agreement with literature reports, dehydrated Sn-Beta (Figure 1a) features resonances corresponding to tetrahedrally coordinated framework Sn sites in the closed (−443 ppm) and open (−420 ppm) configurations.2c,9 Under hydrated conditions the Sn site complexes with two water molecules leading to a pseudo-octahedral geometry and corresponding resonances centered at −700 ppm (Figure 1b). The 1H detected enhancements were 36 and 52 for the dehydrated and hydrated 119Sn-Beta materials. These enhancements led to spectra with high S/N ratios in <2 h using 25 μL of sample. For comparison, a typical MAS NMR experiment without DNP requires 58 h of acquisition time at 300 K and using 80 μL of sample to obtain comparable S/N ratios (see Figure 1c).
Figure 1.
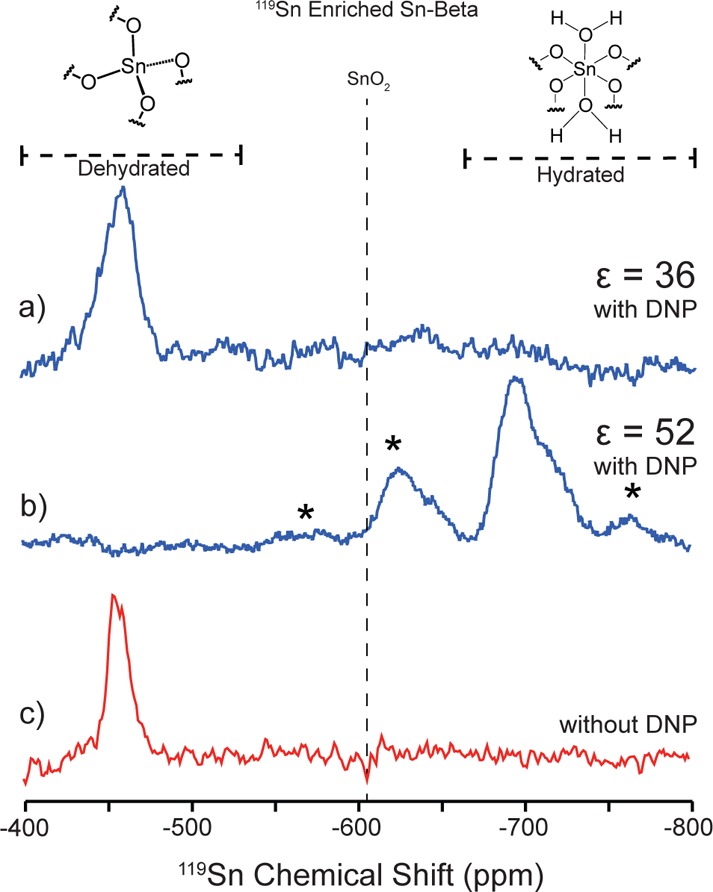
DNP-enhanced 119Sn spectra of dehydrated (a) and hydrated (b) enriched 119Sn-Beta zeolite. Spectra were acquired at 100 K for 3.8 h. 119Sn MAS NMR spectrum of 119Sn enriched dehydrated Sn-Beta zeolite (c) was acquired at 300 K for 58 h. * denotes spinning sidebands.
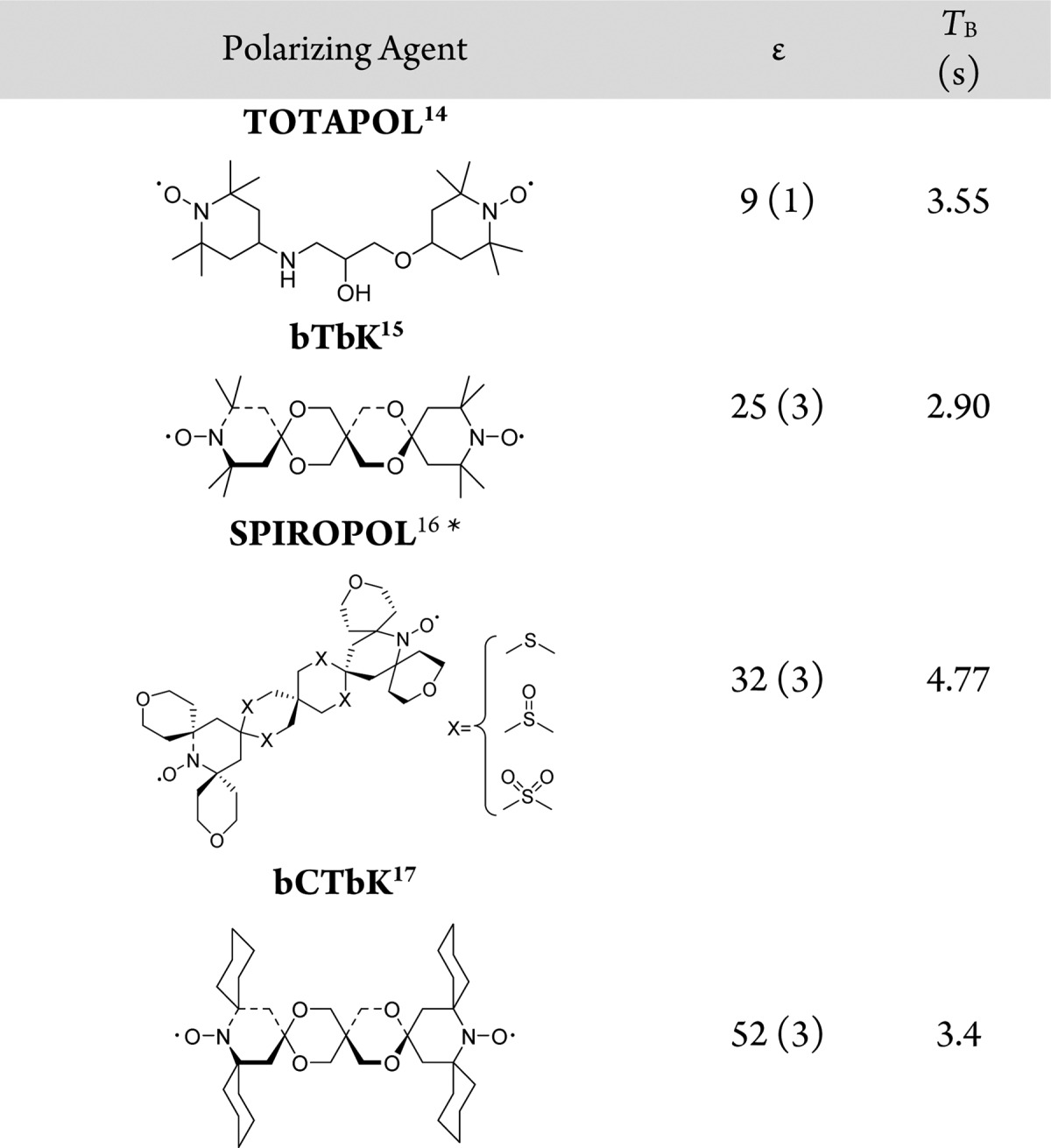
The strong Lewis acid sites in Sn-Beta readily coordinate with functional groups containing lone pairs of electrons. To determine and minimize potential undesirable interactions of the radical with the Lewis acid center, four biradical polarizing agents with varying kinetic diameters were impregnated on the Sn-Beta sample (Table 1). The corresponding 119Sn MAS NMR spectra showed that the radicals 1-(TEMPO-4-oxy)-3-(TEMPO-4-amino)propan-2-ol (TOTAPOL) and bis-TEMPO-bis-ketal (bTbK) entered the zeolite pores and complexed with the Sn sites, leading to multiple resonances in the range of −600 to −800 ppm. Conversely, the radicals bis-TEMPO-bis-thioketal-tetra-tetrahydropyran (SPIROPOL) and bCTbK did not affect the resonances of the dehydrated Sn site (Figure S4). We hypothesize that SPIROPOL and bCTbK are too bulky (Table 1) to enter the pores of zeolite Beta (ring pores of 7.7 × 6.6 Å and 5.6 × 5.6 Å) and thereby rely exclusively on the protons in the solvent to transfer polarization to internal sites. Note that even after prolonged exposure (>48 h, 298 K) of both bCTbK and SPIROPOL with the zeolite, no adsorption on the Sn site was detected.
Table 1. Experimentally Determined Enhancements (ε) and Build-Up Times (TB) for Hydrated, Isotopically-Enriched 119Sn-Beta Samples Analyzed with 10 mM Nitroxide-Based Biradical Polarizing Agents (20 mM electrons in TCE).
The NMR magnetic field was swept ∼50 kHz to the left-edge of the maximum SPIROPOL enhancement.16
Although both small radicals interacted with the Sn site, they still provided a gain in sensitivity. As shown in Table 1, enhancements of 9 and 25, were observed for TOTAPOL and bTbK, respectively. The sterically hindered SPIROPOL and bCTbK proved to be optimal polarizing agents, offering consistent enhancements >30 at 100 K in TCE. Ultimately, bCTbK was chosen for subsequent studies because it generated slightly better enhancements and shorter build-up times, allowing for faster recycling during acquisition. Note that bCTbK 1H enhancements range from 28 to 75 depending on the sample (see Table 2), which we attribute to the glass-forming behavior of the solvent.
Table 2. Enhancements and Build-Up Times of Dehydrated and Hydrated Sn-Beta Zeolites with 10 mM bCTbK in TCE.
| Sn site | TB (sec) | ε | ε† |
|---|---|---|---|
| dehydrated (119Sn - enriched) | 2.7 | 36 | 108 |
| hydrated (119Sn enriched) | 3.4 | 52 | 156 |
| dehydrated/hydrated (119Sn - enriched) | 3.2 | 36 | 108 |
| dehydrated (119Sn natural abundance) | 5.5 | 75 | 225 |
| hydrated (119Sn natural abundance) | 4.2 | 28 | 84 |
Although control experiments in the absence of radical showed that the TCE solvent does not interfere with the dehydrated Sn signal, neat TCE is not an ideal glass-forming medium as is the case for glycerol/water or DMSO/water mixtures. For this reason, TCE is typically doped with a small percentage of another solvent (e.g., methanol or ethanol) to assist in the glass-forming process.10 Although the use of TCE dopants or glycerol/water or DMSO/water mixtures was avoided in this study to prevent interaction with the Sn site, appropriate glass formation at low temperatures is a critical parameter to enhance transfer polarization processes.11 For instance, drastically different enhancements for 13C DNP NMR spectra of glucose bound to Sn-Beta were obtained when performing DNP with 10 mM TOTAPOL dissolved in either TCE, DMSO/water or glycerol/water (Figure S5). TCE generated an enhancement of 3, whereas DMSO/water and glycerol/water generated enhancements of 70 and 100, respectively. Further studies are currently underway to better understand solvent-radical-zeolite interactions.
The use of 1H–1H spin diffusion to polarize crystalline materials in an organic medium was first demonstrated by van der Wel et al. for amyloidgenic peptide cores (dimension, ∼0.2 × 0.2 × 1 μm).12 Similar approaches have been used in other chemical systems, including amorphous silica frameworks, wherein the large abundance of intrinsic protons within the matrix governs the 1H nuclear spin-diffusion across the sample.13 In contrast, dehydrated Sn-Beta zeolite crystals (dimensions, ∼0.45 × 7 × 7 μm based on Figure S1) are severely proton deficient, thus limiting intrinsic 1H spin-diffusion.
Indeed, a polarization gradient can evoke differences in the polarization transfer between the solvent and the crystalline solid system. These effects are mainly dependent on the width of the smallest dimension of the crystal, the nuclear T1, and the nuclear spin diffusion.12 The extremely long contact times required during analysis (i.e., 6–8 ms) indicate that a considerable distance exists between the Sn site and the nearest proton. Since the radical is confined to the external surface of the zeolite, the solvent provides the sole medium for 1H spin diffusion and is therefore responsible for the observed DNP enhancements.18 The physical barrier between the internal Sn sites and the radical minimizes paramagnetic broadening, while the protonated solvent allows for effective 1H–1H dipolar couplings for polarization transfer. Importantly, since the 1H–119Sn CP transfer efficiency depends on the local proton density, collected spectra can only provide qualitative estimates of Sn resonance intensities.
Tin features a significant shell of electron density, making its inherent chemical shift very sensitive to the local environment. Resolution is very sensitive to the degree of crystallinity, site dynamics, and surrounding environment. Cryogenic temperatures and the TCE solvent impart additional broadening, as evidenced by the spectra of the hydrated samples wherein the isotropic chemical shift is heterogeneously broadened. The spinning side bands result from a larger chemical shift anisotropy generated by the bound water molecules. Thus, although cross-polarization of 1H to 119Sn is more efficient in the hydrated state, broader isotropic resonance and larger chemical shift anisotropy (CSA) are observed when compared to the dehydrated state due to complex interaction of the TCE solvent and pseudo-octahedral Sn geometry.
Optimized DNP conditions were implemented on the analysis of natural abundance Sn-Beta samples. Figure 2 shows 119Sn resonances for a natural abundance sample (Si/Sn = 89) in both hydrated and dehydrated states. Both spectra were acquired in <24 h using a rotor with a 25 μL fill volume. The natural abundance hydrated and dehydrated Sn-Beta DNP enhancements were 28 and 75, respectively (Figure 2a,b). In stark contrast, a regular MAS NMR experiment performed on the dehydrated sample using a rotor with a 80 μL fill volume showed no evidence of 119Sn resonances after 246 h of continuous analysis (see Figure 2c). These data demonstrate that DNP enables the analysis of Sn-Beta zeolite without 119Sn isotopic enrichment. Table 2 summarizes the enhancements obtained with 10 mM bCTbK in TCE for different Sn-Beta samples. Water molecules increase the proximity of protons to the Sn site, thereby improving CP efficiency. As a result, the effective contact times for 119Sn CP varied drastically between the dehydrated (τc = 6–8 ms) and hydrated (τc = 2–3 ms) sites.5b
Figure 2.
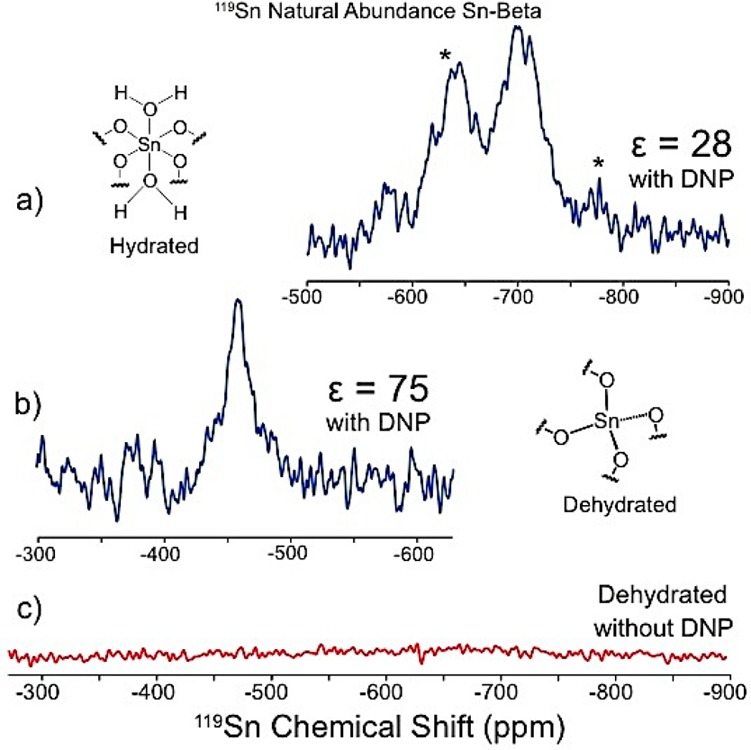
DNP-enhanced 119Sn spectra of hydrated (a) and dehydrated (b) natural abundance Sn-Beta zeolite. Spectra were acquired at 100 K for 18 and 21 h, respectively. 119Sn MAS NMR spectrum of natural abundant dehydrated Sn-Beta zeolite was acquired at 300 K for 246 h. * denotes spinning sidebands.
In conclusion, indirect DNP of 119Sn allows the observation of NMR signals from natural abundance Sn atoms in microporous materials without the need for isotopic enrichment. This technique reduces acquisition times more than ∼2 orders of magnitude, allowing for a difficult analysis to be completed in a matter of hours. The framework incorporation of Sn in porous materials can be readily studied using appropriate radical/solvent combinations; specifically, we show that the radicals bCTbK or SPIROPOL generate extraordinary gains in sensitivity without interacting directly with intrapore Sn sites. Optimizing methods to resolve local environments of the tetrahedral sites is one of our current objectives. With the development of high-frequency, high-power microwave sources and advances in cryogenic MAS probes,19 DNP NMR has emerged as the method of choice for chemical structure determination in extremely challenging systems.
Acknowledgments
This work was supported by the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy (DE-FG02-12ER16352). R.G.G. is supported through the NIH (grants EB-002804, EB-001960, and EB-002026). V.K.M. was partially funded by the NSERC-PDF. We thank Drs Werner Maas and Melanie Rosay at Bruker BioSpin for providing access to the DNP-NMR spectrometer. We thank Dr. Prasomsri for the TOC figure.
Supporting Information Available
Sample preparation and experimental methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
# These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Roman-Leshkov Y.; Davis M. E. ACS Catal. 2011, 1, 1566. [Google Scholar]
- a Corma A.; Domine M. E.; Nemeth L.; Valencia S. J. Am. Chem. Soc. 2002, 124, 3194. [DOI] [PubMed] [Google Scholar]; b Bui L.; Luo H.; Gunther W. R.; Roman-Leshkov Y. Angew. Chem., Int. Ed. 2013, 52, 8022. [DOI] [PubMed] [Google Scholar]; c Corma A.; Nemeth L. T.; Renz M.; Valencia S. Nature 2001, 412, 423. [DOI] [PubMed] [Google Scholar]
- a Moliner M.; Roman-Leshkov Y.; Davis M. E. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 6164. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Roman-Leshkov Y.; Moliner M.; Labinger J. A.; Davis M. E. Angew. Chem., Int. Ed. 2010, 49, 8954. [DOI] [PubMed] [Google Scholar]; c de Clippel F.; Dusselier M.; Van Rompaey R.; Vanelderen P.; Dijkmans J.; Makshina E.; Giebeler L.; Oswald S.; Baron G. V.; Denayer J. F. M.; Pescarmona P. P.; Jacobs P. A.; Sels B. F. J. Am. Chem. Soc. 2012, 134, 10089. [DOI] [PubMed] [Google Scholar]; d Holm M. S.; Saravanamurugan S.; Taarning E. Science (Washington, DC, U.S.) 2010, 328, 602. [DOI] [PubMed] [Google Scholar]; e Li L.; Stroobants C.; Lin K.; Jacobs P. A.; Sels B. F.; Pescarmona P. P. Green Chem. 2011, 13, 1175. [Google Scholar]; f Gunther W. R.; Wang Y.; Ji Y.; Michaelis V. K.; Hunt S. T.; Griffin R. G.; Roman-Leshkov Y. Nat. Commun. 2012, 3, 1109. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Luo H. Y.; Bui L.; Gunther W. R.; Min E.; Roman-Leshkov Y. ACS Catal. 2012, 2, 2695. [Google Scholar]
- a Moliner M. Dalton Trans. 2014, 43, 4197. [DOI] [PubMed] [Google Scholar]; b Bermejo-Deval R.; Gounder R.; Davis M. E. ACS Catal. 2012, 2, 2705. [Google Scholar]; c Boronat M.; Concepción P.; Corma A.; Renz M.; Valencia S. J. Catal. 2005, 234, 111. [Google Scholar]; d Dusselier M.; Van Wouwe P.; Dewaele A.; Makshina E.; Sels B. F. Energy Environ. Sci. 2013, 6, 1415. [Google Scholar]
- a Hammond C.; Conrad S.; Hermans I. Angew. Chem., Int. Ed. 2012, 51, 11736. [DOI] [PubMed] [Google Scholar]; b Chang C.-C.; Wang Z.; Dornath P.; Cho H. J.; Fan W. RSC Adv. 2012, 2, 10475. [Google Scholar]; c Dijkmans J.; Gabriels D.; Dusselier M.; de C. F.; Vanelderen P.; Houthoofd K.; Malfliet A.; Pontikes Y.; Sels B. F. Green Chem. 2013, 15, 2777. [Google Scholar]; d Li P.; Liu G.; Wu H.; Liu Y.; Jiang J.-g.; Wu P. J. Phys. Chem. C 2011, 115, 3663. [Google Scholar]
- Kishor Mal N.; Ramaswamy V.; Ganapathy S.; Ramaswamy A. Appl. Catal., A 1995, 125, 233. [Google Scholar]
- a Lafon O.; Rosay M.; Aussenac F.; Lu X.; Trebosc J.; Cristini O.; Kinowski C.; Touati N.; Vezin H.; Amoureux J.-P. Angew. Chem., Int. Ed. 2011, 50, 8367. [DOI] [PubMed] [Google Scholar]; b Bayro M. J.; Debelouchina G. T.; Eddy M. T.; Birkett N. R.; MacPhee C. E.; Rosay M.; Maas W. E.; Dobson C. M.; Griffin R. G. J. Am. Chem. Soc. 2011, 133, 13967. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Linden A. H.; Lange S.; Franks W. T.; Akbey U.; Specker E.; van Rossum B.-J.; Oschkinat H. J. Am. Chem. Soc. 2011, 133, 19266. [DOI] [PubMed] [Google Scholar]; d Rossini A. J.; Zagdoun A.; Lelli M.; Gajan D.; Rascon F.; Rosay M.; Maas W. E.; Coperet C.; Lesage A.; Emsley L. Chem. Sci. 2012, 3, 108. [DOI] [PubMed] [Google Scholar]; e Hall D. A.; Maus D. C.; Gerfen G. J.; Inati S.; Becerra L. R.; Dahlquist F. W.; Griffin R. G. Science (Washington, DC, U.S.) 1997, 276, 930. [DOI] [PubMed] [Google Scholar]; f Gelis I.; Vitzthum V.; Dhimole N.; Caporini M. A.; Schedlbauer A.; Carnevale D.; Connell S. R.; Fucini P.; Bodenhausen G. J. Biomol. NMR 2013, 56, 85. [DOI] [PubMed] [Google Scholar]; g Renault M.; Pawsey S.; Bos M. P.; Koers E. J.; Nand D.; Tommassen-van Boxtel R.; Rosay M.; Tommassen J.; Maas W. E.; Baldus M. Angew. Chem., Int. Ed. 2012, 51, 2998. [DOI] [PubMed] [Google Scholar]
- Protesescu L.; Rossini A. J.; Kriegner D.; Valla M.; de Kergommeaux A.; Walter M.; Kravchyk K. V.; Nachtegaal M.; Stangl J.; Malaman B.; Reiss P.; Lesage A.; Emsley L.; Coperet C.; Kovalenko M. V. ACS Nano 2014, 8, 2639. [DOI] [PubMed] [Google Scholar]
- Bermejo-Deval R.; Assary R. S.; Nikolla E.; Moliner M.; Roman-Leshkov Y.; Hwang S.-J.; Palsdottira A.; Silverman D.; Lobo R. F.; Curtiss L. A.; Davis M. E. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Eaton G. R.; Eaton S. S.; Barr D. P.; Weber R. T.. Quantitative EPR; SpringerWien: New York, 2010. [Google Scholar]; b Wada T.; Yamanaka M.; Fujihara T.; Miyazato Y.; Tanaka K. Inorg. Chem. 2006, 45, 8887. [DOI] [PubMed] [Google Scholar]
- Zagdoun A.; Rossini A. J.; Gajan D.; Bourdolle A.; Ouari O.; Rosay M.; Maas W. E.; Tordo P.; Lelli M.; Emsley L. Chem. Commun. 2012, 48, 654. [DOI] [PubMed] [Google Scholar]
- Van der Wel P. C. A.; Hu K.-N.; Lewandowski J.; Griffin R. G. J. Am. Chem. Soc. 2006, 128, 10840. [DOI] [PubMed] [Google Scholar]
- a Rossini A. J.; Zagdoun A.; Hegner F.; Schwarzwalder M.; Gajan D.; Coperet C.; Lesage A.; Emsley L. J. Am. Chem. Soc. 2012, 134, 16899. [DOI] [PubMed] [Google Scholar]; b Rossini A. J.; Zagdoun A.; Lelli M.; Canivet J.; Aguado S.; Ouari O.; Tordo P.; Rosay M.; Maas W. E.; Coperet C.; Farrusseng D.; Emsley L.; Lesage A. Angew. Chem., Int. Ed. 2012, 51, 123. [DOI] [PubMed] [Google Scholar]; c Kobayashi T.; Lafon O.; Lilly Thankamony A. S.; Slowing I. I.; Kandel K.; Carnevale D.; Vitzthum V.; Vezin H.; Amoureux J.-P.; Bodenhausen G.; Pruski M. Phys. Chem. Chem. Phys. 2013, 15, 5553. [DOI] [PubMed] [Google Scholar]; d Lelli M.; Gajan D.; Lesage A.; Caporini M. A.; Vitzthum V.; Mieville P.; Heroguel F.; Rascon F.; Roussey A.; Thieuleux C.; Boualleg M.; Veyre L.; Bodenhausen G.; Coperet C.; Emsley L. J. Am. Chem. Soc. 2011, 133, 2104. [DOI] [PubMed] [Google Scholar]; e Lesage A.; Lelli M.; Gajan D.; Caporini M. A.; Vitzthum V.; Miéville P.; Alauzun J.; Roussey A.; Thieuleux C.; Mehdi A. J. Am. Chem. Soc. 2010, 132, 15459. [DOI] [PubMed] [Google Scholar]
- Song C.; Hu K.-N.; Joo C.-G.; Swager T. M.; Griffin R. G. J. Am. Chem. Soc. 2006, 128, 11385. [DOI] [PubMed] [Google Scholar]
- Matsuki Y.; Maly T.; Ouari O.; Karoui H.; Le Moigne F.; Rizzato E.; Lyubenova S.; Herzfeld J.; Prisner T.; Tordo P. Angew. Chem. 2009, 121, 5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiesewetter M. K.; Corzilius B.; Smith A. A.; Griffin R. G.; Swager T. M. J. Am. Chem. Soc. 2012, 134, 4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagdoun A.; Casano G.; Ouari O.; Lapadula G.; Rossini A. J.; Lelli M.; Baffert M.; Gajan D.; Veyre L.; Maas W. E. J. Am. Chem. Soc. 2012, 134, 2284. [DOI] [PubMed] [Google Scholar]
- Lafon O.; Thankamony A. S. L.; Kobayashi T.; Carnevale D.; Vitzthum V.; Slowing I. I.; Kandel K.; Vezin H.; Amoureux J.-P.; Bodenhausen G.; Pruski M. J. Phys. Chem. C 2013, 117, 1375. [DOI] [PubMed] [Google Scholar]
- a Becerra L. R.; Gerfen G. J.; Temkin R. J.; Singel D. J.; Griffen R. G. Phys. Rev. Lett. 1993, 71, 3561. [DOI] [PubMed] [Google Scholar]; b Barnes A. B.; Markhasin E.; Daviso E.; Michaelis V. K.; Nanni E. A.; Jawla S. K.; Mena E. L.; DeRocher R.; Thakkar A.; Woskov P. P.; Herzfeld J.; Temkin R. J.; Griffin R. G. J. Magn. Reson. 2012, 224, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bajaj V. S.; Farrar C. T.; Hornstein M. K.; Mastovsky I.; Vieregg J.; Bryant J.; Elena B.; Kreischer K. E.; Temkin R. J.; Griffin R. G. J. Magn. Reson. 2011, 213, 404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.