

Abstract

The addition of ynamides to acyl chlorides and N-heterocycles activated in situ with ethyl chloroformate has been accomplished at room temperature using copper iodide as catalyst. This economical and practical carbon–carbon bond formation provides convenient access to a variety of 3-aminoynones from aliphatic and aromatic acyl chlorides in up to 99% yield. The addition to pyridines and quinolines occurs under almost identical conditions and proceeds with good to high regioselectivity, producing the corresponding 1,2-dihydro-N-heterocycles in up to 95% yield.
The unique chemistry of ynamines has received continuous attention due to the huge synthetic potential of these remarkably versatile building blocks. In particular, C-substituted ynamines exhibiting an internal triple bond have found widespread use in a variety of reactions and in the total synthesis of natural compounds.1 The reaction scope of ynamines and derivatives thereof differs considerably from that of enamines and alkynes as the reactivity of the electron-rich triple bond is dominated by the adjacent, strongly polarizing amine moiety. Because ynamines are very reactive and therefore of limited practical use, ynamides that can be isolated and stored have become more popular in recent years. The increasing availability of terminal ynamides, ynesulfonamides, and ynecarbamates based on practical procedures developed by Witulski,2 Bruckner,3 Saa,4 and others has further extended the general utility of ynamine chemistry, Figure 1.5 Among the most noteworthy reactions are cycloadditions,6 cycloisomerizations,7 homo- and cross-couplings,8 ring-closing metathesis,9 radical additions,10 and titanium-mediated carbon–carbon bond formations.11
Figure 1.

Structures of terminal ynamines and less reactive ynamide and ynesulfonamide analogues.
Surprisingly, few examples of nucleophilic additions of terminal ynamides, ynesulfonamides, and ynecarbamates to aldehydes, ketones, and other electrophiles, all requiring strongly basic conditions, can be found in the literature.12 The absence of a catalytic procedure that allows mild carbon–carbon bond formation with acyl chlorides and N-heterocycles is in stark contrast to the wealth of reports on this reaction with terminal alkynes. Encouraged by our previous finding that indole-derived ynamines undergo zinc-catalyzed additions with aldehydes toward N-substituted propargylic alcohols, we decided to search for a catalytic variant that is applicable to other electrophiles.13 We now wish to report the copper-catalyzed nucleophilic addition of a readily available terminal ynesulfonamide to acyl chlorides and activated pyridines and quinolines furnishing 3-aminoynones and the corresponding 1,2-dihydro-2-(3-aminoethynyl) N-heterocycles.
Propargylic ketones are key intermediates for the preparation of natural products and heterocyclic compounds and most conveniently prepared through catalytic alkynylation of acyl chlorides14 or via carbonylative Sonogashira coupling.15 Many procedures require heating and long reaction times and are not applicable to ynamides, which lack the thermal stability of alkynes.16 We therefore investigated the possibility of carbon–carbon bond formation with the readily available N-ethynyl-N-phenyl-4-tolylsulfonamide, 1, under mild reaction conditions. Following a literature procedure, we synthesized gram amounts of 1 from N-tosyl aniline, Scheme 1.3 Initial analysis of the reaction between ynesulfonamide 1 and benzoyl chloride showed that copper(I) salts were superior over both zinc and palladium complexes commonly used in alkynylation reactions. Using 10 mol % of cuprous iodide and 2 equiv of diisopropylethylamine in THF, we obtained the desired N-(3-phenyl-3-oxoprop-1-ynyl)-N-phenyl-4-tolylsulfonamide, 2, in 50% yield after 20 h. The screening of various copper(I) salts, organic solvents, base, and temperature revealed that 2 can be isolated in 90% yield when the reaction is performed in the presence of 10 mol % of copper iodide in chloroform at 30 °C; see entry 1 in Table 1. To the best of our knowledge, this is the first catalytic addition of an ynamide to an acyl chloride. It is noteworthy that the order of addition of the reagents is important for this reaction. The best yields were obtained when the catalyst, base, and the ynamide were stirred for 30 min prior to addition of the acyl chloride. The reaction also proceeds with high yields when other aromatic substrates are employed, and we obtained ynones 3–7 in 79–99% yield, entries 2–6. In contrast to the impressive number of high-yielding catalytic cross-couplings of aromatic acyl chlorides with terminal alkynes, very few examples with aliphatic electrophiles typically producing ynones in only moderate yields have been reported.14a,14e This can probably be attributed to fast ketene formation and subsequent side reactions when acyl chlorides exhibiting α-hydrogens are used in the presence of base. While the reaction with pivaloyl chloride gave the corresponding propargylic ketone 8 in high yield as expected, we were very pleased to find that the ynone formation with 2-methylpropanoyl chloride proceeds smoothly at 15 °C providing 9 in 70% yield, entries 7 and 8.
Scheme 1. Synthesis of Ynesulfonamide 1 (Top) and Targeted Catalytic 1,2-Additions (Bottom).

Table 1. Copper(I)-Catalyzed Addition to Acyl Chlorides.


Isolated yields.
20 °C.
15 °C.
As discussed above, the properties and reactivity of ynamines and ynamides are influenced by the amine moiety, which strongly polarizes the triple bond. We therefore decided to investigate if the sulfonamide unit has a similar effect on the ynone unit. A single crystal of 2 was obtained by slow evaporation of a solution in CDCl3. Crystallographic analysis of this compound and a survey of representative C-substituted propargylic ketones from the Cambridge Structural Database showed that the bond lengths of the carbonyl group, the adjacent C(sp2)–C(sp) bond, and the triple bond in the α,β-unsaturated ketone functionalities are almost identical, Figure 2. Similarly, IR analysis of 2 shows the alkyne and the carbonyl stretchings at 2202 and 1637 cm–1, respectively, which suggests that push–pull conjugation plays a minor role in this 3-aminoynone.17
Figure 2.

Crystal structure of 2. Selected crystallographic separations [Å]: N1···C3, 1.345; C3···C2, 1.197; C2···C1, 1.448; C1···O1, 1.224.
In contrast to the results obtained with acyl chlorides, we did not observe any reaction when we applied methyl or ethyl chloroformate in our copper-catalyzed ynamide addition procedure. This led us to investigate the possibility of a catalytic ynamide addition to pyridines by a one-pot procedure in which the heterocycle is activated toward a nucleophilic attack through formation of an N-acylpyridinium intermediate. Substituted 1,2-dihydropyridines and the corresponding 1,2-dihydroquinolines are important N-heterocycles that serve as key intermediates in organic synthesis and are ubiquitous units in many biologically active compounds. The direct incorporation of versatile functionalities into readily available, inexpensive pyridine and quinoline compounds has therefore received increasing attention in recent years. While several reports on regioselective 1,2-additions of organometallic species to pyridine and its analogues exist, the nucleophilic attachment of an ynamide moiety has not been accomplished to date.18
With the mild protocol for the ynamide addition to acyl chlorides in hand, the optimization of the reaction between 1 and pyridine toward N-ethoxycarbonyl-1,2-dihydro-2-(N-phenyl-N-tosylaminoethynyl)pyridine, 10, was straightforward. We systematically changed solvents, temperature, and base, screened zinc and copper catalysts, and tested different chloroformates at varying amounts to activate the pyridine ring for a nucleophilic ynamide attack. We found that quantitative conversion can be achieved for the reaction between pyridine and ynesulfonamide 1 using copper(I) iodide as catalyst and 2 equiv of diisopropylethylamine in dichloromethane at room temperature. The heterocycle activation requires the presence of 2 equiv of ethyl chloroformate; the overall reaction is significantly faster when 5 equiv is used, but this has no effect on the isolated yields. Replacement of ethyl chloroformate with the methyl or benzyl derivative proved detrimental to the conversion. Using our optimized procedure with ethyl chloroformate and 2 equiv of base, we were able to isolate 10 in 71% yield after 2.5 h at room temperature; see entry 1 in Table 2.
Table 2. Copper(I)-Catalyzed Ynamide Addition to Activated Pyridines and Quinolones.

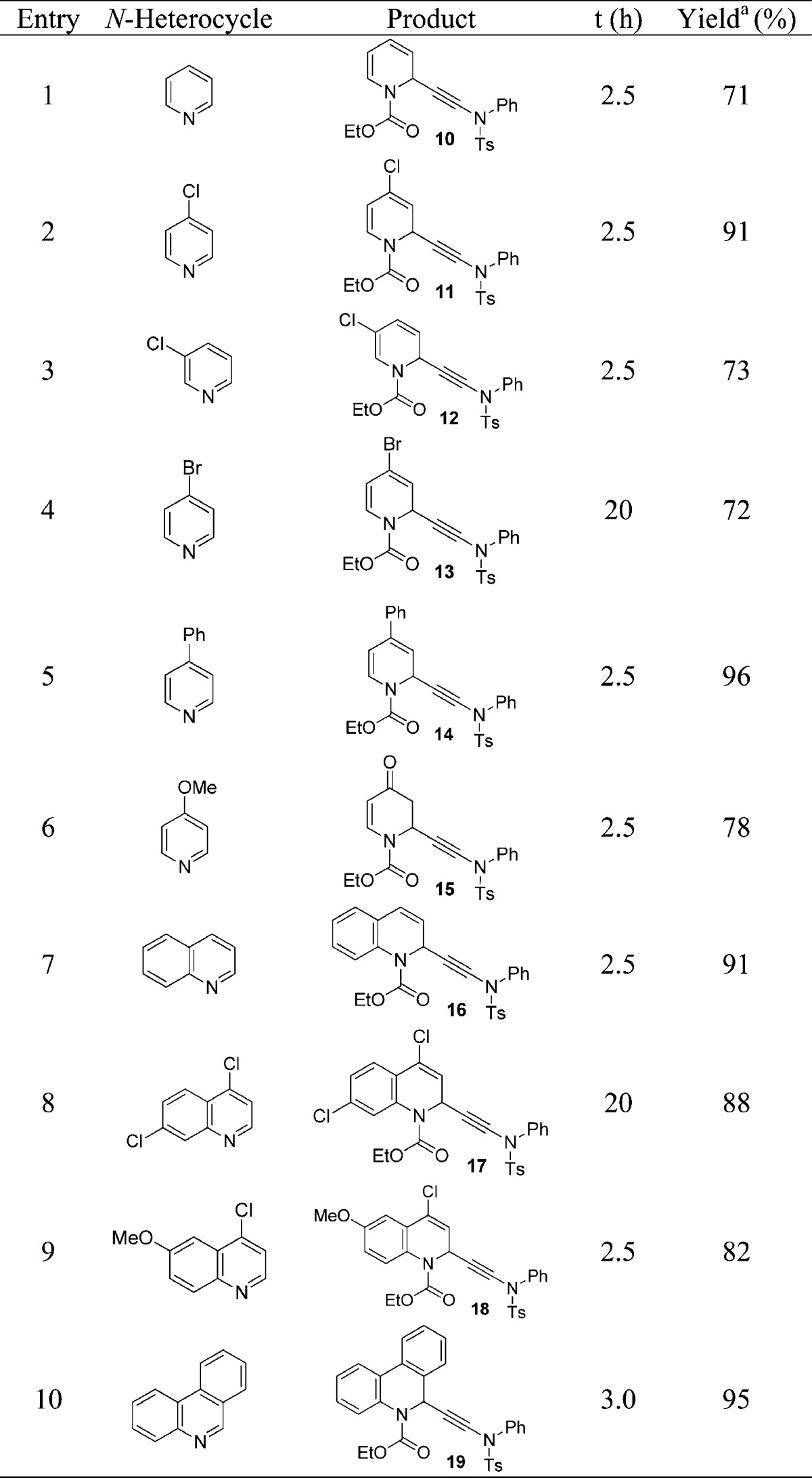
Isolated yield.
We then applied our catalytic procedure to several pyridine analogues and obtained the corresponding 1,2-dihydropyridines 11–14 in 72–96% yield, entries 2–5. The copper-catalyzed ynamide addition to activated pyridines and quinolines typically shows quantitative conversion, but the yield of the desired 1,2-dihydro-2-(2-aminoethynyl)heterocycles is in some cases compromised by concomitant formation of noticeable amounts of the 1,4-regioisomer. With pyridine substrates we observed that the ratio of the 1,2- versus the 1,4-addition product varied between 3:1 and 7:1 unless the para-position was blocked, while solvents (acetonitrile, N-methylpyrrolidinone, acetone, nitromethane, tetrahydrofuran, chloroform, and dichloromethane) and temperature changes (−78 to 25 °C) had literally no impact on the regioselectivity but affected the conversion of this reaction.19 The 1,2-dihydropyridine generated from 4-methoxypyridine rapidly hydrolyses upon acidic workup and careful chromatographic purification on basic alumina gave ketone 15 in 78% yield, entry 6. It is noteworthy that the synthesis of functionalized piperidinones such as 15 has become increasingly important due to the use of these versatile intermediates in medicinal chemistry.18a We were pleased to find that our method can also be applied to quinolines. The ynamide addition to quinoline gave N-ethoxyarbonyl-1,2-dihydro-2-(N-phenyl-N-tosylaminoethynyl)quinoline, 16, in 91% yield, entry 7 in Table 2. In contrast to pyridines, the reaction with quinolines apparently occurs with high 1,2-regioselectivity and no sign of the 1,4-addition product was observed. Finally, 4,7-dichloro- and 4-chloro-6-methoxyquinoline were converted to 17 and 18 with 82–88% yield and 19 was obtained in 95% yield from phenanthridine, entries 8–10.
In analogy to metal-catalyzed nucleophilic additions with alkynes, we believe that side-on coordination of the ynamide to copper(I) increases the acidity of the terminal CH bond. Deprotonation by the tertiary amine base then produces a copper complex that reacts with the electrophilic acyl chloride or activated N-heterocycle and regenerates the catalyst, Figure 3. The ynamide additions are sluggish in the absence of CuI. We found that the synthesis of aminoynone, 2, from 1 and benzoyl chloride is almost complete after 10 h, but less than 50% ynamide consumption and formation of unidentified byproducts were observed when the reaction was performed without the catalyst. NMR monitoring of the catalytic ynamide addition to the activated quinoline ring showed quantitative conversion to 1,2-dihydro-2-aminoethynylquinoline, 16, within 20 min, whereas no product was isolated when the reaction was carried out in the absence of CuI for 2.5 h.
Figure 3.

(Left) Proposed mechanism of the CuI-catalyzed formation of aminoynone, 2, and 1,2-dihydro-2-aminoethynylquinoline, 16, and (right) conversion of the ynamide to 2 and 16 vs time.
In conclusion, we have developed the first catalytic addition of a readily available ynesulfonamide to aliphatic and aromatic acyl chlorides. A slightly modified procedure has been successfully used for regioselective 1,2-addition of ynamides to pyridines and quinolines. Both reactions occur under mild conditions and provide unprecedented access to a variety of 3-aminoynones and 1,2-dihydro-N-heterocycles in good to high yields. The convenient access to these synthetically versatile ynamide derivatives is expected to prove invaluable to medicinal chemistry and natural product synthesis.
Experimental Section
Commercially available reagents and solvents were used without further purification. Anhydrous solvents were used as purchased and not dried any further. NMR spectra were obtained at 400 MHz (1H NMR) and 100 MHz (13C NMR) in deuterated chloroform. Chemical shifts are reported in ppm relative to TMS.
General Procedure for the Copper-Catalyzed Ynamide Addition to Acyl Chlorides
Copper iodide (2.3 mg, 12 μmol), N-ethynyl-N-phenyl-4-tolylsulfonamide (32.5 mg, 0.12 mmol), and N,N-diisopropylethylamine (31.0 mg, 0.24 mmol) were dissolved in chloroform (0.15 mL) under nitrogen. After 30 min an acyl chloride (0.18 mmol) was added, and the mixture was stirred until completion as determined by TLC. Solvents were evaporated under a stream of nitrogen, and the crude residue was purified by flash chromatography on silica gel (particle size 40–63 μm) as described below.
General Procedure for the Copper-Catalyzed Ynamide Addition to Pyridines and Quinolines
The ynamide (54.2 mg, 0.20 mmol), CuI (3.8 mg, 0.02 mmol), and N,N-diisopropylethylamine (70 μL, 0.40 mmol) were dissolved in 1 mL of anhydrous dichloromethane. Then, a solution of the N-heterocycle (0.24 mmol) and ethyl chloroformate (38 μL, 0.40 mmol) in 1 mL of anhydrous dichloromethane was added. The mixture was stirred under nitrogen until the reaction was completed based on NMR and TLC analysis. Solvents were then removed, and the crude residue was directly loaded onto a silica gel column (particle size 32–63 μm) and purified by flash chromatography as described below unless stated otherwise.
N-(3-Phenyl-3-oxoprop-1-ynyl)-N-phenyl-4-tolylsulfonamide, 2
The reaction with benzoyl chloride (25.1 mg, 0.18 mmol) and the ynamide (32.5 mg, 0.12 mmol) was performed at 30 °C for 22 h. The concentrated crude residue was purified by column chromatography (2:1 dichloromethane/hexanes) to give 40.5 mg (0.108 mmol, 90%) of a white solid. 1H NMR (400 MHz): δ 8.19 (d, J = 6.9 Hz, 2H), 7.67–7.57 (m, 3H), 7.52 (dd, J = 8.4 Hz, 6.9 Hz, 2H), 7.41–7.34 (m, 3H), 7.30–7.22 (m, 4H), 2.42 (s, 3H). 13C NMR (100 MHz): δ 176.8, 145.9, 137.2, 136.9, 133.6, 132.9, 129.9, 129.5, 129.17, 129.15, 128.6, 128.1, 126.5, 90.1, 74.9, 21.6. Anal. Calcd For C22H17NO3S: C, 70.38; H, 4.56; N, 3.73. Found: C, 70.51; H, 4.73; N, 3.86. Mp 139–140 °C.
N-(3-(2-Chlorophenyl)-3-oxoprop-1-ynyl)-N-phenyl-4-tolylsulfonamide, 3
The reaction with 2-chlorobenzoyl chloride (32.6 mg, 0.186 mmol) and the ynamide (32.5 mg, 0.12 mmol) was performed at 30 °C for 18 h. The concentrated crude residue was purified by column chromatography (5:2 dichloromethane/hexanes) to give 45 mg (0.11 mmol, 92%) of a white solid. 1H NMR (400 MHz): δ 8.06 (d, J = 7.6 Hz, 1H), 7.60 (d, J = 8.3 Hz, 2H), 7.45 (d, J = 3.6 Hz, 2H), 7.45–7.31 (m, 4H), 7.31–7.21 (m, 4H), 2.43 (s, 3H). 13C NMR (100 MHz): δ 175.3, 145.9, 137.1, 135.4, 133.1, 133.0, 132.9, 132.4, 131.4, 129.9, 129.4, 129.2, 128.2, 126.8, 126.5, 91.0, 76.3, 21.7. Anal. Calcd For C22H16ClNO3S: C, 64.47; H, 3.93; N, 3.42. Found: C, 64.65; H, 4.07; N, 3.41. Mp >105 °C (decomp)
N-(3-(4-Chlorophenyl)-3-oxoprop-1-ynyl)-N-phenyl-4-tolylsulfonamide, 4
The reaction with 4-chlorobenzoyl chloride (31.4 mg, 0.18 mmol) and the ynamide (32.5 mg, 0.12 mmol) was performed at 30 °C for 20 h. The concentrated crude residue was purified by column chromatography (2:1 dichloromethane/hexanes) to give 47 mg (0.115 mmol, 96%) of a white solid. 1H NMR (400 MHz): δ 8.09 (d, J = 8.6 Hz, 2H), 7.57 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 8.5 Hz, 2H), 7.39–7.28 (m, 3H), 7.28–7.18 (m, 4H), 2.39 (s, 3H). 13C NMR (100 MHz): δ 175.4, 146.0, 140.2, 137.0, 135.4, 132.9, 130.5, 129.9, 129.5, 129.3, 128.9, 128.1, 126.4, 90.7, 74.7, 21.6. Anal. Calcd For C22H16ClNO3S: C, 64.47; H, 3.93; N, 3.42. Found: C, 64.38; H, 4.05; N, 3.46. Mp 105–107 °C.
N-(3-(4-Cyanophenyl)-3-oxoprop-1-ynyl)-N-phenyl-4-tolylsulfonamide, 5
The reaction with 4-cyanobenzoyl chloride (30.0 mg, 0.18 mmol) and the ynamide (32.7 mg, 0.12 mmol) was performed at 30 °C for 18 h. The concentrated crude residue was purified by column chromatography (3:1 dichloromethane/hexanes) to give 46.5 mg (0.116 mmol, 97%) of a white solid. 1H NMR (400 MHz): δ 8.29 (d, J = 8.3 Hz, 2H), 7.82 (d, J = 8.1 Hz, 2H), 7.60 (d, J = 8.3 Hz, 2H), 7.47–7.34 (m, 3H), 7.29 (d, J = 8.1 Hz, 2H), 7.27–7.23 (m, 2H), 2.43 (s, 3H). 13C NMR (100 MHz): δ 174.8, 146.2, 139.8, 136.8, 136.7, 132.8, 132.4, 130.0, 129.6, 129.4, 128.1, 126.4, 117.9, 116.8, 92.3, 75.1, 21.7. Anal. Calcd For C23H16N2O3S: C, 68.98; H, 4.03; N, 7.00. Found: C, 68.67; H, 4.14; N, 6.92. Mp >155 °C (decomp)
N-(3-(2-Naphthyl)-3-oxoprop-1-ynyl)-N-phenyl-4-tolylsulfonamide, 6
The reaction with 2-naphthoyl chloride (35.0 mg, 0.18 mmol and the ynamide (32.9 mg, 0.121 mmol) was performed at 30 °C for 12 h. The concentrated crude residue was purified by column chromatography (1:1 dichloromethane/hexanes) to give 51.5 mg (0.12 mmol, 99%) of a white solid. 1H NMR (400 MHz): δ 8.88 (s, 1H), 8.19 (dd, J = 8.6, 1.7 Hz, 1H), 8.06 (d, J = 8.1 Hz, 1H), 7.94 (d, J = 8.7 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.65 (d, J = 8.2 Hz, 2H), 7.65–7.54 (m, 2H), 7.44–7.34 (m, 3H), 7.35–7.27 (m, 2H), 7.28–7.22 (m, 2H), 2.41 (s, 3H). 13C NMR (100 MHz): δ 176.8, 145.9, 137.1, 135.9, 134.5, 132.9, 132.6, 132.5, 130.0, 129.9, 129.5, 129.2, 128.8, 128.4, 128.1, 127.8, 126.8, 126.6, 123.6, 90.2, 75.0, 21.7. Anal. Calcd For C26H19NO3S: C, 73.39; H, 4.50; N, 3.29. Found: C, 73.32; H, 4.77; N, 3.32.
N-(3-(1-Naphthyl)-3-oxoprop-1-ynyl)-N-phenyl-4-tolylsulfonamide, 7
The reaction with 1-naphthoyl chloride (55.0 mg, 0.28 mmol) and the ynamide (54.5 mg, 0.20 mmol) was performed at 20 °C for 38 h. The concentrated crude residue was purified by column chromatography (1:1 dichloromethane/hexanes) to give 67 mg (0.16 mmol, 79%) of a colorless oil. 1H NMR (400 MHz): δ δ 9.21 (d, J = 8.5 Hz, 1H), 8.56 (d, J = 7.1 Hz, 1H), 8.06 (d, J = 8.2 Hz, 1H), 7.89 (d, J = 8.1 Hz, 1H), 7.68–7.57 (m, 4H), 7.54 (dd, J = 7.6, 7.4 Hz, 1H), 7.41- 7.33 (m, 3H), 7.32–7.26 (m, 2H), 7.23 (d, J = 8.1 Hz, 2H), 2.38 (s, 3H). 13C NMR (100 MHz): δ 178.7, 145.9, 137.2, 134.6, 134.0, 133.9, 132.8, 132.7, 130.7, 129.9, 129.5, 129.2, 128.7, 128.5, 128.2, 126.6, 126.5, 125.9, 124.7, 88.7, 76.0, 21.7. Anal. Calcd For C26H19NO3S: C, 73.39; H, 4.50; N, 3.29. Found: C, 73.30; H, 4.89; N, 3.30.
N-(4,4-Dimethyl-3-oxopent-1-ynyl)-N-phenyl-4-tolylsulfonamide, 8
The reaction with pivaloyl chloride (21.6 mg, 0.179 mmol) and the ynamide (33.5 mg, 0.124 mmol) was performed at 30 °C for 18 h. The concentrated crude residue was purified by column chromatography (2:1 dichloromethane/hexanes) to give 39.5 mg (0.111 mmol, 90%) of a white solid. 1H NMR (400 MHz): δ 7.56 (d, J = 7.9 Hz, 2H), 7.36–7.28 (m, 3H), 7.26 (d, J = 8.1 Hz, 2H), 7.21–7.13 (m, 2H), 2.40 (s, 3H), 1.19 (d, J = 1.3 Hz, 9H). 13C NMR (100 MHz): δ 193.0, 145.8, 137.4, 133.1, 129.8, 129.3, 129.0, 128.1, 126.4, 89.2, 73.6, 44.6, 26.2, 21.6. Anal. Calcd For C20H21NO3S: C, 67.58; H, 5.95; N, 3.94. Found: C, 67.68; H, 6.29; N, 3.86. Mp 98–101 °C.
N-(4-Methyl-3-oxopent-1-ynyl)-N-phenyl-4-tolylsulfonamide, 9
The reaction with isobutyryl chloride (30.4 mg, 0.28 mmol) and the ynamide (54.0 mg, 0.20 mmol) was performed at 15 °C for 52 h. The concentrated crude residue was purified by column chromatography (2:1 dichloromethane/hexanes) to give 47.3 mg (0.14 mmol, 70%) of a colorless oil.1H NMR (400 MHz): δ 7.58 (d, J = 7.9 Hz, 2H), 7.40–7.26 (m, 5H), 7.23–7.14 (m, 2H), 2.63 (hept, J = 7.1 Hz, 1H), 2.42 (s, 3H), 1.20 (d, J = 7.1 Hz, 6H). 13C NMR (100 MHz): δ 190.9, 145.9, 137.3, 132.9, 129.9, 129.4, 129.1, 128.1, 126.5, 89.1, 74.3, 42.7, 21.7, 18.1. Anal. Calcd For C19H19NO3S: C, 66.84; H, 5.61; N, 4.10. Found: C, 66.59; H, 5.86; N, 4.00.
N-Ethoxycarbonyl-1,2-dihydro-2-(N-phenyl-N-tosylaminoethynyl)pyridine, 10
The reaction between the ynamide (54.2 mg, 0.20 mmol) and pyridine (20 μL, 0.24 mmol) was completed after 2.5 h. Chromatographic purification (1:7 Et2O/hexanes) gave 60.3 mg (0.14 mmol, 71%) of a slightly yellow oil. 1H NMR (400 MHz): δ 7.42–7.72 (m, 2H), 7.21–7.33 (m, 5H), 7.14–7.21 (m, 2H), 6.76 (m, 1H), 5.99 (dd, J = 9.3, 5.6 Hz, 1H), 5.43–5.82 (m, 2H), 5.35 (d, J = 7.3 Hz, 1H), 4.18–4.34 (m, 2H), 2.43 (s, 3H), 1.24–1.36 (m, 3H). 13C NMR (100 MHz): δ 153.8, 153.0, 145.0, 144.8, 138.7, 132.6, 129.4, 128.9, 128.5, 128.2, 128.1, 126.0, 125.1, 124.7, 122.5, 122.2, 118.4, 117.9, 105.2, 69.1, 62.6, 44.1, 43.5, 21.7, 14.5. Anal. Calcd for C23H22N2O4S: C, 65.38; H, 5.25; N, 6.63. Found: C, 65.17; H, 5.36; N, 6.51.
N-Ethoxycarbonyl-1,2-dihydro-2-(N-phenyl-N-tosylaminoethynyl)-4-chloropyridine, 11
The ynamide (54.2 mg, 0.20 mmol), CuI (3.8 mg, 0.02 mmol), and N,N-diisopropylethylamine (70 μL, 0.40 mmol) were dissolved in 1 mL of anhydrous dichloromethane. Then, a solution of 4-bromopyridine hydrochloride (46.7 mg, 0.24 mmol), N,N-diisopropylethylamine (70 μL, 0.40 mmol), and ethyl chloroformate (38 μL, 0.40 mmol) in 1 mL of anhydrous dichloromethane was added. The reaction was completed after 2.5 h. Chromatographic purification (3:8 Et2O/hexanes) gave 83.0 mg (0.18 mmol, 91%) of a slightly yellow oil. 1H NMR (400 MHz): δ 7.54 (d, J = 8.0 Hz, 2H), 7.22–7.35 (m, 5H), 7.11–7.21 (m, 2H), 6.82 (m, 1H), 5.71 (s, 1H), 5.56 (m, 1H), 5.31 (dd, J = 8.0, 2.1 Hz, 1H), 4.19–4.34 (m, 2H), 2.44 (s, 3H), 1.27–1.34 (m, 3H). 13C NMR (100 MHz): δ 144.9, 138.5, 132.7, 129.4, 129.0, 128.4, 128.1, 126.6, 126.0, 113.6, 106.6, 78.5, 68.2, 63.0, 45.3, 21.6, 14.3. Anal. Calcd for C23H21ClN2O4S: C, 60.46; H, 4.63; N, 6.13. Found: C, 60.28; H, 4.90; N, 6.13.
N-Ethoxycarbonyl-1,2-dihydro-2-(N-phenyl-N-tosylaminoethynyl)-5-chloropyridine, 12
The reaction between the ynamide (54.2 mg, 0.20 mmol) and 3-chloropyridine (23 μL, 0.24 mmol) was completed after 2.5 h. Chromatographic purification (1:7 Et2O/hexanes) gave 67.1 mg (0.15 mmol, 73%) of a slightly yellow oil. 1H NMR (400 MHz): δ 7.44–7.59 (m, 2H), 7.22–7.33 (m, 5H), 7.14–7.21 (m, 2H), 6.77 (m, 1H), 6.10 (d, J = 6.2 Hz, 1H), 5.69 (m, 1H), 5.33 (m, 1H), 4.19–4.35 (m, 2H), 2.43 (s, 3H), 1.31 (m, 3H). 13C NMR (100 MHz): δ 144.9, 138.5, 132.7, 129.4, 128.9, 128.3, 128.1, 126.0, 123.8, 123.3, 120.7, 120.4, 104.4, 104.1, 78.3, 66.9, 63.0, 50.0, 49.4, 34.1, 21.6, 14.3. Anal. Calcd for C23H21ClN2O4S: C, 60.46; H, 4.63; N, 6.13. Found: C, 60.64; H, 4.46; N, 5.96.
N-Ethoxycarbonyl-1,2-dihydro-2-(N-phenyl-N-tosylaminoethynyl)-4-bromopyridine, 13
The ynamide (54.2 mg, 0.20 mmol), CuI (3.8 mg, 0.02 mmol) and N,N-diisopropylethylamine (70 μL, 0.40 mmol) were dissolved in 1 mL of anhydrous acetonitrile. Then, a solution of 4-bromopyridine hydrochloride (46.7 mg, 0.24 mmol), N,N-diisopropylethylamine (70 μL, 0.40 mmol), and ethyl chloroformate (38 μL, 0.40 mmol) in 2 mL of anhydrous acetonitrile was added. The reaction was completed after 20 h. Chromatographic purification (1:4 EtOAc/hexanes) gave 72.4 mg (0.14 mmol, 72%) of a slightly yellow oil. 1H NMR (400 MHz): δ 7.47–7.61 (m, 2H), 7.23–7.36 (m, 5H), 7.14–7.22 (m, 2H), 6.82 (m, 1H), 5.71 (m, 1H), 5.57 (d, J = 6.8 Hz, 1H), 5.31 (dd, J = 8.0, 2.1 Hz, 1H), 4.20–4.34 (m, 2H), 2.44 (s, 3H), 1.30 (s, 3H). 13C NMR (100 MHz): δ 144.9, 138.4, 132.5, 129.4, 129.0, 128.4, 128.2, 127.1, 126.6, 126.0, 113.8, 113.3, 106.6, 78.4, 68.2, 63.0, 45.5, 45.1, 21.7, 14.3. Anal. Calcd for C23H21BrN2O4S: C, 55.10; H, 4.22; N, 5.59. Found: C, 54.73; H, 4.63; N, 5.28.
N-Ethoxycarbonyl-1,2-dihydro-2-(N-phenyl-N-tosylaminoethynyl)-4-phenylpyridine, 14
The reaction between the ynamide (54.2 mg, 0.20 mmol) and 4-phenylpyridine (37.2 mg, 0.24 mmol) was completed after 2.5 h. Chromatographic purification (1:4 EtOAc/hexanes) gave 95.5 mg (0.19 mmol, 96%) of a slightly yellow oil. 1H NMR (400 MHz): δ 7.46–7.58 (m, 2H), 7.23–7.46 (m, 8H), 7.16–7.23 (m, 2H), 7.02–7.16 (m, 2H), 6.92 (m, 1H), 5.69–5.91 (m, 3H), 4.22–4.37 (m, 2H), 2.36 (s, 3H), 1.27–1.40 (m, 3H). 13C NMR (100 MHz): δ 153.6, 152.8, 144.8, 144.7, 138.6, 138.3, 134.6, 134.3, 132.6, 129.3, 128.9, 128.6, 128.4, 128.2, 128.1, 128.0, 126.0, 125.8, 125.5, 113.9, 113.4, 106.2, 77.7, 69.1, 62.7, 44.5, 44.0, 21.6, 14.5. Anal. Calcd for C29H26N2O4S: C, 69.86; H, 5.26; N, 5.62. Found: C, 69.76; H, 5.47; N, 5.56.
N-Ethoxycarbonyl-2-(N-phenyl-N-tosylaminoethynyl)-2,3-dihydro-4-pyridone, 15
The reaction between the ynamide (54.2 mg, 0.20 mmol) and 4-methoxypyridine (25 μL, 0.24 mmol) was completed after 2.5 h. Acidic workup (1 M aqueous HCl) followed by chromatographic purification (1:1 EtOAc/hexanes, Al2O3) gave 68.2 mg (0.16 mmol, 78%) of a slightly yellow oil. 1H NMR (400 MHz): δ 7.75 (d, J = 8.4 Hz, 1H), 7.47 (d, J = 8.3 Hz, 2H), 7.22–7.34 (m, 5H), 7.08–7.18 (m, 2H), 5.51 (d, J = 6.3 Hz, 1H), 5.43 (dd, J = 8.4, 1.3 Hz, 1H), 4.27–4.41 (m, 2H), 2.88 (dd, J = 16.3, 6.5 Hz, 1H), 2.59 (ddd, J = 16.3, 1.6, 1.6 Hz, 1H), 2.44 (s, 3H), 1.35 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz): δ 191.3, 152.1, 145.2, 141.3, 138.2, 132.5, 129.6, 129.1, 128.3, 128.2, 126.0, 107.5, 77.9, 66.9, 63.9, 45.5, 41.6, 21.7, 14.3. Anal. Calcd for C23H22N2O5S: C, 63.00; H, 5.06; N, 6.39. Found: C, 62.74; H, 4.99; N, 6.09.
N-Ethoxycarbonyl-1,2-dihydro-2-(N-phenyl-N-tosylaminoethynyl)quinoline, 16
The reaction between the ynamide (54.2 mg, 0.20 mmol) and quinoline (29 μL, 0.24 mmol) was completed after 2.5 h. Chromatographic purification (3:8 Et2O/hexanes) gave 86.0 mg (0.18 mmol, 91%) of a slightly yellow oil. 1H NMR (400 MHz): δ 7.59 (d, J = 8.3 Hz, 1H), 7.11–7.30 (m, 8H), 7.06–7.11 (m, 2H), 6.93–7.00 (m, 2H), 6.55 (d, J = 9.1 Hz, 1H), 6.06 (dd, J = 9.2, 6.2 Hz, 1H), 5.98 (d, J = 6.1 Hz, 1H), 4.19–4.41 (m, 2H), 2.41 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz): δ 153.6, 144.6, 138.5, 134.5, 132.5, 129.2, 128.9, 128.1, 128.0, 127.7, 126.8, 126.6, 125.9, 125.9, 125.2, 124.4, 124.2, 78.0, 67.8, 62.5, 44.3, 21.7, 14.5. Anal. Calcd for C27H24N2O4S: C, 68.62; H, 5.12; N, 5.93. Found: C, 68.82; H, 5.36; N, 5.66.
N-Ethoxycarbonyl-1,2-dihydro-2-(N-phenyl-N-tosylaminoethynyl)-4,7-dichloroquinoline, 17
The reaction between the ynamide (54.2 mg, 0.20 mmol) and 4,7-dichloroquinoline (47.5 mg, 0.24 mmol) was completed after 20 h. Chromatographic purification (1:6 Et2O/hexanes) gave 95.0 mg (0.18 mmol, 88%) of a slightly yellow oil. 1H NMR (400 MHz): δ 7.63 (s, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.20–7.30 (m, 6H), 7.15–7.20 (m, 2H), 6.92–6.97 (m, 2H), 6.20 (d, J = 6.9 Hz, 1H), 6.00 (d, J = 6.8 Hz, 1H), 4.19–4.46 (m, 2H), 2.45 (s, 3H), 1.35 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz) δ 152.7, 144.9, 138.3, 135.8, 134.7, 132.5, 129.3, 128.9, 128.2, 128.1, 125.9, 125.8, 124.6, 124.2, 123.8, 122.4, 79.3, 66.4, 63.1, 45.3, 21.7, 14.4. Anal. Calcd for C27H22Cl2N2O4S: C, 59.89; H, 4.10; N, 5.17. Found: C, 60.08; H, 4.49; N, 5.28.
N-Ethoxycarbonyl-1,2-dihydro-2-(N-phenyl-N-tosylaminoethynyl)-4-chloro-6-methoxyquinoline, 18
The reaction between the ynamide (54.2 mg, 0.20 mmol) and 4-chloro-6-methoxyquinoline (46.5 mg, 0.24 mmol) was completed after 2.5 h. Chromatographic purification (1:4 Et2O/hexanes) gave 88.1 mg (0.16 mmol, 82%) of a slightly yellow oil. 1H NMR (400 MHz): δ 7.47 (s, 1H), 7.13–7.32 (m, 8H), 6.92–7.00 (m, 2H), 6.90 (dd, J = 8.9, 3.0 Hz, 1H), 6.21 (d, J = 6.8 Hz, 1H), 6.01 (d, J = 7.0 Hz, 1H), 4.18–4.36 (m, 2H), 3.87 (s, 3H), 2.43 (s, 3H), 1.32 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz): δ 156.6, 144.7, 138.5, 132.7, 130.0, 129.3, 128.9, 128.2, 128.1, 126.5, 125.9, 114.9, 109.7, 78.7, 66.7, 62.7, 55.7, 45.2, 21.7, 14.5. Anal. Calcd for C28H25ClN2O5S: C, 62.62; H, 4.69; N, 5.22. Found: C, 62.69; H, 5.02; N, 5.28.
N-Ethoxycarbonyl-5,6-dihydro-6-(N-phenyl-N-tosylaminoethynyl)phenanthridine, 19
The reaction between the ynamide (54.2 mg, 0.20 mmol) and phenanthridine (43.0 mg, 0.24 mmol) was completed after 3 h. Chromatographic purification (1:3 Et2O/pentanes) gave 99.5 mg (0.19 mmol, 95%) of a colorless oil. 1H NMR (400 MHz): δ 7.76 (dd, J = 7.0 Hz, 7.0 Hz, 2H), 7.58 (m, 1H), 7.42 (m, 1H), 7.24–7.37 (m, 4H), 7.00–7.21 (m, 7H), 6.71 (d, J = 7.7 Hz, 2H), 6.48 (s, 1H), 4.16–4.37 (m, 2H), 2.41 (s, 3H), 1.32 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz): δ 153.1, 144.4, 138.5, 135.0, 132.2, 130.7, 129.2, 128.8, 128.7, 128.1, 128.0, 127.8, 125.7, 125.6, 125.1, 123.9, 78.9, 68.5, 62.5, 48.1, 21.7, 14.5. Anal. Calcd for C31H26N2O4S: C, 71.24; H, 5.01; N, 5.36. Found: C, 70.89; H, 5.26; N, 5.57.
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (GM106260).
Supporting Information Available
NMR spectra and crystallographic details. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For excellent reviews, see:; a Zificsak C. A.; Mulder J. A.; Hsung R. P.; Rameshkumar C.; Wei L.-L. Tetrahedron 2001, 57, 7575–7606. [Google Scholar]; b Evano G.; Coste A.; Jouvin K. Angew. Chem., Int. Ed. 2010, 49, 2840–2859. [DOI] [PubMed] [Google Scholar]; c DeKorver K. A.; Li H.; Lohse A. G.; Hayashi R.; Lu Z.; Zhang Y.; Hsung R. P. Chem. Rev. 2010, 110, 5064–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witulski B.; Goessmann M. Chem. Commun. 1999, 1879–1880. [Google Scholar]
- a Bruckner D. Synlett 2000, 1402–1404. [Google Scholar]; b Bruckner D. Tetrahedron 2006, 62, 3809–3814. [Google Scholar]
- Rodriguez D.; Castedo L.; Saa C. Synlett 2007, 1963–1965. [Google Scholar]
- Selected examples. Hydroboration:; a Witulski B.; Buschmann N.; Bergstraesser U. Tetrahedron 2000, 56, 8473–8480. [Google Scholar]; Oxoarylations and cyclizations:; b Bhunia S.; Chang C.-J.; Liu R.-S. Org. Lett. 2012, 14, 5522–5525. [DOI] [PubMed] [Google Scholar]; c Yang L.-Q.; Wang K.-B.; Li C.-Y. Eur. J. Org. Chem. 2013, 2775–2779. [Google Scholar]; d Couty S.; Meyer C.; Cossy J. Synlett 2007, 2819–282. [Google Scholar]; e Al-Rashid Z. F.; Hsung R. P. Org. Lett. 2008, 10, 661–663. [DOI] [PubMed] [Google Scholar]; Nucleophilic alkylations with lithiated ynamides:; f Frederick M. O.; Mulder J. A.; Tracey M. R.; Hsung R. P.; Huang J.; Kurtz K. C. M.; Shen L.; Douglas C. J. J. Am. Chem. Soc. 2003, 125, 2368–2369. [DOI] [PubMed] [Google Scholar]; Homologation with aldehydes or ketones:; g You L.; Al-Rashid Z. F.; Figueroa R.; Ghosh S. K.; Ti G.; Lu T.; Hsung R. P. Synlett 2007, 1656–1662. [Google Scholar]
- [3 + 2] Cycloadditions:; a IJsselstijn M.; Cintrat J.-C. Tetrahedron 2006, 62, 3837–3842. [Google Scholar]; b Li H.; You L.; Zhang X.; Johnson W. L.; Figueroa R.; Hsung R. P. Heterocycles 2007, 74, 553–568. [Google Scholar]; c Li H.; Hsung R. P. Org. Lett. 2009, 11, 4462–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zhang X.; Li H.; You L.; Tang Y.; Hsung R. P. Adv. Synth. Catal. 2006, 348, 2437–2442. [Google Scholar]; e Zhang X.; Hsung R. P.; Li H. Chem. Commun. 2007, 2420–2422. [DOI] [PubMed] [Google Scholar]; f Kim J. Y.; Kim S. H.; Chang S. Tetrahedron Lett. 2008, 49, 1745–1749. [Google Scholar]; g Zhang X.; Hsung R. P.; Li H.; Zhang Y.; Johnson W. L.; Figueroa R. Org. Lett. 2008, 10, 3477–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]; [4 + 2] Cycloadditions:; h Martinez-Esperon M. F.; Rodriguez D.; Castedo L.; Saa C. Org. Lett. 2005, 7, 2213–2216. [DOI] [PubMed] [Google Scholar]; [2 + 2+1] Cycloadditions:; i Witulski B.; Goessmann M. Synlett 2000, 1793–1797. [Google Scholar]; [2 + 2+2] Cycloadditions:; j Witulski B.; Stengel T. Angew. Chem., Int. Ed. 1999, 38, 2426–2430. [PubMed] [Google Scholar]; k Witulski B.; Stengel T.; Fernandez-Hernandez J. Chem. Commun. 2000, 1965–1966. [Google Scholar]; l Witulski B.; Alayrac C. Angew. Chem., Int. Ed. 2002, 41, 3281–3284. [DOI] [PubMed] [Google Scholar]; m Dateer R. B.; Shaibu B. S.; Liu R.-S. Angew. Chem., Int. Ed. 2012, 51, 113–117. [DOI] [PubMed] [Google Scholar]
- a Marion F.; Coulomb J.; Courillon C.; Fensterbank L.; Malacria M. Org. Lett. 2004, 6, 1509–1511. [DOI] [PubMed] [Google Scholar]; b Marion F.; Coulomb J.; Servais A.; Courillon C.; Fensterbank L.; Malacria M. Tetrahedron 2006, 62, 3856–3871. [Google Scholar]; c Hashmi A. S.; Rudolph M.; Bats J.; Frey W.; Rominger F.; Oeser T. Chem.—Eur. J. 2008, 14, 6672–6678. [DOI] [PubMed] [Google Scholar]; d Couty S.; Meyer C.; Cossy J. Tetrahedron 2009, 65, 1809–1832. [Google Scholar]
- Homocoupling reactions:; a Rodriguez D.; Castedo L.; Saa C. Synlett 2004, 377–379. [Google Scholar]; Negishi cross-couplings:; b Rodriguez D.; Castedo L.; Saa C. Synlett 2004, 783–786. [Google Scholar]; c Martinez-Esperon M. F.; Rodriguez D.; Castedo L.; Saa C. Tetrahedron 2006, 62, 3843–3855. [Google Scholar]; Sonogashira couplings:; d Tracey M. R.; Zhang Y.; Frederick M. O.; Mulder J. A.; Hsung R. P. Org. Lett. 2004, 6, 2209–2212. [DOI] [PubMed] [Google Scholar]; e Dooleweerdt K.; Ruhland T.; Skrydstrup T. Org. Lett. 2009, 11, 221–224. [DOI] [PubMed] [Google Scholar]; Heck reactions:; f Couty S.; Liegault B.; Meyer C.; Cossy J. Org. Lett. 2004, 6, 2511–2514. [DOI] [PubMed] [Google Scholar]; g Couty S.; Liegault B.; Meyer C.; Cossy J. Tetrahedron 2006, 62, 3882–3895. [Google Scholar]
- Saito N.; Sato Y.; Mori M. Org. Lett. 2002, 4, 803–805. [DOI] [PubMed] [Google Scholar]
- Banerjee B.; Litvinov D. N.; Kang J.; Bettale J. D.; Castle S. L. Org. Lett. 2010, 12, 2650–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tanaka R.; Hirano S.; Urabe H.; Sato F. Org. Lett. 2003, 5, 67–70. [DOI] [PubMed] [Google Scholar]; b Tanaka R.; Yuza A.; Watai Y.; Suzuki D.; Takayama Y.; Sato F.; Urabe M. J. Am. Chem. Soc. 2005, 127, 7774–7780. [DOI] [PubMed] [Google Scholar]; c Tanaka D.; Sato Y.; Mori M. J. Am. Chem. Soc. 2007, 129, 7730–7731. [DOI] [PubMed] [Google Scholar]
- a Joshi R. V.; Xu Z.-Q.; Ksebati M. B.; Kessel D.; Corbett T. H.; Drach J. C.; Zemlicka J. J. Chem. Soc., Perkin Trans. 1 1994, 10, 1089–1098. [Google Scholar]; b Rodriguez D.; Castedo L.; Saa C. Synlett 2007, 1963–1965. [Google Scholar]; c Egi M.; Yamaguchi Y.; Fujiwara N.; Akai S. Org. Lett. 2008, 10, 1867–1870. [DOI] [PubMed] [Google Scholar]; d Wang X.-N.; Winston-McPherson G. N.; Walton M. C.; Zhang Y.; Hsung R. P.; DeKorver K. A. J. Org. Chem. 2013, 78, 6233–6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cook A. M.; Wolf C. Chem. Commun. 2014, 50, 3151–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]; For the synthesis of indole-derived ynamines, see:; b Hamada T.; Ye X.; Stahl S. S. J. Am. Chem. Soc. 2008, 130, 833–835. [DOI] [PubMed] [Google Scholar]
- Selected examples:; a Tohda Y.; Sonogashira K.; Hagihara N. Synthesis 1977, 777–778. [Google Scholar]; b Chowdhury C.; Kundu N. G. Tetrahedron 1999, 55, 7011–7016. [Google Scholar]; c Guo M.; Li D.; Zhang Z. J. Org. Chem. 2003, 68, 10172–10174. [DOI] [PubMed] [Google Scholar]; d Chen L.; Li C.-J. Org. Lett. 2004, 6, 3151–3153. [DOI] [PubMed] [Google Scholar]; e Likhar P. R.; Subhas M. S.; Roy M.; Roy S.; Kantam M. L. Helv. Chim. Acta 2008, 91, 259–264. [Google Scholar]; f Yuan H.; Jin H.; Li B.; Shen Y.; Yue R.; Shan L.; Sun Q.; Zhang W. Can. J. Chem. 2013, 91, 333–337. [Google Scholar]
- Selected examples:; a Kang S.-K.; Lim K.-H.; Ho P.-S.; Kim W.-Y. Synthesis 1999, 874–876. [Google Scholar]; b Liang B.; Huang M.; You Z.; Xiong Z.; Lu K.; Fathi R.; Chen J.; Yang Z. J. Org. Chem. 2005, 70, 6097–6100. [DOI] [PubMed] [Google Scholar]; c Fusano A.; Fukuyama T.; Nishitani S.; Inouye T.; Ryu I. Org. Lett. 2010, 12, 2410–2413. [DOI] [PubMed] [Google Scholar]; d Wu X.-F.; Neumann H.; Beller M. Chem.—Eur. J. 2010, 16, 12104–12107. [DOI] [PubMed] [Google Scholar]
- We observed that the ynesulfonamide used in this study undergoes thermal decomposition at 60 °C.
- For comparison we prepared 1,3-diphenylprop-2-yn-1-one from phenylacetylene and benzoyl chloride. This C-substituted ynone has the alkyne and the carbonyl stretchings at 2199 and 1640 cm–1, respectively.
- Selected examples:; a Fernandez-Ibanez M.; Macia B.; Pizzuti M. G.; Minnaard A. J.; Feringa B. L. Angew. Chem. 2009, 48, 9339–9341. [DOI] [PubMed] [Google Scholar]; b Sun Z.; Yu S.; Ding Z.; Ma D. J. Am. Chem. Soc. 2007, 129, 9300–9301. [DOI] [PubMed] [Google Scholar]; c Black D. A.; Beveridge R. E.; Arndtsen B. A. J. Org. Chem. 2008, 736, 1906–1910. [DOI] [PubMed] [Google Scholar]; d Chen Q.; du Jourdin X. M.; Knochel P. J. Am. Chem. Soc. 2013, 135, 4958–4961. [DOI] [PubMed] [Google Scholar]; e Chau S. T.; Lutz P.; Wu K.; Doyle A. G. Angew. Chem. 2013, 51, 9153–9156. [DOI] [PMC free article] [PubMed] [Google Scholar]; For recent reviews, see:; f Bull J. A.; Mousseau J. J.; Pelletier G.; Charette A. B. Chem. Rev. 2012, 112, 2642–2713. [DOI] [PubMed] [Google Scholar]; g Silva E. M. P.; Varandas P. A. M. M.; Silva A. M. S. Synthesis 2013, 45, 3053–3089. [Google Scholar]; h Larionov O. V.; Stephens D.; Mfuh A.; Chavez G. Org. Lett. 2014, 16, 864–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- We observed exclusive 1,2-addition of the ynesulfonamide to quinoline, but slow conversion was observed when acridine and isoquinolines were applied in this procedure. It is noteworthy that this protocol is not limited to ynesulfonamide 1, and we found that ynecarbamates or indole-derived ynamines react smoothly with pyridine under the same conditions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.