

Abstract
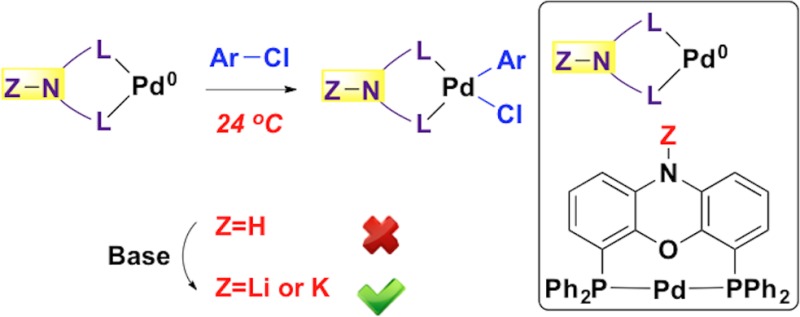
Although the past 15 years have witnessed the development of sterically bulky and electron-rich alkylphosphine ligands for palladium-catalyzed cross-couplings with aryl chlorides, examples of palladium catalysts based on either triarylphosphine or bidentate phosphine ligands for efficient room temperature cross-coupling reactions with unactivated aryl chlorides are rare. Herein we report a palladium catalyst based on NiXantphos, a deprotonatable chelating aryldiphosphine ligand, to oxidatively add unactivated aryl chlorides at room temperature. Surprisingly, comparison of an extensive array of ligands revealed that under the basic reaction conditions the resultant heterobimetallic Pd–NiXantphos catalyst system outperformed all the other mono- and bidentate ligands in a deprotonative cross-coupling process (DCCP) with aryl chlorides. The DCCP with aryl chlorides affords a variety of triarylmethane products, a class of compounds with various applications and interesting biological activity. Additionally, the DCCP exhibits remarkable chemoselectivity in the presence of aryl chloride substrates bearing heteroaryl groups and sensitive functional groups that are known to undergo 1,2-addition, aldol reaction, and O-, N-, enolate-α-, and C(sp2)–H arylations. The advantages and importance of the Pd–NiXantphos catalyst system outlined herein make it a valuable contribution for applications in Pd-catalyzed arylation reactions with aryl chlorides.
1. Introduction
The past 15 years have witnessed the development of phosphine ligands used for palladium-catalyzed cross-couplings with aryl chlorides.1 Among them, sterically bulky and electron-rich alkylphosphine ligands have proved particularly successful and have been widely applied in synthesis.2 Simple triarylphosphine ligands are generally ineffective for oxidative addition of unactivated aryl chlorides to Pd(0), in part due to their reduced electron-donating ability. Yet, aryl chlorides are arguably the most useful substrates among aryl halides and pseudohalides, because of their low cost and wide availability. To date, few reports have documented the use of triarylphosphine ligands for cross-couplings with unactivated aryl chlorides, most of which require higher temperatures (≥60 °C), and typically give moderate yields with narrow substrate scope.3a−3i In 2005, the Buchwald group reported that a sterically hindered monodentate triarylphosphine ligand, 2-diphenylphosphino-2′,4′,6′-triisopropylbiphenyl, promoted Suzuki–Miyaura couplings of aryl chlorides at room temperature to 40 °C in excellent yields.3j
Mechanistic studies on oxidative addition of aryl chlorides to Pd(0) bearing bulky electron-rich phosphine ligands have been recently reported: the Hartwig group, as well as others, demonstrated that oxidative addition of aryl chlorides to Pd(0) proceeds through a monoligated palladium species, LPd(0) (L = monodentate phosphine).4 This mechanistic picture is consistent with the inability of chelating phosphines to oxidatively add aryl chlorides at low temperature. The two reasonable pathways for oxidative addition of aryl chlorides with bidentate phosphines are dissociation of one of the phosphorus centers of a bidentate chelating phosphine or direct oxidative addition without dissociation of a phosphorus center, both of which are high in energy.4e−4g As a result, palladium complexes with chelating bidentate ligands typically catalyze cross-couplings of unactivated aryl chlorides only at elevated temperatures.5 In 1993, the Milstein group reported the oxidative addition of aryl chlorides to Pd(P̂P), which was generated from Pd(P̂P)2 [P̂P = a bidentate phosphine, in this case, 1,3-bis(diisopropylphosphino)propane (dippp)].6 Despite the bulky and strongly donating phosphorus centers, the oxidative addition was conducted at 90 °C to achieve complete conversion in 2 h. In 2003, a similar observation was reported by the Hartwig group for the room temperature oxidative addition of aryl tosylates to Pd(P̂P)2 (P̂P = PPFt-Bu and CyPFt-Bu).7 However, we are not aware of examples of palladium catalysts based on bidentate phosphine ligands for efficient room temperature cross-coupling reactions with unactivated aryl chlorides.
Herein we report unprecedented reactivity employing a deprotonatable chelating aryldiphosphine ligand, NiXantphos,8 for the room temperature palladium-catalyzed cross-coupling reaction of unactivated aryl chlorides. Surprisingly, comparison of an extensive array of ligands revealed that the heterobimetallic (M–NiXantphos)Pd catalyst system (M = alkali metal)9 outperformed all the other mono- and bidentate ligands in deprotonative cross-coupling reactions with aryl chlorides.
2. Results and Discussion
2.1. Initial Studies with NiXantphos
We recently initiated a program in the catalytic functionalization of weakly acidic sp3-hybridized C–H bonds. We have categorized these reactions as deprotonative cross-coupling processes (DCCP), because they involve initial reversible deprotonation of the C–H by base without the participation of the catalyst. The catalyst promotes the subsequent functionalization of the deprotonated species. Thus, DCCP is mechanistically distinct from C–H activation/functionalization processes.
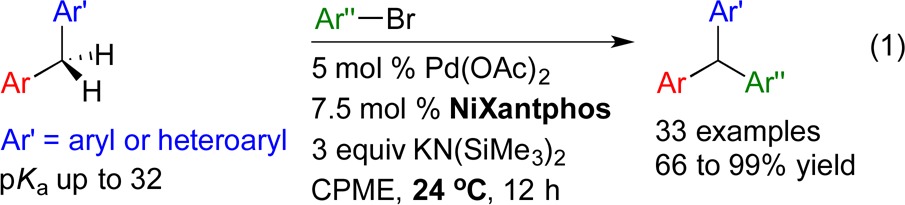
Substrates we reported to undergo DCCP include diarylmethanes, allyl benzenes, sulfoxides, sulfones, amides and η6-arene complexes of toluene derivatives and benzylic amines.10−12 Diphenylmethane (pKa = 32.313) and diarylmethane derivatives were arylated at room temperature with aryl bromides in the presence of KN(SiMe3)2 and a palladium catalyst bearing van Leeuwen’s NiXantphos ligand8 (eq 1, see Scheme 1 for the structure of NiXantphos).11a This method facilitates rapid access to a wide variety of sterically and electronically diverse triarylmethanes, a class of compounds with various applications and interesting biological activity.14−18
 |
1 |
Scheme 1. Selected HTE Results of the Cross-Coupling of 1a with 1-Bromo-4-tert-butylbenzene (AY= assay yield).
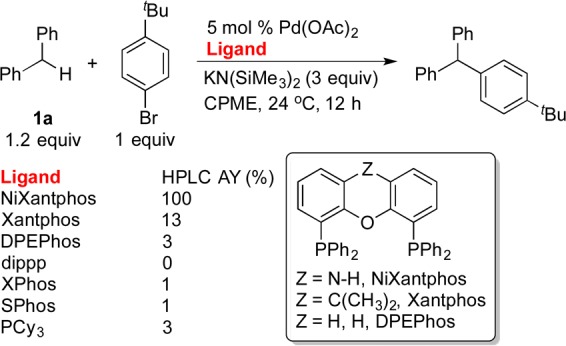
HTE conducted on a 10 μmol scale at 0.1 M.
Interestingly, when we examined various ligands for the cross-coupling of diphenylmethane (1a) with 1-bromo-4-tert-butylbenzene at room temperature using microscale high-throughput experimentation (HTE, Scheme 1),19 the NiXantphos-based catalyst showed superior performance over dippp as well as the other bidentate ligands sharing similar bite angles (see Supporting Information [SI] for a complete list of ligands).8 The NiXantphos-based catalyst also outperformed monodentate alkylphosphine ligands. The dominance of NiXantphos over the structurally similar Xantphos begged the question: Why is NiXantphos so active under these reaction conditions? We hypothesized that the presence of a somewhat acidic N–H moiety under the basic reaction conditions would result in deprotonation and that the resultant heterobimetallic system might exhibit cooperative reactivity. We then set out to determine the validity of this hypothesis, starting with a deprotonation study.
2.2. Deprotonation of NiXantphos
2.2.1. Solution Studies
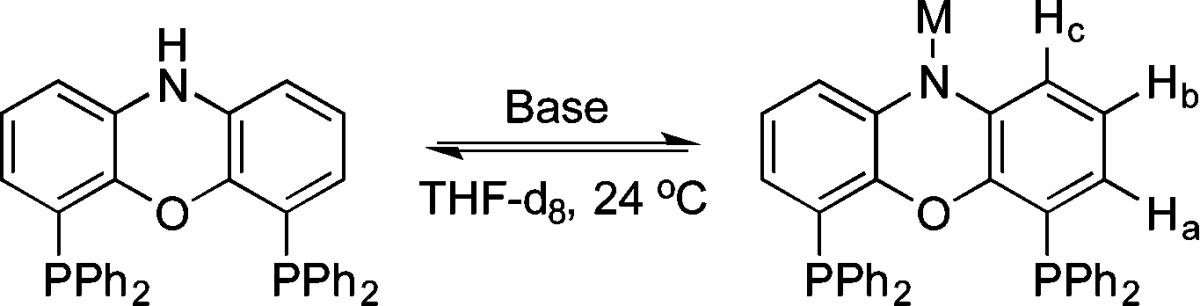
NiXantphos has a phenoxazine core, with a pKa around 22,13 and should be deprotonated by KN(SiMe3)2 under the reaction conditions in Scheme 1. The 1H and 31P{1H} NMR spectra of NiXantphos were recorded in THF-d8 at room temperature before and after combination with 1.5–6 equiv of MN(SiMe3)2 (M = Li, K). After 1.5 equiv of MN(SiMe3)2 were added, the 1H NMR spectrum of the resulting solution displayed significant shifts in the phenoxazine hydrogens (Ha, Hb, and Hc, Table 1). A distinct upfield shift, Δ, of −0.75 ppm of Ha was observed in the presence of 1.5 equiv of KN(SiMe3)2 (entry 2 vs 1). Similarly, Hc exhibited a smaller shift (Δ=–0.59 ppm) as did Hb (Δ=–0.46 ppm). 1H NMR chemical shifts did not change upon further addition of KN(SiMe3)2 to NiXantphos (entries 3 and 4 vs 2). Similar shifts were observed in the 1H NMR spectrum of the phenoxazine hydrogens of NiXantphos in the presence of 1.5 equiv of LiN(SiMe3)2 (entry 5). These observations indicated that deprotonation was complete with 1.5 equiv of MN(SiMe3)2 (M = Li, K). Interestingly, only a very small shift was observed in the 31P{1H} NMR spectra of the PPh2 moiety of NiXantphos after addition of MN(SiMe3)2 (Table 1), suggesting (1) the countercation is not bound to the phosphorus centers (also see Figure 1a), and (2) the anionic phenoxazine backbone does not render the phosphorus centers significantly more electron-rich (see section 2.2.2 for DFT calculations).
Table 1. 1H and 31P{1H} NMR Studies of NiXantphos Deprotonated by Basea.
| entry | base (equiv) | Ha | Hb | Hc | PPh2 |
|---|---|---|---|---|---|
| 1 | none | 5.91 | 6.52 | 6.32 | –18.99 |
| 2 | KN(SiMe3)2 (1.5) | 5.16 | 6.06 | 5.73 | –18.69 |
| 3 | KN(SiMe3)2 (3) | 5.14 | 6.03 | 5.70 | –18.69 |
| 4 | KN(SiMe3)2 (6) | 5.15 | 6.04 | 5.70 | –18.68 |
| 5 | LiN(SiMe3)2 (1.5) | 5.22 | 6.07 | 5.75 | –18.45 |
Reactions conducted on a 0.04 mmol scale with 1 equiv of NiXantphos and 1.5–6 equiv of MN(SiMe3)2 (M = Li, K) in 0.75 mL of THF-d8 in a J. Young NMR tube at room temperature, chemical shifts reported in ppm, referenced to the proteo internal standard.
Figure 1.

(a) ORTEP diagram of [K(THF)3–NiXantphos]2 with 50% probability thermal ellipsoids displayed. Hydrogen atoms omitted for clarity. N1–K1 = 2.808(3) Å, N1–K1′ = 2.857(3) Å, P1···P2 distance = 4.245 Å. (b) ORTEP diagram of K(THF)(18-crown-6)–NiXantphos with 50% probability thermal ellipsoids displayed. Hydrogen atoms omitted for clarity. N1–K1 = 2.885(2) Å, P1···P2 distance = 4.301 Å.
2.2.2. DFT Calculations with NiXantphos and Related Bis(phosphines)
In reported work, the electron-donating ability of mono- and bidentate phosphine ligands has been assessed using density functional theory (DFT). For example, Spokoyny and Buchwald correlated computed partial charges of B9-connected trivalent aryl and alkyl phosphinoboranes with the electronic properties of their resulting carbonyl complexes.20 In order to probe the experimental observations for the Pd–NiXantphos system, DFT calculations were performed to assess the electronic structures of the ligands. Gas-phase geometry optimizations were performed at the B3LYP level of theory with the 6-31 G* basis set for all atoms using Gaussian ’09. Natural bond orbital (NBO) analyses of the resulting structures afforded comparisons of atomic natural charges (Table 2 and SI).
Table 2. DFT Calculated Natural Charges for Members of the Xantphos Ligand Family and Relevant Bidentate Phosphines.
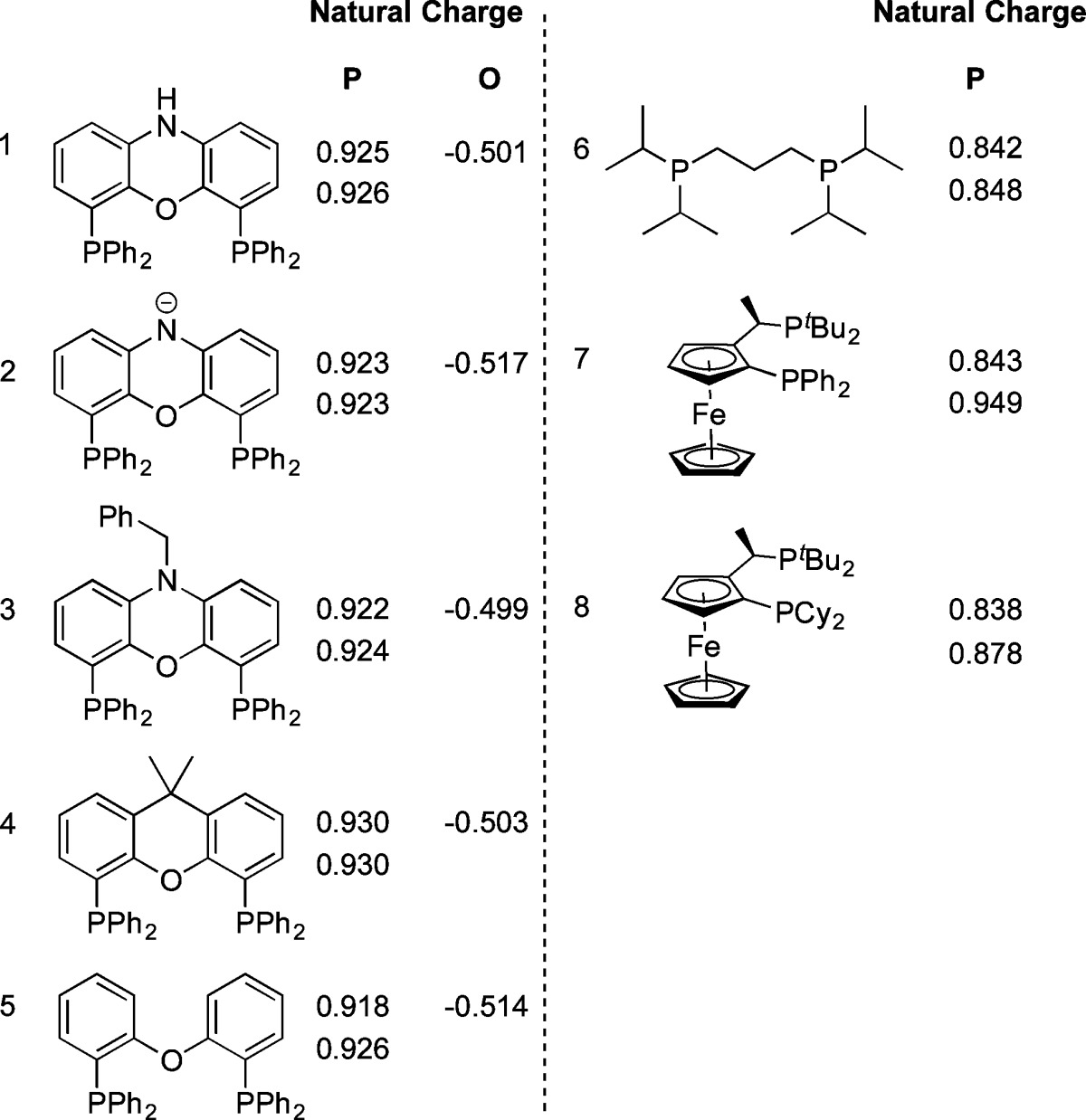
Computations for the five ligands: neutral NiXantphos, the deprotonated NiXantphos anion, N-Bn NiXantphos, Xantphos, and DPEPhos, and three related bidentate phosphines, P̂P = dippp, PPFt-Bu, and CyPFt-Bu, were performed. The results suggested that both the phosphorus atoms and the oxygen atom in the deprotonated NiXantphos anion exhibited natural charges comparable to those in the other four structurally similar ligands (Table 2). In particular, only small ranges of natural charges, qP = 0.918 to 0.930 and qO = −0.499 to −0.517, were observed, suggesting the electronic structure at the phosphorus and oxygen atoms is largely unchanged upon deprotonation of NiXantphos.
The calculations also suggested the three alkyl-substituted bidentate phosphine ligands: P̂P, PPFt-Bu, and CyPFt-Bu, were significantly more electron-rich than the Xantphos family of ligands. Overall, the calculations disfavor the possibility that the oxygen atom in the deprotonated NiXantphos is electron-rich, suggesting it is not likely to play a critical role in the oxidative addition of aryl chlorides (see section 2.3).
2.2.3. Structures of Deprotonated NiXantphos
We were curious how deprotonation would impact the structure of the NiXantphos ligand and how the main group metal would interact with the ligand framework. Bala and co-workers reported the crystal structure of neutral NiXantphos, where the phenoxazine is essentially planar.21 We synthesized the metalated ligand, K–NiXantphos by combination of 1 equiv of NiXantphos with 1 equiv of KN(SiMe3)2 in Et2O at room temperature under a nitrogen atmosphere. Upon addition of KN(SiMe3)2, K–NiXantphos rapidly precipitated from Et2O as a yellow solid, and was isolated by vacuum filtration. 1H and 31P{1H} NMR spectra of the isolated K–NiXantphos in THF-d8 were identical to those obtained in situ from combination of NiXantphos with KN(SiMe3)2 in THF-d8 (Table 1, entry 3).
We obtained diffraction-quality single crystals of K–NiXantphos in the absence and presence of 18-crown-6 for a structural study. As illustrated in Figure 1, [K(THF)3–NiXantphos]2 is a dimer in the solid state, and the deprotonated phenoxazine ring exhibits a dihedral angle of 1.9° between the two benzo groups, similar to that observed in neutral NiXantphos. The reddish-orange crown ether adduct, K(THF)(18-crown-6)–NiXantphos, was found to be a monomer in the solid state (Figure 1b). A change in geometry of the phenoxazine ring was observed with deprotonation: a dihedral angle of 16.4° between the two benzo rings. The K–N distances were 2.808(3) Å and 2.857(3) Å in the dimer (Figure 1a), and 2.885(2) Å in the monomer (Figure 1 b).
2.2.4. Probing NiXantphos N-Arylation
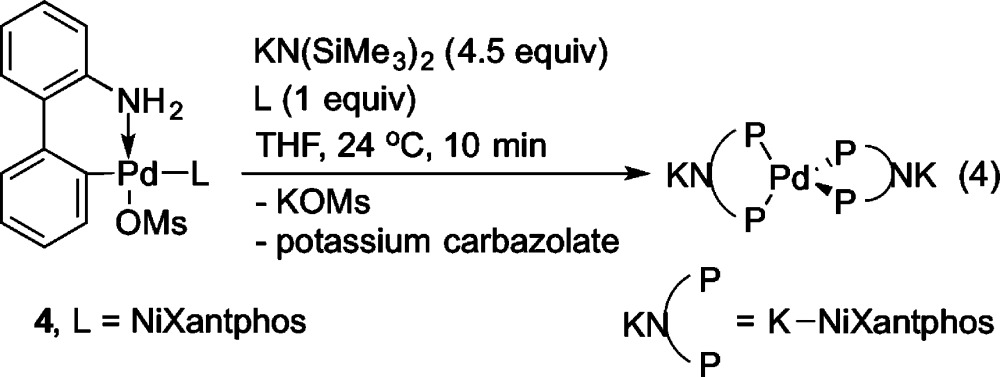
The above studies confirm that NiXantphos is deprotonated in the presence of MN(SiMe3)2 (M = Li, K). Thus, under the DCCP reaction conditions (Scheme 1) we were concerned that the deprotonated NiXantphos could be N-arylated via Buchwald–Hartwig amination of aryl halides.22 To address this concern, we needed to displace the NiXantphos-derived products from palladium after the DCCP. For this purpose, an excess of 1,2-bis(diethylphosphino)ethane (depe), a more basic and stronger binding phosphine, was examined for this ligand exchange/NiXantphos recovery experiment. As shown in Scheme 2A, palladium acetate was stirred with NiXantphos for 12 h at room temperature, resulting in coordination of NiXantphos, as determined by 31P{1H} NMR spectroscopy (see SI). Treatment of the complex with 8 equiv depe followed by filtration over a pad of silica and flash chromatography resulted in 88% recovered NiXantphos (Scheme 2A). In order to determine whether the deprotonated NiXantphos was N-arylated after the cross-coupling reaction with aryl bromide, we performed the DCCP in Scheme 2B and then carried out a ligand exchange/recovery procedure. We recovered 80% of NiXantphos after flash chromatography (average of two runs). No N-arylated NiXantphos was observed (Scheme 2B), suggesting the catalyst does not undergo N-arylation.
Scheme 2. Ligand Exchange and Recovery of NiXantphos.

2.3. Oxidative Addition with Aryl Chlorides
On the basis of the studies in sections 2.1 and 2.2, NiXantphos was deprotonated to generate the metalated ligand, K–NiXantphos, but it was not N-arylated under the DCCP reaction conditions. While the potassium atom was positioned away from the oxygen and the phosphorus atoms in the solid-state structures, we wondered if its presence could facilitate the activation of less reactive aryl chlorides, either by cooperative reactivity with the palladium9,23 or by an electrostatic effect caused by the presence of the charged potassium and nitrogen atoms near the site of oxidative addition.24 Such interactions could be envisioned to facilitate oxidative additions with less reactive aryl chlorides. On the basis of this supposition, we set out to study the oxidative addition of chlorobenzene to our heterobimetallic catalyst system.
The oxidative addition of aryl bromides to (Xantphos)Pd(dba) (dba = dibenzylideneacetone) has been previously studied.25 In these studies, combination of 1 equiv of Pd(dba)2 (or 0.5 equiv of Pd2dba3) with 1 equiv of Xantphos generated (Xantphos)Pd(dba) in situ.26 In the presence of 4-bromobenzonitrile, this intermediate reacted at room temperature to afford (Xantphos)Pd(4-C6H4CN)(Br) in 80% yield.25a Higher temperature was required (80 °C) for reaction with less reactive bromobenzene to afford (Xantphos)Pd(C6H5)(Br).25b We used the same procedure for the oxidative addition studies with chlorobenzene (2b, 5 equiv) to in situ-generated (Xantphos)Pd(dba) and neutral (NiXantphos)Pd(dba) at 24 and 80 °C. Under these conditions no oxidative addition products were detected. These results suggested that Xantphos and neutral NiXantphos were not effective ligands for oxidative addition of aryl chlorides to palladium(0). We also mixed in situ-generated (NiXantphos)Pd(dba) with 1 equiv of KN(SiMe3)2 and 5 equiv of chlorobenzene (2b) at 24 °C, but the reaction gave multiple products, as judged by 31P{1H} NMR spectroscopy, probably because of interference with dba.
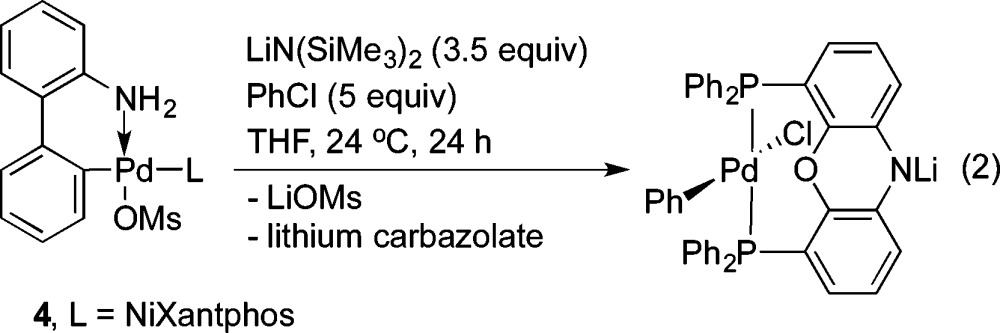
The Buchwald group demonstrated an oxidative addition of chlorobenzene to an LPd(0) (L = a monodentate phosphine) complex, which was in situ-generated by combination of their first-generation chloride precatalyst with 1 equiv of base.27 Other groups also reported on palladacycles used as precatalysts for cross-coupling reactions with aryl chlorides.28 The Buchwald group recently reported their third-generation precatalysts, wherein a dimeric 2-aminobiphenylpalladium methanesulfonate complex can be treated with a range of phosphine ligands (including sterically bulky ligands and bidentate ligands) to provide methanesulfonate precatalysts.29 To determine the reactivity of our palladium catalyst system based on deprotonated NiXantphos in the absence of dba, we combined 1 equiv of the methanesulfonate precatalyst 4 (see eq 2 for the structure) with 3.5 equiv of LiN(SiMe3)2 and 5 equiv of chlorobenzene 2b in THF at 24 °C (eq 2). We propose that the first equiv of LiN(SiMe3)2 deprotonates the NH2 moiety to induce the reductive elimination affording carbazole and (NiXantphos)Pd(0).27 The second and third equivalents of LiN(SiMe3)2 deprotonate carbazole (pKa = 19.9) and (NiXantphos)Pd(0) (pKa ≈ 22).13 The room-temperature oxidative addition of chlorobenzene reached about 75% conversion in 6 h and near completion in 24 h, as judged by a singlet at 2.6 ppm in 31P{1H} NMR spectrum for the oxidative addition product. The singlet is due to the rapid exchange between cis- and trans-chelation modes of the wide bite angle NiXantphos ligand (114°)8 in solution, as also observed with (Xantphos)Pd(Ar)(Br).30 The oxidative addition product, (Li–NiXantphos)Pd(Ph)(Cl), is generated along with byproducts lithium mesylate and lithium carbazoleate, rendering the isolation of the Pd-containing product challenging (eq 2). Nevertheless, single crystals of the neutral (NiXantphos)Pd(Ph)(Cl) were obtained by vapor diffusion of pentane into a concentrated THF solution of the reaction mixture (Figure 2). The solid-state structure shows a slightly distorted square planar geometry with trans phosphorus atoms. This trans-chelating mode of NiXantphos is similar to that of Xantphos: Xantphos is trans-chelating in all reported solid-state structures of (Xantphos)Pd(Ar)(halide) complexes to date.30 A larger dihedral angle between the two benzo groups of the phenoxazine ring system was observed in (NiXantphos)Pd(Ph)(Cl): 27.8° vs 1.9° in Figure 1a and 16.4° in Figure 1b.
 |
2 |
Figure 2.

ORTEP diagram of (NiXantphos)Pd(Ph)(Cl) with 50% probability thermal ellipsoids displayed. Hydrogen atoms omitted for clarity. Pd1–P1 = 2.3168(9) Å, Pd1–P2 = 2.3083(8) Å, Pd1–C37 = 2.048(2) Å, Pd1–Cl1 = 2.4348(8) Å, P1···P2 distance =4.454 Å, Pd1···O1 distance = 2.686 Å, C37–Pd1–Cl1 angle = 178.70°, P1–Pd1–P2 angle = 148.76°.
The same reactivity was also observed when KN(SiMe3)2 was used in place of LiN(SiMe3)2 in the oxidative addition of chlorobenzene: the reaction neared completion after 24 h, as judged by a singlet at 2.8 ppm in 31P{1H} NMR spectrum. The results of these experiments are counterintuitive given that oxidative addition of aryl chlorides has been shown to proceed via a PdL1 pathway. Bidentate ligands typically require significantly higher temperatures because they react via the less favorable PdL2 pathway. Thus, the observation that a bidentate triarylphosphine derivative can activate aryl chlorides at room temperature is unexpected and highlights the novel characteristics of this heterobimetallic catalyst system under the basic DCCP conditions. The oxidative addition studies lay the foundation for aryl chlorides to undergo room temperature cross-coupling reactions catalyzed by Pd–NiXantphos.
By directly employing K–NiXantphos as a ligand, attempts to synthesize the corresponding palladium compounds (K–NiXantphos)Pd(dba), (K–NiXantphos)PdCl2 and (K–NiXantphos)Pd(OAc)2 were unsuccessful. Upon mixing 1 equiv of K–NiXantphos with the corresponding palladium sources [0.5 equiv of Pd2dba3, 1 equiv of (norbornadiene)PdCl2, or 1 equiv of Pd(OAc)2] we observed multiple products as judged by 31P{1H} NMR spectroscopy. It is possible that K–NiXantphos could undergo rapid transmetalation with Pd(II) or react with dba. Furthermore, combination of 0.5 equiv of Pd2dba3 with 2 equiv of neutral NiXantphos in THF (or toluene) resulted in precipitation of a greenish yellow solid. The identity of the precipitate was confirmed as Pd(NiXantphos)2 by HRMS analysis. Unfortunately, Pd(NiXantphos)2 was insoluble in common organic solvents. A similar synthetic route was reported for Pd(Xantphos)2 by the Buchwald group, and its poor solubility was also noted.26
2.4. Development of DCCP with Aryl Chlorides
On the basis of the preliminary studies in section 2.3, we carried out a ligand comparison experiment employing 1-tert-butyl-4-chlorobenzene (2a) as the cross-coupling partner under reaction conditions related to our previous DCCP of aryl bromides (Scheme 3). The comparison was performed on a 10 μmol scale. To probe our hypothesis that the deprotonation of NiXantphos is responsible for the exceptional reactivity in the oxidative addition of aryl chlorides, we synthesized and tested N-benzyl-NiXantphos, which cannot undergo deprotonation.8
Scheme 3. Selected Results of the Cross-Coupling of 1a with 2a.
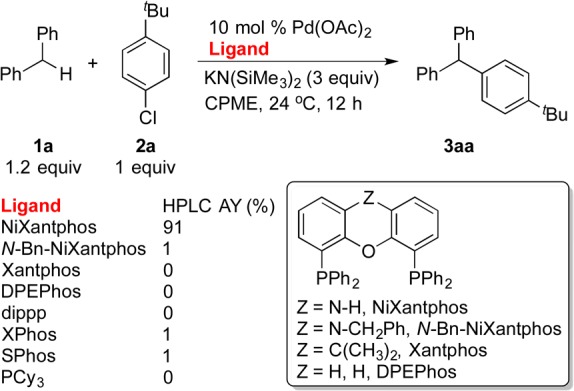
Conducted on a 10 μmol scale at 0.1 M.
In agreement with our hypothesis, excellent HPLC assay yield (AY) of the product 3aa was observed using 10 mol % Pd–NiXantphos. In contrast, catalysts generated from N-Bn-NiXantphos showed <2% conversion and Xantphos did not generate detectable amounts of products (Scheme 3). Other ligands known to participate in coupling reactions with aryl chlorides showed little or no reactivity in this cross-coupling.
Considering the structural similarity of NiXantphos, N-Bn-NiXantphos, and Xantphos, as well as the fact that NiXantphos was deprotonated by KN(SiMe3)2 under the DCCP reaction conditions, the data in Scheme 3 support the hypothesis that K–NiXantphos forms the active palladium catalyst to “turn on” the cross-couplings with aryl chloride 2a at room temperature, while the neutral N-Bn-NiXantphos and Xantphos do not (Figure 3).
Figure 3.

Proposed “turn on” form of palladium(0) bearing a deprotonated NiXantphos and the “turn off” form of palladium(0) bearing a neutral N-Bn-NiXantphos or Xantphos for the DCCP with aryl chlorides.
2.5. Optimization of DCCP with Diphenylmethane and Aryl Chloride 2a
The results in Scheme 3 provided an excellent starting point for the C–C bond formation from diphenylmethane and aryl chloride 2a at room temperature. A 91% assay yield (HPLC) was obtained at 10 mol % catalyst loading on microscale. This microscale reaction was successfully translated to laboratory scale (0.1 mmol) under the same conditions (Table 3, entry 1, 87% assay yield). In further agreement with the data in Scheme 3, Pd–Xantphos catalyst system did not render any product 3aa under the same conditions either at 24 or 80 °C. Considering 10 mol % catalyst loading was relatively high, further optimization on laboratory scale was desired for the room temperature DCCP.
Table 3. Optimization of Pd–NiXantphos Catalyzed DCCP of 1a with 2aa.
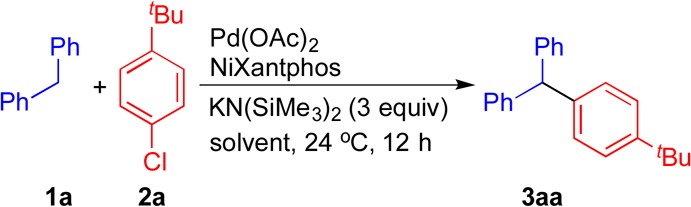
| entry | 1a:2a | Pd (mol %) | solvent | concentration (M) | yield (%)b |
|---|---|---|---|---|---|
| 1 | 1.2:1.0 | 10 | CPME | 0.1 | 87 |
| 2 | 1.2:1.0 | 10 | DME | 0.1 | 61 |
| 3 | 1.2:1.0 | 10 | 2-MeTHF | 0.1 | 78 |
| 4 | 1.2:1.0 | 10 | THF | 0.1 | >95 |
| S | 1.2:1.0 | 10 | dioxane | 0.1 | 25 |
| 6 | 1.2:1.0 | 10 | MTBE | 0.1 | 10 |
| 7 | 1.2:1.0 | 5 | THF | 0.1 | 79 |
| 8 | 1.2:1.0 | 5 | THF | 0.2 | 58 |
| 9c | 1.2:1.0 | 5 | THF | 0.2 | 76 |
| 10 | 1.0:2.0 | 5 | THF | 0.2 | >95 (99)d |
| 11 | 1.0:2.0 | 2.5 | THF | 0.2 | (81)d |
Reactions conducted on a 0.1 mmol scale using 1a, 2a, and 3 equiv of KN(SiMe3)2 at 24 °C.
Yield determined by 1H NMR spectroscopy of the crude reaction mixture.
5 mol % the methanesulfonate precatalyst 4 was used in place of Pd(OAc)2/NiXantphos.
Isolated yield after chromatographic purification.
We next examined five additional ethereal solvents [DME, 2-MeTHF, THF, dioxane, and MTBE (methyl tert-butyl ether)] in the presence of 1.2 equiv of diphenylmethane (1a), 1 equiv of 1-tert-butyl-4-chlorobenzene (2a), and 3 equiv of KN(SiMe3)2 at 0.1 M and 24 °C (entries 2–6). Dioxane and MTBE (entries 5 and 6) afforded <25% yield of product 3aa due to solubility issues of the reactants at 24 °C. The lead result was obtained when THF was used as solvent, where the triarylmethane 3aa was generated in >95% yield (entry 4). Reduction of the catalyst loading from 10 to 5 mol % while increasing the reaction concentration from 0.1 to 0.2 M in THF resulted in only a minor loss in yield (entries 7 and 8 vs 4). Under otherwise identical conditions, a similar yield (76%, entry 9) was obtained using 5 mol % of the methanesulfonate precatalyst 4 in place of Pd(OAc)2/NiXantphos (entry 9 vs 8). Considering the commercial availability of both Pd(OAc)2 and NiXantphos, we continued with the Pd(OAc)2/NiXantphos catalyst system for further optimization. Switching the limiting reagent from aryl chloride 2a to diphenylmethane 1a resulted in >95% yield of the triarylmethane product 3aa with 5 mol % catalyst loading in THF at 0.2 M at 24 °C (entry 10). Compound 3aa was ultimately isolated in 99% yield after flash chromatography. Further reducing the catalyst loading to 2.5 mol % rendered 3aa in 81% isolated yield (entry 11). These optimized conditions were carried forward in the next phase, which focused on the determination of the scope of aryl chlorides in Pd–NiXantphos-catalyzed DCCP.
2.6. Scope of Aryl Chlorides in DCCP with Diphenylmethane
In previous reports, we demonstrated that a large range of sterically and electronically diverse hetero- and nonheteroaryl-containing diarylmethane derivatives readily undergo DCCP with aryl bromides to afford an array of triarylmethane products.11 In the current study, the scope of Pd–NiXantphos catalyzed DCCP using a variety of aryl chlorides with a model diarylmethane substrate (1a) is presented in Table 4.
Table 4. Scope of Aryl Chlorides in Pd–NiXantphos-Catalyzed DCCP with 1aa.
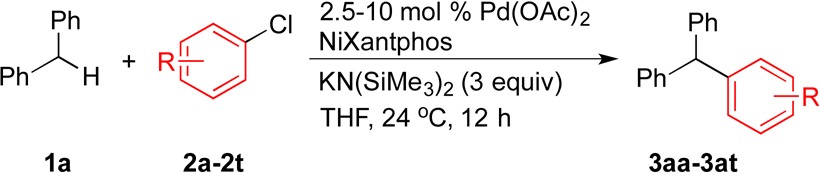
Reactions conducted on a 0.1 mmol scale using 1.2 equiv of 1a, 3 equiv of KN(SiMe3)2, and 1 equiv of 2 (conditions A) or 1 equiv of 1a, 3 equiv of KN(SiMe3)2, and 2 equiv of 2 (conditions B) using 2.5–10 mol % Pd(OAc)2 and NiXantphos (Pd:L = 1:2) in THF at 0.2 M at 24 °C. Isolated yield after chromatographic purification.
1a:KN(SiMe3)2:2 = 4:2:1.
CPME was used as solvent.
80 °C.
1a:KN(SiMe3)2:2 = 1:3:3.
2-MeTHF was used as solvent.
1a:KN(SiMe3)2:2 = 1.2:4:1.
1a:KN(SiMe3)2:2 = 3:4:1.
1a:KN(SiMe3)2:2 = 1:5:2.
In general, fair to excellent reactivity was exhibited by aryl chlorides bearing various substituents (2a–2k). At least one combination from (1) the two ratios of reagents [diphenylmethane (1a):KN(SiMe3)2:aryl chloride (2) = 1.2:3:1 (conditions A) and 1:3:2 (conditions B)] and (2) the three ethereal solvents (THF, CPME, and 2-MeTHF) at 2.5–10 mol % catalyst loading successfully delivered the desired DCCP products in 56–99% yields (average yield: 86% for 11 aryl chlorides, 2a–2k). Both 4-tert-butylphenyl and phenyl chloride furnished 3aa and 3ab in 81% and 96% yield, respectively, at 2.5 mol % palladium loading. Methyl (2c), N,N-dimethylamino (2d), and trifluoromethyl (2e) groups at the meta position were all well-tolerated (3ac–3ae in 82–94% yields). Excellent yields were obtained using aryl chlorides bearing both electron-withdrawing CF3 and 4-F groups (2e, 2f, 2g) and electron-donating 4-OMe and 4-(N-pyrrolyl) groups (2h, 2i). The sterically more hindered 1-chloronaphthalene and 2-chloroanisole (2j and 2k) also participated in DCCP to produce the desired products. These substrates, however, required higher temperature (80 °C) to give 66% (3aj) and 56% (3ak) yield, respectively.
We next tested aryl chloride substrates bearing sensitive functional groups and heteroaryl groups (2l–2t). As shown in Table 4, remarkable chemoselectivity was demonstrated with aryl chlorides containing cyano, keto, acetyl, phenol, phenothiazine, acetamide, and 1H-indole moieties, all of which underwent DCCP delivering the corresponding functionalized triarylmethane products without observation of byproducts (3al–3at). Nitriles and ketones are well-known to undergo 1,2-addition reactions with reactive organometallics, while 4′-chloroacetophenone can participate in competitive aldol31 and α-arylation32 reactions under the basic reaction conditions (pKa of acetophenone: 24.713). Yet the triarylmethane products (3al, 3am, 3an) were produced in 59–94% yields. Challenging classes of substrates bearing acidic O–H and N–H bonds (2o–2t) were next examined. Phenols are known to undergo O– and C(sp2)–H arylation,33 while 1H-indoles (and anilines) have been reported to react via N-arylation (Buchwald–Hartwig coupling)22,34 and C-2- and C-3-arylation35 in the presence of palladium catalysts and bases. Our method exhibits orthogonal chemoselectivity with DCCP taking place exclusively at the benzylic C(sp3)–H of diphenylmethane (1a) and the C(sp2)–Cl of aryl chlorides 2o–2t. These functional groups and heteroaryl groups present opportunities to further functionalize the triarylmethane products. Note that for each equivalent of aryl chloride bearing acidic protons (2n–2t), an extra equivalent of KN(SiMe3)2 was employed. For substrates giving less than 80% yield (2n, 2p, 2r–2t), 1H NMR of the reaction mixture after workup showed only remaining starting materials and product. No byproduct formation was observed. Attempts to push these reactions to completion, however, were unsuccessful. Unfortunately, 3-chloropyridine and 2- and 3-chlorothiophene failed to give the DCCP products in reasonable yield (<10%).
After demonstrating that aryl chloride substrates bearing sensitive functional groups and heteroaryl groups (2l–2t) were viable cross-coupling partners in Pd–NiXantphos-catalyzed DCCP with diphenylmethane (1a), an unactivated alkenyl chloride 2u was tested (eq 3). Vinyl chloride 2u participated in DCCP to afford 3au in 99% yield at 2.5 mol % catalyst loading at 80 °C. Even at 1.25 mol % catalyst loading under identical conditions, 3au was produced in 80% yield. These results indicate the Pd–NiXantphos catalyst can oxidatively add unactivated alkenyl chlorides. It is also interesting that the product did not isomerize, suggesting that deprotonation of the product did not occur.
 |
3 |
As shown in Table 4, both the synthetic utility and the remarkable chemoselectivity of the Pd–NiXantphos-catalyzed C–C cross-coupling method with a large variety of aryl chlorides have been demonstrated. Several aspects are noteworthy: (1) 3- and 4-chlorophenols (2o, 2p) reacted with 1a to give the DCCP products in 84% and 40% yield, respectively. Given that these phenols are deprotonated by KN(SiMe3)2 to give phenoxides under the reaction conditions, these results suggest that the Pd–NiXantphos catalyst system is able to undergo oxidative addition with very electron-rich C(sp2)–Cl bonds (such as those in phenoxides). (2) 2-Chlorophenothiazine (2q) produced the DCCP product in 86% yield without observation of N-arylation byproduct. Since 2q has a phenothiazine core, which is structurally similar to the phenoxazine core in NiXantphos, this result supports our earlier conclusion based on the ligand exchange/recovery experiments that NiXantphos was not N-arylated during the reaction.
2.7. Scope of Diarylmethanes in DCCP
The scope of the DCCP with respect to diarylmethanes was next explored with aryl chlorides (2a, 2b) (Table 5).
Table 5. Scope of Diarylmethanes in Pd–NiXantphos-Catalyzed DCCPa.
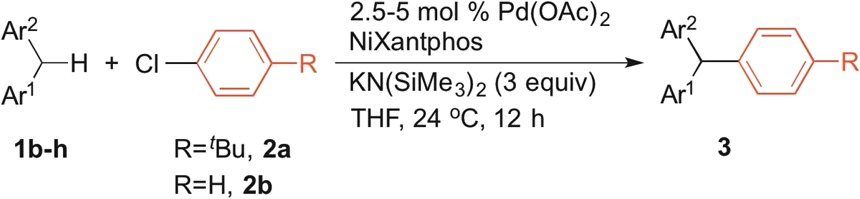
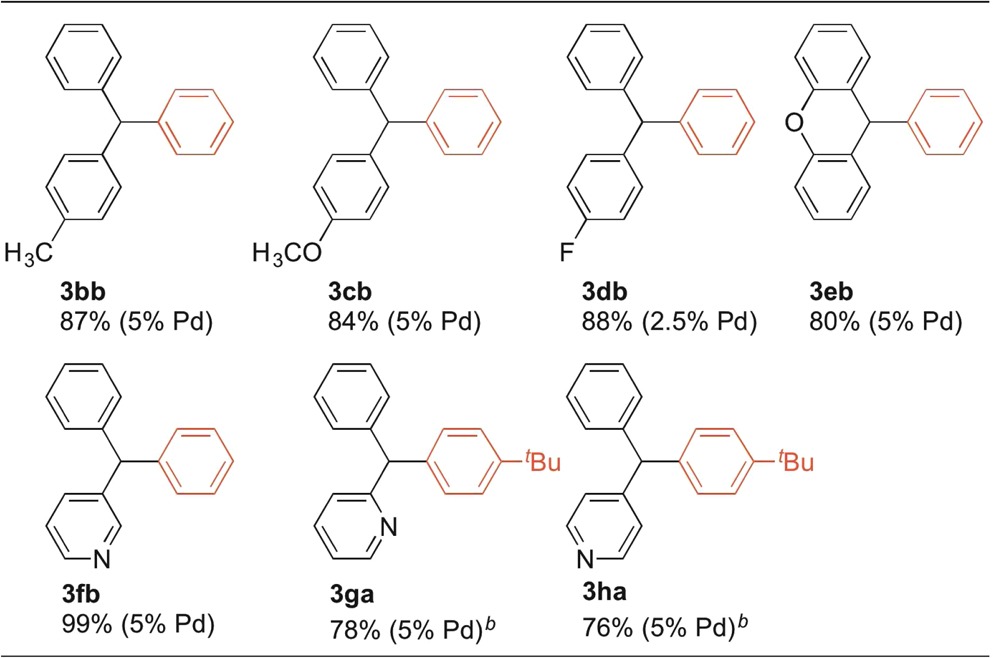
Reactions conducted on a 0.1 mmol scale using 1 equiv of 1, 3 equiv of KN(SiMe3)2, and 2 equiv of 2 with 2.5–5 mol % Pd(OAc)2 and NiXantphos (Pd:L = 1:2) in THF at 0.2 M at 24 °C. Isolated yield after chromatographic purification.
LiN(SiMe3)2 was used as base instead of KN(SiMe3)2.
Substrates bearing various substituents on the diarylmethane exhibited excellent reactivity at 2.5–5 mol % catalyst loading at room temperature. Alkyl (3bb), electron-donating (3cb), and electron-withdrawing groups (3db) were all well-tolerated. Heteroaryl-containing diarylmethane analogues proved to be good substrates, with corresponding products isolated in 76–99% isolated yields (3eb, 3fb, 3ga, 3ha). Note that, although 3-benzylpyridine required KN(SiMe3)2 as base to promote DCCP (3fb), a weaker base, LiN(SiMe3)2, successfully promoted DCCP with the more acidic 2- and 4-benzylpyridines (pKa = 28.2 and 26.7, respectively)13 to deliver 3ga and 3ha in good yields.
2.8. Identification of the Catalyst Resting State and Countercation Effects
To determine the resting state of the palladium catalyst in our DCCP reactions, we conducted the catalytic reaction with diphenylmethane (1a), chlorobenzene (2b), and KN(SiMe3)2 as base in the presence of 10 mol % of the methanesulfonate precatalyst 4 in THF in a sealed J. Young NMR tube at room temperature. The only species observed by 31P{1H} NMR spectroscopy over the course of the DCCP (12 h) was Pd(K–NiXantphos)2, as judged by a singlet at −1.3 ppm in 31P{1H} NMR spectrum. Using 10 mol % Pd(OAc)2 and 20 mol % NiXantphos gave the same dominant catalyst resting state 10 min after addition of 1a, 2b, and KN(SiMe3)2 at room temperature (see SI). The catalyst resting state suggests that ligand dissociation and/or oxidative addition is turnover limiting. The Hartwig group has reported the identification of Pd(P̂P)2 complexes (P̂P = a chelating diphosphine ligand such as BINAP and DPPF) as the dominant resting state in the Pd–P̂P catalyzed amination of aryl bromides.7b The turnover-limiting step was dissociation of BINAP from Pd(BINAP)2 when BINAP was used as the ligand, while the combination of ligand dissociation and irreversible oxidative addition were the turnover-limiting steps in the catalytic process when DPPF was used as ligand.
Pd(K–NiXantphos)2 was independently prepared from combination of 1 equiv of the methanesulfonate precatalyst 4 with 4.5 equiv of KN(SiMe3)2 and 1 equiv of NiXantphos ligand in THF at 24 °C under a nitrogen atmosphere (eq 4). We propose that the in situ-generated coordinatively unsaturated (K–NiXantphos)Pd(0) reacts rapidly with 1 equiv of K–NiXantphos. The formation of Pd(K–NiXantphos)2 was complete in 10 min as judged by the disappearance of 4 and appearance of a new singlet at −1.3 ppm in the 31P{1H} NMR spectrum. Single-crystals of Pd(K–NiXantphos)2 were obtained by crystallization from THF at 24 °C and found to be extremely sensitive, and sample decomposition during mounting could not be avoided (see SI). Although the quality of the structure precludes detailed discussion of the metrics, it does serve to establish the connectivity {see Figure 4 for a drawing of the structure and Figure 5 for the structure of [PdK2(THF)4(NiXantphos)2]∞}. In the solid state Pd(K–NiXantphos)2 exhibits a polymeric structure in which the Pd(0) center adopts a slightly distorted tetrahedral geometry. The PdL2 units are linked together by bridging potassium–nitrogen interactions, similar to those observed in the structure of K–NiXantphos (Figure 1a). The P–Pd–P angle containing two phosphorus atoms of the same K–NiXantphos ligand is 113.22(7)°. The other P–Pd–P angles are 102.91(12)°, 106.78(7)°, and 113.54(12)°. The deprotonated phenoxazine ring exhibits a dihedral angle of 23.1° between the two benzo groups of the heterocyclic framework. In contrast to the solid-state polymeric structure, the hydrodynamic radius (rH) of Pd(K–NiXantphos)2, as measured by diffusion-ordered 1H NMR spectroscopy (DOSY) in THF-d8, was consistent with a monomer (see SI).
Figure 4.

Drawing of the solid-state structure of polymeric Pd(K–NiXantphos)2 illustrating the connectivity.
Figure 5.
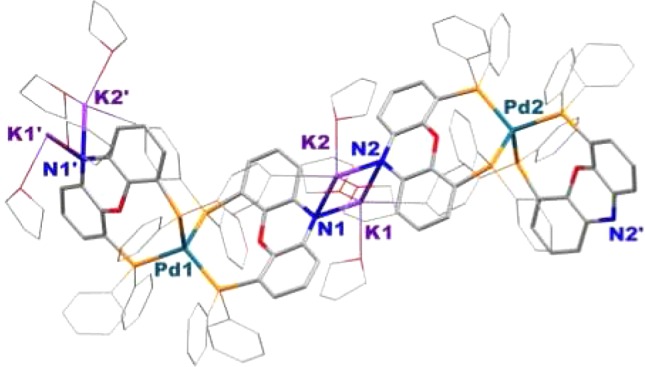
Structure of polymeric [PdK2(THF)4(NiXantphos)2]∞. Coordinated THF molecules and phenyl groups from NiXanphos are shown in wireframe, and hydrogen atoms are omitted for clarity.
 |
4 |
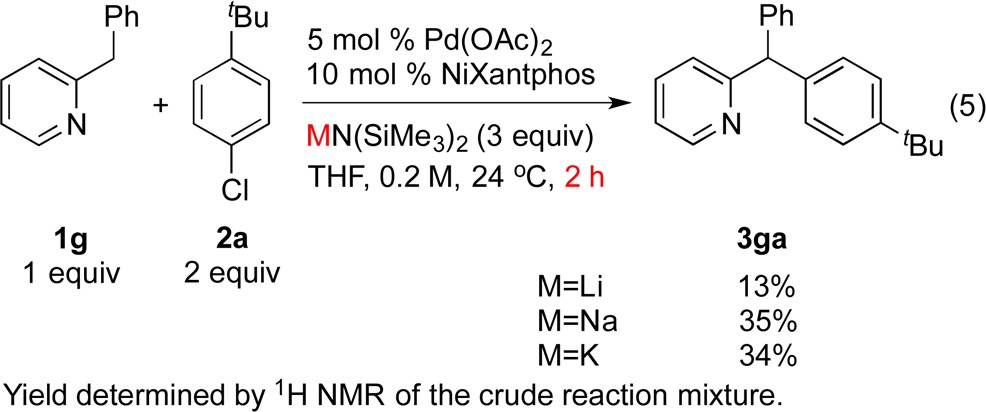
To compare the catalytic reactivity using different countercations (Li, Na vs K), we carried out our DCCP reactions under standard conditions with 2-benzylpyridine (1g) and 1-tert-butyl-4-chlorobenzene (2a) using the following 3 bases: LiN(SiMe3)2, NaN(SiMe3)2, and KN(SiMe3)2. We have previously demonstrated that DCCP of diphenylmethane (1a) with aryl bromides is readily promoted by KN(SiMe3)2, but the reaction fails when NaN(SiMe3)2 or LiN(SiMe3)2 is used in the absence of additives. In contrast, DCCP of the more acidic substrate 2-benzylpyridine (1g) can be promoted by MN(SiMe3)2 (M = Li, Na, K).11b Assay yields of the product 3ga at 2 h from the DCCP reactions employing 3 bases MN(SiMe3)2 (M = Li, Na, K) are illustrated in eq 5 (average of two runs). The impact of the countercation on the catalytic reaction follows the trend: K ≈ Na > Li. At this point, it is difficult to draw conclusions from these data because the main group metal is involved in the deprotonation of the substrate and catalyst and in each step of the catalytic cycle.
 |
5 |
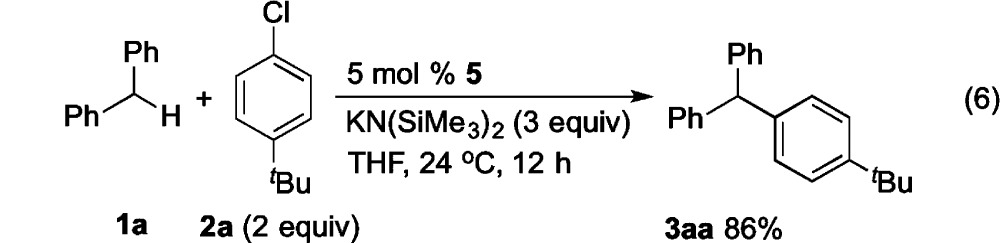
Finally, we wanted to determine if the oxidative addition product was a competent intermediate in the catalytic reaction. As mentioned in eq 2, isolation of (Li–NiXantphos)Pd(Ph)(Cl) from the byproducts of its synthesis was challenging. Instead, we synthesized a neutral oxidative addition species, (NiXantphos)Pd(4-C6H4CN)(Br) (5) in 88% yield, following the procedure for the preparation of (Xantphos)Pd(4-C6H4CN)(Br).25a Subjecting 5 mol % 5 to the catalytic reaction resulted in 86% isolated yield of the DCCP product 3aa (eq 6), suggesting that the oxidative addition species is a competent intermediate.
 |
6 |
3. Summary and Outlook
On the basis of a large number of studies, it was widely accepted that palladium complexes based on bidentate triarylphosphines would not oxidatively add unactivated aryl chlorides at room temperature and could not, therefore, catalyze coupling processes with aryl chloride substrates. In this report we have disclosed an exception to this paradigm by demonstrating that under basic reaction conditions the heterobimetallic Pd(M-NiXantphos)-based catalyst system readily activates aryl chlorides at room temperature and successfully promotes the arylation of diphenylmethane derivatives.
The advantages of the Pd–NiXantphos catalyst system are (1) mild conditions (room temperature) for cross-coupling reactions with unactivated aryl chlorides, (2) greater air- and oxidative-stability of NiXantphos relative to alkylphosphines (many of which are of high sensitivity and/or pyrophoric), (3) superior catalytic performance to all the other mono- and bidentate ligands examined in this report, and (4) commercial availability of palladium source and ligand.
A dramatic difference in the catalytic performance was observed between NiXantphos (91% AY) and its structurally similar analogues N-Bn-NiXantphos (1% AY) and Xantphos (0% AY), supporting our hypothesis that the oxidative addition is facilitated with NiXantphos because the heterobimetallic Pd–ligand catalyst system exhibits greatly enhanced reactivity due to the presence of the main group metal. The DCCP with aryl chlorides affords a variety of triarylmethane products, a class of compounds with applications and biological activity. Additionally, the DCCP exhibits remarkable chemoselectivity in the presence of aryl chloride substrates bearing heteroaryl groups and sensitive functional groups that are known to undergo 1,2-addition, aldol reaction, O–, N–, enolate-α–, and C(sp2)–H arylation reactions.
The advantages and importance of the Pd–NiXantphos catalyst system outlined herein make it a valuable contribution to applications in Pd-catalyzed arylation reactions with aryl chlorides under mild conditions. Future work will focus on structural modification of NiXantphos to increase its reactivity and efficiency in catalytic processes.
4. Experimental Section
Representative procedures are described herein. Full experimental details and characterization of all compounds are provided in the SI.
4.1. General Methods
All reactions were performed under nitrogen using oven-dried glassware and standard Schlenk or vacuum line techniques. Air- and moisture-sensitive solutions were handled under nitrogen and transferred via syringe. THF was freshly distilled from Na/benzophenone ketyl under nitrogen. Anhydrous CPME, 2-MeTHF, dioxane, and MTBE were purchased from Sigma-Aldrich and used as solvent without further purification. Unless otherwise stated, reagents were commercially available and used as purchased without further purification. Chemicals were obtained from Sigma-Aldrich, Acros, TCI America or Alfa Aesar, and solvents were purchased from Fisher Scientific. The progress of the reactions was monitored by thin-layer chromatography using Whatman Partisil K6F 250 μm precoated 60 Å silica gel plates and visualized by short-wavelength ultraviolet light as well as by treatment with ceric ammonium molybdate (CAM) stain or iodine. Silica gel (230–400 mesh, Silicycle) was used for flash chromatography. The 1H NMR and 13C{1H} NMR spectra were obtained using a Brüker AM-500 Fourier-transform NMR spectrometer at 500 and 125 MHz, respectively. Chemical shifts are reported in units of parts per million (ppm) downfield from tetramethylsilane (TMS), and all coupling constants are reported in hertz. The 31P{1H} NMR spectra were obtained using a Brüker DMX-360 NMR spectrometer at 145.8 MHz, with chemical shifts reported with respect to calibration with an external standard of phosphoric acid (0 ppm). The infrared spectra were obtained with KBr plates using a PerkinElmer Spectrum 100 series FTIR spectrometer. High-resolution mass spectrometry (HRMS) data were obtained on a Waters LC-TOF mass spectrometer (model LCT-XE Premier) using chemical ionization (CI) or electrospray ionization (ESI) in positive or negative mode, depending on the analyte. Melting points were determined on a Unimelt Thomas-Hoover melting point apparatus and are uncorrected.
4.2. General Procedure A. Pd-Catalyzed DCCP of Diarylmethanes with Aryl Chlorides
An oven-dried 10 mL reaction vial equipped with a stir bar was charged with KN(SiMe3)2 (59.8 mg, 0.30 mmol, 3 equiv) under a nitrogen atmosphere. A solution (from a stock solution) of Pd(OAc)2 (0.56 mg, 0.0025 mmol, 2.5 mol %) and NiXantphos (2.76 mg, 0.0050 mmol, 5 mol %) in 0.5 mL of dry THF was taken up by syringe and added to the reaction vial. After stirring for 5 min at 24 °C, diphenylmethane (16.7 μL, 0.1 mmol, 1 equiv) was added to the reaction mixture followed by 1-tert-butyl-4-chlorobenzene (33.4 μL, 0.2 mmol, 2 equiv). Note that the aryl chloride in a solid form was added to the reaction vial prior to KN(SiMe3)2. The reaction mixture was stirred for 12 h at 24 °C, quenched with three drops of H2O, diluted with 3 mL of ethyl acetate, and filtered over a pad of MgSO4 and silica. The pad was rinsed with additional ethyl acetate, and the solution was concentrated in vacuo. The crude material was loaded onto a silica gel column and purified by flash chromatography.
4.3. General Procedure B. Pd-Catalyzed DCCP Followed by Ligand Exchange and Recovery of NiXantphos
The experiments were set up inside a glovebox under a nitrogen atmosphere. An 8 mL reaction vial equipped with a stir bar was charged with KN(SiMe3)2 (329 mg, 1.65 mmol, 3 equiv). A solution of Pd(OAc)2 (6.2 mg, 0.028 mmol, 5 mol %) and NiXantphos (30.0 mg, 0.054 mmol, 10 mol %) in 6 mL of dry CPME was taken up by syringe and added to the reaction vial. After stirring for 5 min at 24 °C, diphenylmethane (110 μL, 0.66 mmol, 1.2 equiv) was added to the reaction mixture followed by 1-bromo-4-tert-butylbenzene (95 μL, 0.55 mmol, 1 equiv). The reaction mixture was stirred for 12 h at 24 °C, before 1,2-bis(diethylphosphino)ethane (depe, 103 μL, 0.44 mmol, depe:NiXantphos = 8:1) was added into the reaction mixture. The reaction mixture was stirred for another 40 min at 24 °C. The reaction was quenched with H2O, diluted with ethyl acetate, and filtered over a pad of silica. The pad was rinsed with additional ethyl acetate, and the solution was concentrated in vacuo. The crude material was loaded onto a silica gel column and purified by flash chromatography to afford the triarylmethane product 3aa (91% yield) and NiXantphos (80% recovery).
4.4. General Procedure C. HTE for Pd-Catalyzed DCCP of Diphenylmethane with 1-tert-Butyl-4-chlorobenzene
Experiments were set up inside a glovebox under a nitrogen atmosphere. A 96-well aluminum block containing 1 mL glass vials was predosed with Pd(OAc)2 (1 μmol) and Ligand (Ligand was used in a 4:1 ratio relative to Pd for monodentate ligands and 2:1 ratio for bidentate ligands) in THF. The solvent was evacuated to dryness using a Genevac vacuum centrifuge, and KN(SiMe3)2 (30 μmol) in THF was added to the ligand/catalyst mixture. The solvent was removed on the Genevac, and a parylene stir bar was then added to each reaction vial. 1-tert-Butyl-4-chlorobenzene (10 μmol/reaction), diphenylmethane (12 μmol/reaction) and biphenyl (1 μmol/reaction) (used as an internal standard to measure HPLC yields) were then dosed together into each reaction vial as a solution in CPME (100 μL, 0.1 M). The 96-well plate was then sealed and stirred for 18 h at 24 °C. Upon opening the plate to air, 500 μL of acetonitrile was pipetted into each vial. The plate was then covered again and the vials stirred for 20 min to extract the product and to ensure good homogenization. Into a separate 96-well LC block was added 700 μL of acetonitrile, followed by 40 μL of the diluted reaction mixtures. The LC block was then sealed with a silicon-rubber storage mat, and mounted on an HPLC instrument for analysis.
4.5. General Procedure D. Synthesis of K–NiXantphos
Experiments were set up inside a glovebox under a nitrogen atmosphere. To a 20 mL vial containing NiXantphos (110 mg, 0.2 mmol, 1 equiv) dissolved in 10 mL of Et2O was slowly added a solution of KN(SiMe3)2 (40 mg, 0.2 mmol, 1 equiv) in 2 mL of Et2O, resulting in rapid precipitation of a yellow solid. After stirring for 2 h, the slurry was filtered through a fritted filter, and the solid was washed with 3 × 5 mL Et2O. Drying the solid under reduced pressure yielded a yellow powder. X-ray diffraction-quality single crystals were obtained by layering a concentrated THF solution of K–NiXantphos with hexanes at −21 °C. X-ray diffraction-quality single crystals of the reddish-orange crown ether adduct, K(THF)(18-crown-6)–NiXantphos, were obtained by vapor diffusion of pentane into a concentrated THF solution of K–NiXantphos and 18-crown-6 (1:1) at −21 °C.
4.6. General Procedure E. Oxidative Addition of Chlorobenzene to (M–NiXantphos)Pd(0)
Experiments were set up inside a glovebox under a nitrogen atmosphere. Precatalyst 4 (9.2 mg, 0.01 mmol, 1 equiv) was added to a J. Young NMR tube followed by chlorobenzene (5.1 μL, 0.05 mmol, 5 equiv). LiN(SiMe3)2 (5.9 mg, 0.035 mmol, 3.5 equiv) was weighed in a vial, dissolved in THF (500 μL), and transferred to the NMR tube. The solution became reddish-orange immediately. The progress of the reaction was monitored by 31P{1H} NMR spectroscopy. X-ray diffraction-quality single crystals of the protonated (NiXantphos)Pd(Ph)(Cl) were obtained by vapor diffusion of pentane into a concentrated THF solution of the reaction mixture at −21 °C.
4.7. General Procedure F. Synthesis of Pd(K–NiXantphos)2
Experiments were set up inside a glovebox under a nitrogen atmosphere. NiXantphos (5.5 mg, 0.01 mmol, 1 equiv) and precatalyst 4 (9.2 mg, 0.01 mmol, 1 equiv) were added to a J. Young NMR tube. KN(SiMe3)2 (9.0 mg, 0.045 mmol, 4.5 equiv) was weighed in a vial, dissolved in THF (500 μL), and transferred to the NMR tube. The solution became reddish-orange immediately. The progress of the reaction was monitored by 31P{1H} NMR spectroscopy. The formation of Pd(K–NiXantphos)2 was complete in 10 min, as judged by disappearance of 4 and appearance of a new singlet at −1.3 ppm in 31P{1H} NMR spectrum. The reaction mixture was set undisturbed for 12 h. X-ray diffraction-quality single crystals of Pd[K(THF)2(NiXantphos)]2 were obtained under these conditions. The crystalline product was then filtered and washed with 3 × 10 mL Et2O. Drying under reduced pressure yielded the product as a yellow crystalline solid. The product is highly sensitive to trace oxygen.
Acknowledgments
We thank the NSF (CHE-1152488 and 0848460) and NIH (NIGMS 104349) for partial support of this research. We would also like to thank Dr. Jun Gu for assistance with the DOSY measurements.
Supporting Information Available
Procedures, full characterization of new compounds, and crystallographic data for (NiXantphos)Pd(Ph)(Cl), [K(THF)3–NiXantphos]2, K(THF)(18-crown-6)–NiXantphos, and [PdK2(THF)4(NiXantphos)2]∞.This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Littke A. F.; Fu G. C. Angew. Chem., Int. Ed. 2002, 41, 4176. [DOI] [PubMed] [Google Scholar]; b Whitcombe N. J.; Hii K. K.; Gibson S. E. Tetrahedron 2001, 57, 7449. [Google Scholar]; c Bedford R. B.; Cazin C. S. J.; Holder D. Coord. Chem. Rev. 2004, 248, 2283. [Google Scholar]
- a Fu G. C. Acc. Chem. Res. 2008, 41, 1555. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Martin R.; Buchwald S. L. Acc. Chem. Res. 2008, 41, 1461. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Surry D. S.; Buchwald S. L. Angew. Chem., Int. Ed. 2008, 47, 6338. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Christmann U.; Vilar R. N. Angew. Chem., Int. Ed. 2005, 44, 366. [DOI] [PubMed] [Google Scholar]
- Suzuki coupling at 70 °C:; a Liu S.-Y.; Choi M. J.; Fu G. C. Chem. Commun. 2001, 2408. [PubMed] [Google Scholar]; Suzuki coupling at 60 °C, five examples with moderate yields (≤ 57% by GC):; b Pickett T. E.; Richards C. J. Tetrahedron Lett. 2001, 42, 3767. [Google Scholar]; c Pickett T. E.; Roca F. X.; Richards C. J. J. Org. Chem. 2003, 68, 2592. [DOI] [PubMed] [Google Scholar]; Suzuki coupling at 90 °C, two examples with moderate yields (≤57% by GC):; d Pramick M. R.; Rosemeier S. M.; Beranek M. T.; Nickse S. B.; Stone J. J.; Stockland R. A.; Baldwin S. M.; Kastner M. E. Organometallics 2003, 22, 523. [Google Scholar]; Suzuki coupling at ≥110 °C:; e Kwong F. Y.; Chan K. S.; Yeung C. H.; Chan A. S. C. Chem. Commun. 2004, 2336. [DOI] [PubMed] [Google Scholar]; Suzuki coupling at ≥50 °C and Heck reaction at ≥110 °C:; f Iwasawa T.; Komano T.; Tajima A.; Tokunaga M.; Obora Y.; Fujihara T.; Tsuji Y. Organometallics 2006, 25, 4665. [Google Scholar]; Suzuki coupling at ≥75 °C (2 examples) and Heck reaction at 120 °C (1 example):; g Iwasawa T.; Kamei T.; Watanabe S.; Nishiuchi M.; Kawamura Y. Tetrahedron Lett. 2008, 49, 7430. [Google Scholar]; Suzuki coupling at 60 °C:; h Fujihara T.; Yoshida S.; Terao J.; Tsuji Y. Org. Lett. 2009, 11, 2121. [DOI] [PubMed] [Google Scholar]; Suzuki coupling at ≥110 °C:; i So C. M.; Chow W. K.; Choy P. Y.; Lau C. P.; Kwong F. Y. Chem.–Eur. J. 2010, 16, 7996. [DOI] [PubMed] [Google Scholar]; j Barder T. E.; Walker S. D.; Martinelli J. R.; Buchwald S. L. J. Am. Chem. Soc. 2005, 127, 4685. [DOI] [PubMed] [Google Scholar]
- a Barrios-Landeros F.; Hartwig J. F. J. Am. Chem. Soc. 2005, 127, 6944. [DOI] [PubMed] [Google Scholar]; b Barrios-Landeros F.; Carrow B. P.; Hartwig J. F. J. Am. Chem. Soc. 2009, 131, 8141. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lewis A. K.; de K.; Caddick S.; Cloke F. G. N.; Billingham N. C.; Hitchcock P. B.; Leonard J. J. Am. Chem. Soc. 2003, 125, 10066. [DOI] [PubMed] [Google Scholar]; d Galardon E.; Ramdeehul S.; Brown J. M.; Cowley A.; Hii K. K.; Jutand A. Angew. Chem., Int. Ed. 2002, 41, 1760. [DOI] [PubMed] [Google Scholar]; See references on computational studies of oxidative addition of aryl chlorides to Pd(0):; e Schoenebeck F.; Houk K. N. J. Am. Chem. Soc. 2010, 132, 2496. [DOI] [PubMed] [Google Scholar]; f Senn H. M.; Ziegler T. Organometallics 2004, 23, 2980. [Google Scholar]; g Kozuch S.; Amatore C.; Jutand A.; Shaik S. Organometallics 2005, 24, 2319. [Google Scholar]; h Ahlquist M.; Norrby P.-O. Organometallics 2007, 26, 550. [Google Scholar]
- Reference (1a) and references therein.
- Portnoy M.; Milstein D. Organometallics 1993, 12, 1665.Note that the oxidative addition of 10-fold excess of PhCl to Pd(dippp)2 required 90 °C to go to completion in 2 h in dioxane. The temperature range of the kinetic measurements was 30–70 °C. [Google Scholar]
- Aryl tosylates oxidatively add to Pd(chelate)2:; a Roy A. H.; Hartwig J. F. J. Am. Chem. Soc. 2003, 125, 8704. [DOI] [PubMed] [Google Scholar]; Aryl bromides oxidatively add to Pd(chelate)2:; b Alcazar-Roman L. M.; Hartwig J. F.; Rheingold A. L.; Liable-Sands L. M.; Guzei I. A. J. Am. Chem. Soc. 2000, 122, 4618. [Google Scholar]; c Amatore C.; Broeker G.; Jutand A.; Khalil F. J. Am. Chem. Soc. 1997, 119, 5176. [Google Scholar]; d Jutand A.; Hii K. K.; Thornton-Pett M.; Brown J. M. Organometallics 1999, 18, 5367. [Google Scholar]
- van der Veen L. A.; Keeven P. H.; Schoemaker G. C.; Reek J. N. H.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Lutz M.; Spek A. L. Organometallics 2000, 19, 872. [Google Scholar]
- Note that transition metal/main group metal bimetallic activation mode of sp2 C–X bonds has rarely been reported. The Nakamura group disclosed that in the reaction of a lithium diorganocuprate(I) with an alkenyl bromide, the sp2 C–Br bond was activated by Cu(I) and Li(I) synergistically. See:; a Yoshikai N.; Nakamura E. J. Am. Chem. Soc. 2004, 126, 12264. [DOI] [PubMed] [Google Scholar]; b Yoshikai N.; Iida R.; Nakamura E. Adv. Synth. Catal. 2008, 350, 1063. [Google Scholar]; The same group later reported that in nickel-catalyzed cross-coupling reactions of aryl electrophiles and Grignard reagents, the sp2 C–X bonds (X = F, Cl, carbamates, phosphates, etc.) were activated via a nickel/magnesium bimetallic cooperative mechanism. See:; c Yoshikai N.; Matsuda H.; Nakamura E. J. Am. Chem. Soc. 2009, 131, 9590. [DOI] [PubMed] [Google Scholar]
- a McGrew G. I.; Temaismithi J.; Carroll P. J.; Walsh P. J. Angew. Chem., Int. Ed. 2010, 49, 5541. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McGrew G. I.; Stanciu C.; Zhang J.; Carroll P. J.; Dreher S. D.; Walsh P. J. Angew. Chem., Int. Ed. 2012, 51, 11510. [DOI] [PubMed] [Google Scholar]; c Zhang J.; Stanciu C.; Wang B.; Hussain M. M.; Da C.-S.; Carroll P. J.; Dreher S. D.; Walsh P. J. J. Am. Chem. Soc. 2011, 133, 20552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang J.; Bellomo A.; Creamer A. D.; Dreher S. D.; Walsh P. J. J. Am. Chem. Soc. 2012, 134, 13765. [DOI] [PubMed] [Google Scholar]; b Bellomo A.; Zhang J.; Trongsiriwat N.; Walsh P. J. Chem. Sci. 2013, 4, 849. [Google Scholar]; c Hussain N.; Frensch G.; Zhang J.; Walsh P. J. Angew. Chem., Int. Ed. 2014, 53, 3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Jia T.; Bellomo A.; EL Baina K.; Dreher S. D.; Walsh P. J. J. Am. Chem. Soc. 2013, 135, 3740. [DOI] [PubMed] [Google Scholar]; b Zheng B.; Jia T.; Walsh P. J. Org. Lett. 2013, 15, 1690. [DOI] [PubMed] [Google Scholar]; c Zheng B.; Jia T.; Walsh P. J. Org. Lett. 2013, 15, 4190. [DOI] [PubMed] [Google Scholar]; d Zheng B.; Jia T.; Walsh P. J. Adv. Synth. Catal. 2014, 356, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Montel S.; Jia T.; Walsh P. J. Org. Lett. 2014, 16, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Montel S.; Raffier L.; He Y.; Walsh P. J. Org. Lett. 2014, 16, 1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordwell F. G. Acc. Chem. Res. 1988, 21, 456. [Google Scholar]
- a Ryss P.; Zollinger H.. Fundamentals of the Chemistry and Application of Dyes; Wiley-Interscience: New York, 1972. [Google Scholar]; b Katritzky A. R.; Gupta V.; Garot C.; Stevens C. V.; Gordeev M. F. Heterocycles 1994, 38, 345. [Google Scholar]; c Muthyala R.; Katritzky A. R.; Lan X. F. Dyes Pigm. 1994, 25, 303. [Google Scholar]
- a Aldag R.Photochromism: Molecules and Systems; Dürr H., Bouas-Laurent H., Eds.; Elsevier:: London,, 1990. [Google Scholar]; b Irie M. J. Am. Chem. Soc. 1983, 105, 2078. [Google Scholar]; c Herron N.; Johansson G. A.; Radu N. S. U.S. Patent Application 2005/0187364, Aug 25, 2005.; d Xu Y. Q.; Lu J. M.; Li N. J.; Yan F.; Xia X. W.; Xu Q. F. Eur. Polym. J. 2008, 44, 2404. [Google Scholar]
- a Das S. K.; Panda G.; Chaturvedi V.; Manju Y. S.; Galkwad A. K.; Sinha S. Bioorg. Med. Chem. Lett. 2007, 17, 5586. [DOI] [PubMed] [Google Scholar]; b Panda G.; Shagufta; Mishra J. K.; Chaturvedi V.; Srivastava A. K.; Srivastava R.; Srivastava B. S. Bioorg. Med. Chem. 2004, 12, 5269. [DOI] [PubMed] [Google Scholar]; c Parai M. K.; Panda G.; Chaturvedi V.; Manju Y. K.; Sinha S. Bioorg. Med. Chem. Lett. 2008, 18, 289. [DOI] [PubMed] [Google Scholar]; d Shagufta; Srivastava A. K.; Sharma R.; Mishra R.; Balapure A. K.; Murthy P. S. R.; Panda G. Bioorg. Med. Chem. 2006, 14, 1497. [DOI] [PubMed] [Google Scholar]; e Palchaudhuri R.; Hergenrother P. J. Bioorg. Med. Chem. Lett. 2008, 18, 5888. [DOI] [PubMed] [Google Scholar]; f Palchaudhuri R.; Nesterenko V.; Hergenrother P. J. J. Am. Chem. Soc. 2008, 130, 10274. [DOI] [PubMed] [Google Scholar]; g Risberg K.; Guldvik I. J.; Palchaudhuri R.; Xi Y. G.; Ju J. F.; Fodstad O.; Hergenrother P. J.; Andersson Y. J. Immunother. 2011, 34, 438. [DOI] [PubMed] [Google Scholar]; h Al-Qawasmeh R. A.; Lee Y.; Cao M. Y.; Gu X. P.; Vassilakos A.; Wright J. A.; Young A. Bioorg. Med. Chem. Lett. 2004, 14, 347. [DOI] [PubMed] [Google Scholar]
- For reviews:; a Duxbury D. F. Chem. Rev. 1993, 93, 381. [Google Scholar]; b Shchepinov M. S.; Korshun V. A. Chem. Soc. Rev. 2003, 32, 170. [DOI] [PubMed] [Google Scholar]; c Nair V.; Thomas S.; Mathew S. C.; Abhilash K. G. Tetrahedron 2006, 62, 6731. [Google Scholar]
- a Schnitzer R. J.; Hawking F.. Experimental Chemotherapy; Academic Press: New York, 1963; Vol. I. [Google Scholar]; b Greene T. W.; Wuts P. G. M.. Protective Groups in Organic Synthesis, 3rd ed.; Wiley: New York, 1999. [Google Scholar]; c Wulff H.; Miller M. J.; Hansel W.; Grissmer S.; Cahalan M. D.; Chandy K. G. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 8151. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hernandez A. I.; Balzarini J.; Karlsson A.; Camarasa M. J.; Perez-Perez M. J. J. Med. Chem. 2002, 45, 4254. [DOI] [PubMed] [Google Scholar]; e Ellsworth B. A.; Ewing W. R.; Jurica E. U.S. Patent Application 2011/0082165 A1, Apr 7, 2011.; f Mullen L. M. A.; Duchowicz P. R.; Castro E. A. Chemom. Intell. Lab. Syst. 2011, 107, 269. [Google Scholar]; g Rodriguez D.; Ramesh C.; Henson L. H.; Wilmeth L.; Bryant B. K.; Kadavakollu S.; Hirsch R.; Montoya J.; Howell P. R.; George J. M.; Alexander D.; Johnson D. L.; Arterburn J. B.; Shuster C. B. Bioorg. Med. Chem. 2011, 19, 5446. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Bobko A. A.; Dhimitruka I.; Zweier J. L.; Khramtsov V. V. Angew. Chem., Int. Ed. 2014, 53, 2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Reetz M. T.; Kühling K. M.; Deege A.; Hinrichs H.; Belder D. Angew. Chem., Int. Ed. 2000, 39, 3891. [DOI] [PubMed] [Google Scholar]; b Trapp O.; Weber S. K.; Bauch S.; Hofstadt W. Angew. Chem., Int. Ed. 2007, 46, 7307. [DOI] [PubMed] [Google Scholar]; c Dreher S. D.; Dormer P. G.; Sandrock D. L.; Molander G. A. J. Am. Chem. Soc. 2008, 130, 9257. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Davies I. W.; Welch C. J. Science 2009, 325, 701. [DOI] [PubMed] [Google Scholar]; e Robbins D. W.; Hartwig J. F. Science 2011, 333, 1423. [DOI] [PMC free article] [PubMed] [Google Scholar]; f McNally A.; Prier C. K.; MacMillan D. W. C. Science 2011, 334, 1114. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Bellomo A.; Celebi-Olcum N.; Bu X.; Rivera N.; Ruck R. T.; Welch C. J.; Houk K. N.; Dreher S. D. Angew. Chem., Int. Ed. 2012, 51, 6912. [DOI] [PubMed] [Google Scholar]
- a Spokoyny A. M.; Lewis C. D.; Teverovskiy G.; Buchwald S. L. Organometallics 2012, 31, 8478. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Spokoyny A. M.; Machan C. W.; Clingerman D. J.; Rosen M. S.; Wiester M. J.; Kennedy R. D.; Stern C. L.; Sarjeant A. A.; Mirkin C. A. Nat. Chem. 2011, 3, 590. [DOI] [PubMed] [Google Scholar]
- Marimuthu T.; Bala M. D.; Friedrich H. B. Acta Crystallogr. 2008, E64, o711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hartwig J. F. Nature 2008, 455, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Surry D. S.; Buchwald S. L. Chem. Sci. 2011, 2, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hartwig J. F. Acc. Chem. Res. 2008, 41, 1534. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Schlummer B.; Scholz U. Adv. Synth. Catal. 2004, 346, 1599. [Google Scholar]; e Buchwald S. L.; Mauger C.; Mignani G.; Scholz U. Adv. Synth. Catal. 2006, 348, 23. [Google Scholar]
- Poirier V.; Roisnel T.; Carpentier J.-F.; Sarazin Y. Dalton Trans. 2009, 9820. [DOI] [PubMed] [Google Scholar]
- a Hoffman B. M.; Lukoyanov D.; Dean D. R.; Seefeldt L. C. Acc. Chem. Res. 2013, 46, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Horn B.; Pfirrmann S.; Limberg C.; Herwig C.; Braun B.; Mebs S.; Metzinger R. Z. Anorg. Allg. Chem. 2011, 637, 1169. [Google Scholar]; c Ding K.; Pierpont A. W.; Brennessel W. W.; Lukat-Rodgers G.; Rodgers K. R.; Cundari T. R.; Bill E.; Holland P. L. J. Am. Chem. Soc. 2009, 131, 9471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yin J.; Buchwald S. L. J. Am. Chem. Soc. 2002, 124, 6043. [DOI] [PubMed] [Google Scholar]; b Fujita K.-I.; Yamashita M.; Puschmann F.; Alvarez-Falcon M. M.; Incarvito C. D.; Hartwig J. F. J. Am. Chem. Soc. 2006, 128, 9044. [DOI] [PubMed] [Google Scholar]
- Klingensmith L. M.; Strieter E. R.; Barder T. E.; Buchwald S. L. Organometallics 2006, 25, 82. [Google Scholar]
- Biscoe M. R.; Fors B. P.; Buchwald S. L. J. Am. Chem. Soc. 2008, 130, 6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See leading references on palladacycles used as precatalysts for cross-coupling reactions with aryl chlorides:; a Herrmann W. A.; Brossmer C.; Ofele K.; Reisinger C.-P.; Priermeier T.; Beller M.; Fischer H. Angew. Chem., Int. Ed. Engl. 1995, 34, 1844. [Google Scholar]; b Beller M.; Fischer H.; Herrmann W. A.; Ofele K.; Brossmer C. Angew. Chem., Int. Ed. Engl. 1995, 34, 1848. [Google Scholar]; c Schnyder A.; Indolese A. F.; Studer M.; Blaser H.-U. Angew. Chem., Int. Ed. 2002, 41, 3668. [DOI] [PubMed] [Google Scholar]
- a Bruno N. C.; Tudge M. T.; Buchwald S. L. Chem. Sci. 2013, 4, 916. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bruno N. C.; Buchwald S. L. Org. Lett. 2013, 15, 2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Note that due to the coordinatively flexible nature of the Xantphos framework, both cis- and trans-isomers of (Xantphos)Pd(II) complexes are known in literature, depending on the nature of the other substituents bound to the Pd(II) center. In several complexes Xantphos can even have different chelating modes between solid state and solution. Furthermore, (Xantphos)Pd(II) complexes can show equilibrium between cis- and trans-isomers in solution. (Xantphos)Pd(acyl)(halide):; a Martinelli J. R.; Watson D. A.; Freckmann D. M. M.; Barder T. E.; Buchwald S. L. J. Org. Chem. 2008, 73, 7102. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Miloserdov F. M.; Grushin V. V. Angew. Chem., Int. Ed. 2012, 51, 3668. [DOI] [PubMed] [Google Scholar]; (Xantphos)Pd(aryl)(halide) and related (Xantphos)Pd(II) complexes:; c Reference (22a); d Reference (22b); e Zuideveld M. A.; Swenneshuis B. H. G.; Boele M. D. K.; Guari Y.; van Strijdonck G. P. F.; Reek J. N. H.; Kamer P. C. J.; Goubitz K.; Fraanje J.; Lutz M.; Spek A. L.; van Leeuwen P. W. N. M. J. Chem. Soc., Dalton Trans. 2002, 2308. [Google Scholar]; f Grushin V. V.; Marshall W. J. J. Am. Chem. Soc. 2006, 128, 12644. [DOI] [PubMed] [Google Scholar]
- Seminal publications on aldol reaction:; a Kane R. J. Prakt. Chem. 1838, 15, 129. [Google Scholar]; b Kane R. Ann. Phys. Chem., Ser. 2 1838, 44, 475. [Google Scholar]
- For reviews on arylation of activated C(sp3)–H bonds:; a Bellina F.; Rossi R. Chem. Rev. 2010, 110, 1082. [DOI] [PubMed] [Google Scholar]; b Johansson C. C. C.; Colacot T. J. Angew. Chem., Int. Ed. 2010, 49, 676. [DOI] [PubMed] [Google Scholar]; c Culkin D. A.; Hartwig J. F. Acc. Chem. Res. 2003, 36, 234. [DOI] [PubMed] [Google Scholar]; d Novák P.; Martin R. Curr. Org. Chem. 2011, 15, 3233. [Google Scholar]; e Burtoloso A. C. B. Synlett 2009, 320. [Google Scholar]
- For examples of Pd-catalyzed direct arylation of phenols:; a Bates R. W.; Gabel C. J.; Ji J. H.; Ramadevi T. Tetrahedron 1995, 51, 8199. [Google Scholar]; b Kawamura Y.; Satoh T.; Miura M.; Nomura M. Chem. Lett. 1999, 961. [Google Scholar]; c Kataoka N.; Shelby Q.; Stambuli J. P.; Hartwig J. F. J. Org. Chem. 2002, 67, 5553. [DOI] [PubMed] [Google Scholar]; d Burgos C. H.; Barder T. E.; Huang X. H.; Buchwald S. L. Angew. Chem., Int. Ed. 2006, 45, 4321. [DOI] [PubMed] [Google Scholar]; e Hu T. J.; Schulz T.; Torborg C.; Chen X. R.; Wang J.; Beller M.; Huang J. Chem. Commun. 2009, 7330. [DOI] [PubMed] [Google Scholar]; f Beydoun K.; Doucet H. Catal. Sci. Technol. 2011, 1, 1243. [Google Scholar]; g Platon M.; Cui L. C.; Mom S.; Richard P.; Saeys M.; Hierso J. C. Adv. Synth. Catal. 2011, 353, 3403. [Google Scholar]; h Xiao B.; Gong T. J.; Liu Z. J.; Liu J. H.; Luo D. F.; Xu J.; Liu L. J. Am. Chem. Soc. 2011, 133, 9250. [DOI] [PubMed] [Google Scholar]
- For examples of Pd-catalyzed N-arylation of indoles:; a Hartwig J. F.; Kawatsura M.; Hauck S. I.; Shaughnessy K. H.; Alcazar-Roman L. M. J. Org. Chem. 1999, 64, 5575. [DOI] [PubMed] [Google Scholar]; b Old D. W.; Harris M. C.; Buchwald S. L. Org. Lett. 2000, 2, 1403. [DOI] [PubMed] [Google Scholar]; c Grasa G. A.; Viciu M. S.; Huang J. K.; Nolan S. P. J. Org. Chem. 2001, 66, 7729. [DOI] [PubMed] [Google Scholar]
- For examples of Pd-catalyzed C-2- and C-3-arylation of indoles:; a Lane B. S.; Sames D. Org. Lett. 2004, 6, 2897. [DOI] [PubMed] [Google Scholar]; b Lane B. S.; Brown M. A.; Sames D. J. Am. Chem. Soc. 2005, 127, 8050. [DOI] [PubMed] [Google Scholar]; c Deprez N. R.; Kalyani D.; Krause A.; Sanford M. S. J. Am. Chem. Soc. 2006, 128, 4972. [DOI] [PubMed] [Google Scholar]; d Djakovitch L.; Dufaud V.; Zaidi R. Adv. Synth. Catal. 2006, 348, 715. [Google Scholar]; e Toure B. B.; Lane B. S.; Sames D. Org. Lett. 2006, 8, 1979. [DOI] [PubMed] [Google Scholar]; f Wang X.; Gribkov D. V.; Sames D. J. Org. Chem. 2007, 72, 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Bellina F.; Benelli F.; Rossi R. J. Org. Chem. 2008, 73, 5529. [DOI] [PubMed] [Google Scholar]; h Yang S. D.; Sun C. L.; Fang Z.; Li B. H.; Li Y. Z.; Shi Z. J. Angew. Chem., Int. Ed. 2008, 47, 1473. [DOI] [PubMed] [Google Scholar]; i Zhao J. L.; Zhang Y. H.; Cheng K. J. Org. Chem. 2008, 73, 7428. [DOI] [PubMed] [Google Scholar]; j Liang Z. J.; Yao B. B.; Zhang Y. H. Org. Lett. 2010, 12, 3185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.