

Abstract
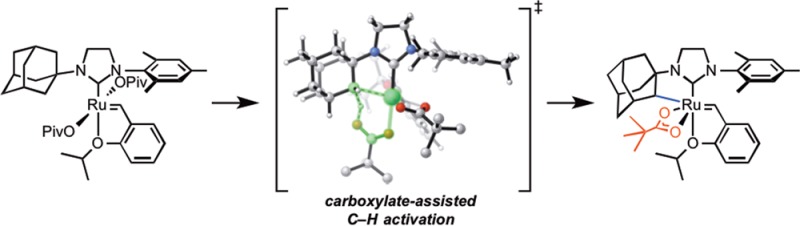
The mechanism of C–H activation at metathesis-relevant ruthenium(II) benzylidene complexes was studied both experimentally and computationally. Synthesis of a ruthenium dicarboxylate at a low temperature allowed for direct observation of the C–H activation step, independent of the initial anionic ligand-exchange reactions. A first-order reaction supports an intramolecular concerted metalation–deprotonation mechanism with ΔG⧧298K = 22.2 ± 0.1 kcal·mol–1 for the parent N-adamantyl-N′-mesityl complex. An experimentally determined ΔS⧧ = −5.2 ± 2.6 eu supports a highly ordered transition state for carboxylate-assisted C(sp3)–H activation. Experimental results, including measurement of a large primary kinetic isotope effect (kH/kD = 8.1 ± 1.7), agree closely with a computed six-membered carboxylate-assisted C–H activation mechanism where the deprotonating carboxylate adopts a pseudo-apical geometry, displacing the aryl ether chelate. The rate of cyclometalation was found to be influenced by both the electronics of the assisting carboxylate and the ruthenium ligand environment.
Introduction
The activation of C–H bonds by transition metal complexes has become an important, growing field in organic synthesis.1 At the core of this field is the need to understand the general mechanisms of the C–H activation step in order to harness the reactivity for synthetic processes. As a result, mechanistic and computational studies have been of great interest to organic and inorganic chemists in order to elucidate the mechanisms of C–H activation reactions by transition metals, including Pd,1b,2 Ir,3 Rh,4 Ru,5 and others.6
Carboxylate-assisted C–H activation has recently garnered attention as a generally mild method for C–H activation at transition metal centers.1a,1c The use of carboxylates in catalytic C–H activation reactions has grown tremendously in recent years, and several reports investigating the mechanism have appeared.7 The majority of these reactions involve activation of aromatic or vinylic C(sp2)–H bonds, and only sparing examples of carboxylate-assisted C(sp3)–H activation have been reported.1c Computational and mechanistic studies have been primarily focused on palladium carboxylate-catalyzed C–H activations, which often involve a concerted metalation–deprotonation (CMD) mechanism with a six-membered transition state.2e,2i,7d,8 Activation of C(sp3)–H bonds with ruthenium complexes is particularly rare9 and typically occurs through C–H oxidative addition to ruthenium9n,10 or C–H radical abstraction with a ruthenium-oxo/nitrenoid.11 The mechanism of carboxylate-mediated C(sp2)–H activation at ruthenium has been studied in only a single family of complexes.5d,12 The CMD C–H activation mechanism, which is particularly common with palladium carboxylate catalysts, is largely unexplored with ruthenium. Furthermore, inner-sphere activation of methylene C(sp3)–H bonds is quite rare and is essentially unprecedented for ruthenium.
We recently reported the synthesis of a new family of cyclometalated ruthenium benzylidene complexes (1–3, Figure 1).13 These ruthenium complexes were found to be highly selective for Z-olefins (typically >90% Z) in a number of olefin metathesis reactions, including macrocyclic ring-closing metathesis,14 cross metathesis,15 asymmetric ring-opening cross metathesis,16 ring-opening metathesis polymerization,13d and ethenolysis reactions.17 These findings were complementary to the reactivity of molybdenum and tungsten catalysts reported by Schrock and Hoveyda.18
Figure 1.

Cyclometalated ruthenium metathesis catalysts.
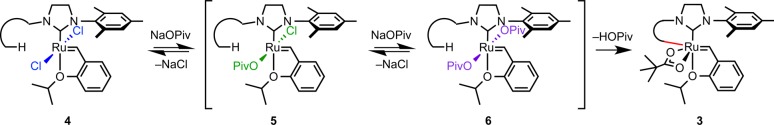
Cyclometalated complexes 1–3 are highly interesting organometallic species. These complexes contain a stable Ru–C bond in the presence of a reactive ruthenium alkylidene. Previous observations of C–H bond activation in ruthenium alkylidene complexes led to decomposition of the Ru–C bond, typically through insertion into the alkylidene followed by hydride elimination reactions.19 The stable cyclometalated complexes are synthesized by the reaction of a ruthenium dichloride complex with an excess of a pivalate salt (Scheme 1). While silver pivalate was found to efficiently provide cyclometalated complexes,13a,13b product stability to the reaction conditions was improved by the use of sodium carboxylate salts.13c In light of this initial advance in cyclometalation methodology, kinetic studies on the cyclometalation of these complexes would enable the synthesis of a broader family of potentially useful ruthenium alkylidene complexes. Furthermore, because of the high stability of complexes 1–3 and the lack of in-depth kinetic studies on the activation of C(sp3)–H bonds, it was envisioned that an investigation into the reactivity of complexes 4 toward cyclometalation would be a valuable addition to the C–H activation literature.
Scheme 1. Synthesis of Cyclometalated Complexes.

This report contains a detailed study of the mechanism of C(sp3)–H activation at ruthenium(II) alkylidenes. By complementing direct observation of the elementary C–H activation step with density functional theory (DFT) studies, unique and important features of a carboxylate-assisted CMD at metathesis-relevant ruthenium(II) complexes could be elucidated.
Results
Dichloride Reactivity
Initial experiments were conducted to investigate the kinetics of C–H activation as it proceeds from the dichloride complex (4, Scheme 2). Complexes 4 were exposed to 10 equiv of sodium pivalate as a solution in 1:1 THF-d8:CD3OD at 40 °C. Both THF and methanol are required for this reaction because of the limited solubility of complexes 4 and 3 in pure methanol and of sodium pivalate in pure THF. The reaction progress could be easily monitored by observation of benzylidene peaks (δ 15–20 ppm) in 1H NMR spectra.
Scheme 2. Proposed Mechanism of C–H Activation of Ruthenium Complex 4a.
These reactions were characterized by a first-order decay of the starting complex, with a buildup of two intermediates identified as the mono- and dicarboxylates resulting from salt exchange with the chloride ligands (Figure 2). This initial ligand exchange resulted in a short induction period prior to the generation of cyclometalated complex 3. In the specific case of N-adamantyl complex 4a, the salt metathesis was slow, generating low concentrations of mono- and dicarboxylate species.
Figure 2.
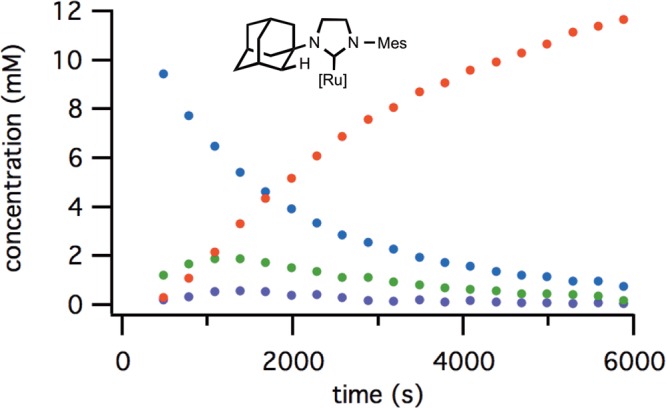
Reaction progress of cyclometalation of complex 4a: blue = [4a], green = [5a], purple = [6a], red = [3a].
The C–H activation of mesityl complex 4b was also followed by 1H NMR spectroscopy (Figure 3). The production of 3b was characterized by the same induction period as observed with complex 3a; however, the equilibrium more heavily favored the carboxylate complexes. In this case, equilibrium was also reached more rapidly, with only 3% of the dichloride complex observable at the first time point.
Figure 3.
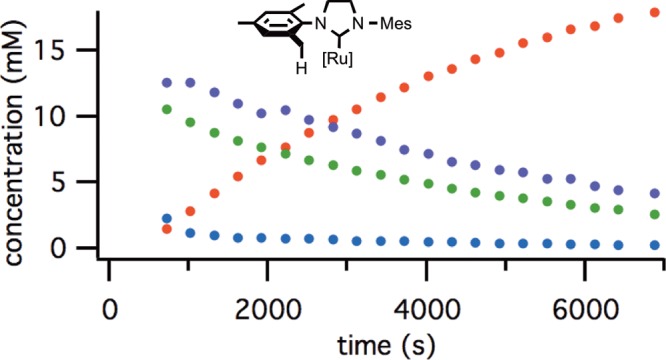
Reaction progress of cyclometalation of complex 4b: blue = [4b], green = [5b], purple = [6b], red = [3b].
Cyclometalation of N-t-Bu-N′-mesityl-substituted NHC ruthenium complex 4c was also monitored by 1H NMR spectroscopy (Figure 4). Much like complex 4a, ligand exchange to provide the dipivalate was slow, and only the monocarboxylate complex could be observed in the reaction mixture. The overall rate was only slightly faster than for the previous complexes.
Figure 4.
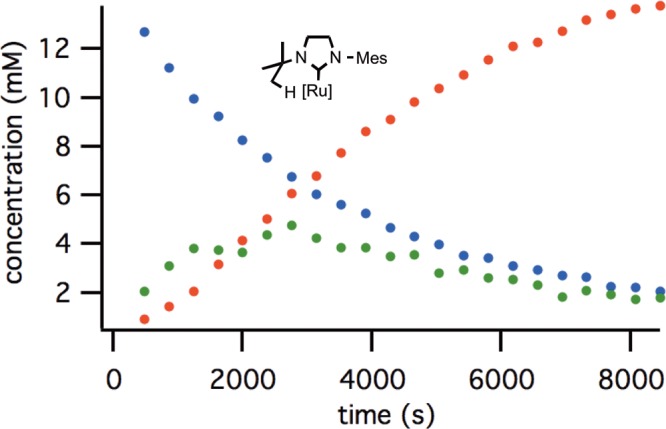
Reaction progress of cyclometalation of complex 4c: blue = [4c], green = [5c], red = [3c]; 6c was not observed.
The C–H activation of a complex with N-adamantyl-N′-2,6-diisopropylphenyl (DIPP, 4d) substitution of the NHC ligand was also monitored. It was found to possess a notably decreased rate when compared to complexes 4a–c. For instance, whereas cyclometalation of complex 4a reaches completion after approximately 4 h, DIPP-substituted complex 4d is not fully consumed until after 96 h. During the course of this experiment, negligible amounts of carboxylate-ligated complexes 5d or 6d were observed.
In order to study the nature of the salt-exchange steps that precede C–H activation, monochloride monopivalate complex 5b was synthesized (Scheme 3).13e Complex 5b is a stable green solid that does not undergo ligand disproportionation in THF/MeOH solution. Furthermore, no cyclometalation was observed upon exposure of complex 5b to the reaction conditions in the absence of excess sodium pivalate. Addition of sodium pivalate (9 equiv) to a THF/MeOH solution of 5b provides cyclometalated product 3b after heating to 40 °C, indicating the intermediacy of monocarboxylate 5b in the formation of 3b. It should also be noted that C(sp2)–H activation occurs competitively at complexes 4 with aryl groups bearing ortho hydrogen atoms; however, these complexes rapidly decompose upon insertion into the benzylidene.19a
Scheme 3. Synthesis and Reactivity of Monopivalate Complex 5b.

Activation of Dicarboxylate Complexes
In order to study the central C–H activation step, a method for the independent synthesis of dipivalates 6 was developed. It was envisioned that replacing the chloride ligands with more rapidly exchanged X-type ligands would allow dicarboxylates to be synthesized at temperatures where C–H activation was sufficiently retarded (Scheme 4). To achieve this, dichlorides 4 were treated with 4 equiv of silver triflate in benzene. Stable bis-triflate complexes 7 were isolated after removal of silver salts by filtration of the reaction mixture. Both N-mesityl complex 7a and N-DIPP complex 7d could be synthesized in this manner. Though the bis-triflate analogue of mesityl complex 4b is known, it was found to be unstable to cyclometalation conditions.13e,20 The bis-triflate analogue of tert-butyl complex 4c could not be synthesized. Immediate general decomposition of 4c was observed upon exposure to silver triflate. Importantly, triflates 7 did not undergo C–H activation upon heating.
Scheme 4. Synthesis and C–H Activation of Dicarboxylate Precursor for Kinetic Studies.

It was found that bis-triflate 7a could be treated with as little as 3 equiv of sodium carboxylate salts to cleanly generate dicarboxylate 6a in situ. Complete conversion to the dicarboxylate was typically achieved in under 30 min at 0 °C, and no cyclometalated products (e.g., 3a) were generated within this time frame. While a THF/MeOH solvent mixture was not necessary to observe C(sp3)–H activation (reaction could be achieved in benzene or pure THF), 1:1 THF/MeOH was chosen for these studies to enable both increased reaction homogeneity and ready comparison to reactions that proceed from dichlorides 4.
The C–H activation of dicarboxylate 6a was then directly observed after the solution was warmed to the reaction temperature. The reaction was found to be first order in dicarboxylate, with rate constant k313K = (1.6 ± 0.1) × 10–3 s–1. A similar procedure was used to study the C–H activation of N-DIPP complex 6d, and the rate constant was determined to be k313K = (1.4 ± 0.2) × 10–3 s–1.
Eyring Analysis
The method described in Scheme 4 was used to conduct an Eyring analysis of the C–H activation step (Figure 5). The reaction was monitored over a temperature range from 15 to 55 °C. A linear Eyring plot was generated with an excellent fit. Analysis of the slope of the plot and the y-intercept provided the energies of activation. It was found that the conversion of dipivalate 6a to cyclometalated ruthenium benzylidene 3a requires ΔH⧧ = 20.6 ± 0.8 kcal·mol–1, with a negative entropy of activation, ΔS⧧ = −5.2 ± 2.6 eu. This provides a Gibbs free energy of activation, ΔG⧧ = 22.2 ± 0.1 kcal·mol–1 at 25 °C.
Figure 5.
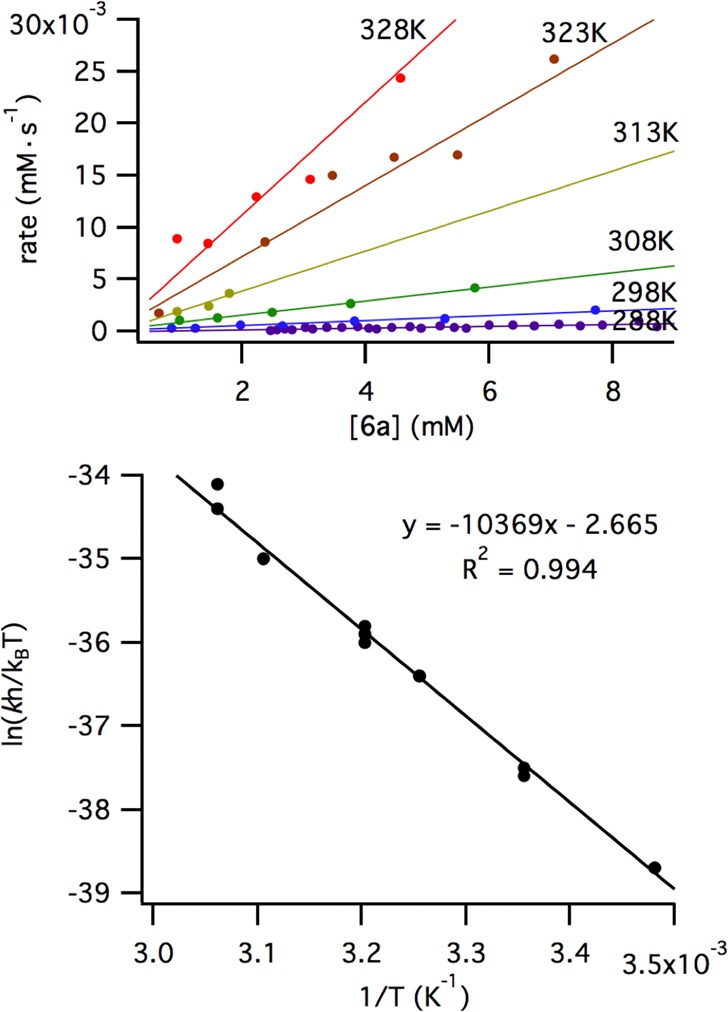
Rate plots (top) and Eyring analysis (bottom) of cyclometalation of ruthenium complex 6a.
Linear Free Energy Relationships
The effect of the electronic properties of the carboxylate was probed by establishing a linear free energy relationship between the Hammett substituent constants and the rate of cyclometalation. Previous computational Hammett studies revealed that C–H σ-bond metathesis with Tp(CO)Ru(II)–X (X = R, NH2, OR, or BOR2) is insensitive to the electronic property of the C–H bond.21 Dicarboxylates 8a–e were synthesized by using the sodium salts of various 4-substituted benzoic acids (Figure 6). While the rate of activation by substituted benzoic acids correlated only moderately with Hammett σ values, the correlation was increased when σ+ values were utilized. A moderately negative ρ value of −0.24 was observed (Figure 6).
Figure 6.

Linear free energy relationships with ruthenium dibenzoates 8a–e.
The electronic influence of substitution of the benzylidene chelate on the rate of C–H activation was also investigated (Figure 7). Substitution of the benzylidene chelate has demonstrated effects on the initiation rates of metathesis catalysts.22 Using the Hammett σpara values, substitution of the 4-position of the benzylidene chelate provided insignificant correlation. The strongest correlation was observed when using the corresponding σmeta values. This observation indicates a much stronger influence of benzylidene substituents on the 2-position of the chelate, inductively to the ruthenium center. The ρ value for this plot was found to be +0.53.
Figure 7.

Linear free energy relationships with substituted ruthenium chelates 10a–d.
Initiation rates of related dichloride complexes (10a–d; OPiv = Cl) were measured by observing the rate of decay of the benzylidene peak after the complex was treated with butyl vinyl ether.23 While the initiation rates were also affected by the electronic nature of the benzylidene chelate, the rate of C–H activation also has little correlation with the initiation rates of the related dichloride complexes (Figure 8). Initiation rates also did not correspond well with Hammett σ values.
Figure 8.

Relationship between initiation rate of dichlorides and rate of cyclometalation of dicarboxylates 3.
Kinetic Isotope Effects
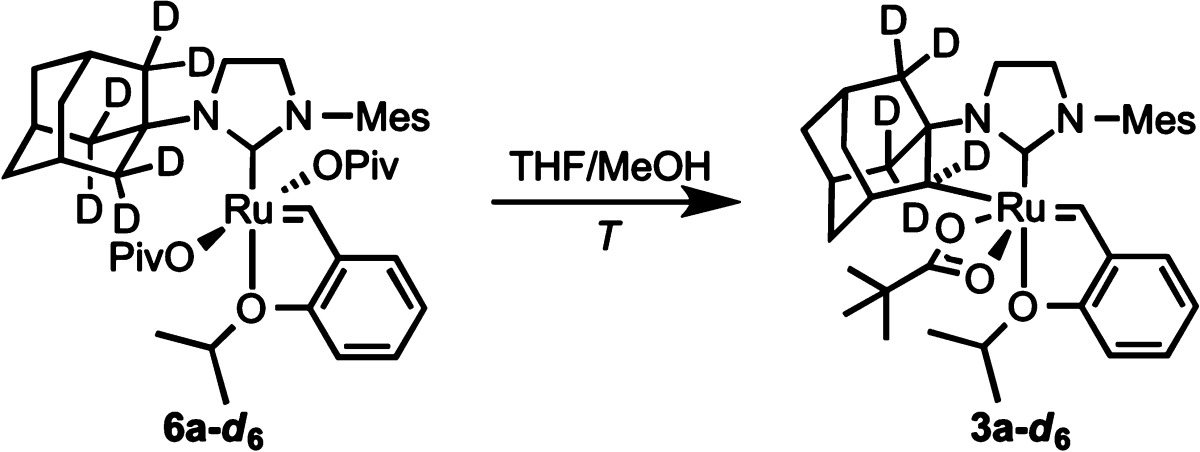
Kinetic isotope effects (KIEs) are a common measurement when studying the mechanism of C–H activation with transition metals.24 In order to measure the KIE (= kH/kD) of C(sp3)–H activation at these ruthenium complexes, deuterated dicarboxylate 6a-d6 was synthesized utilizing d6-1-adamantylamine (Table 1). In separate experiments, the rates of C–H and C–D activation were measured. At 25 °C, KIE = 8.1 ± 1.7 was found. At an elevated temperature of 50 °C, the value was reduced to KIE = 6.4 ± 1.1.
Table 1. Kinetic Isotope Effects.
| entry | k | T (°C) | value (s–1)a | KIEa |
|---|---|---|---|---|
| 1 | kH | 25 | (2.8 ± 0.4) × 10–4 | 8.1 ± 1.7 |
| 2 | kD | 25 | (3.5 ± 0.6) × 10–5 | |
| 3 | kH | 50 | (4.3 ± 0.3) × 10–3 | 6.4 ± 1.1 |
| 4 | kD | 50 | (6.7 ± 1) × 10–4 |
Uncertainty reported with 90% confidence intervals.
Computational Studies on the Mechanism of C–H Activation
We performed DFT calculations to investigate the mechanism of the C–H activation pathways and to explain the effects of N-substituents on the reactivity and selectivity of C–H activation. The calculations were performed using the theoretical method that was found satisfactory in our recent computations with ruthenium metathesis catalysts.17,19a,25 The geometries were optimized with B3LYP and the SDD basis set for Ru and 6-31G(d) for other atoms. Single-point energies were calculated with M06 and the SDD basis set for Ru and the 6-311+G(d,p) basis set for other atoms. The SMD solvation model was employed in the single-point energy calculations. THF was used as the solvent in the calculations. All calculations were performed with Gaussian 09.26
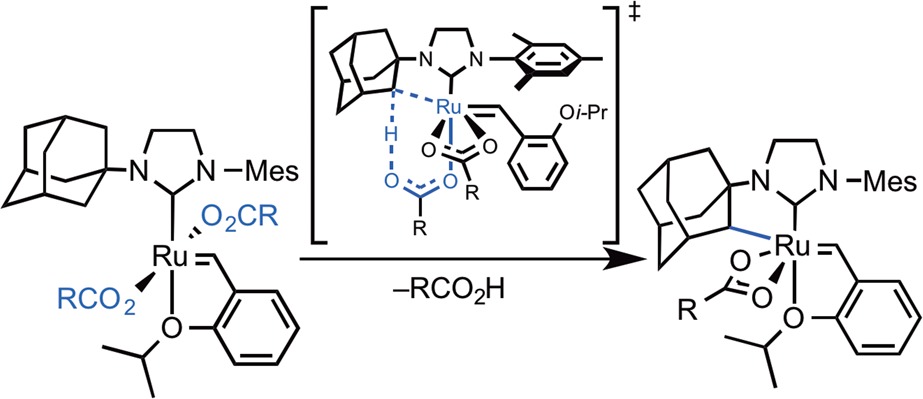
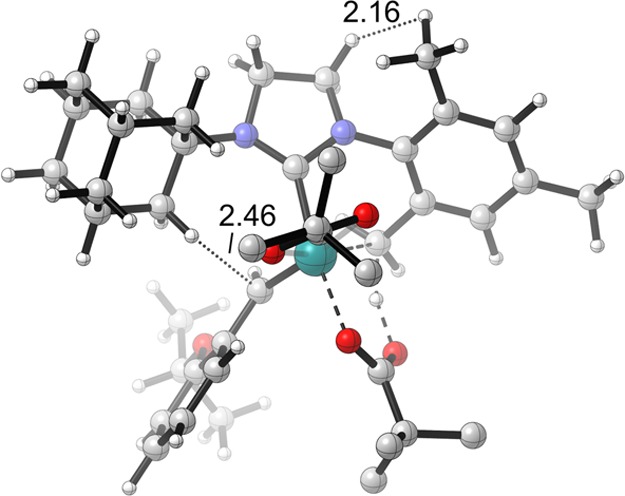
The calculations indicated that the C–H activation of ruthenium dichloride complex 4a occurs through the pathway shown in blue in Figure 9. The monopivalate pathway shown in red is unfavorable (vide infra). The anion-exchange steps to replace both chlorides in 4a with pivalates are exergonic by 1.4 and 4.0 kcal·mol–1, respectively. The reaction with silver pivalate is expected to be even more favorable, driven by the formation of solid silver chloride precipitate. The pivalate in complex 5a and both pivalates in complex 6a are monoligated (Figure 10). The binding site trans to the benzylidene in these 16-electron complexes is blocked by the bulky N-adamantyl group.27 The most favorable C–H activation pathway from the dipivalate complex 6a involves rotation of the o-isopropoxyphenyl group (12a-TS) and dissociation of the Ru–O chelate to form 13a, followed by deprotonation of the adamantyl C–H bond by a bottom-bound pivalate (14a-TS-A) via a CMD mechanism. The C–H activation step leads to a pivalic acid-bound complex 15a, which then liberates pivalic acid and generates Ru–C cyclometalated catalyst 3a. The rate-determining step was found to be the C–H activation via intramolecular CMD (14a-TS-A) and requires an overall free energy barrier of 23.5 kcal·mol–1 (6a → 14a-TS-A).28
Figure 9.

Free energy profile of the C–H activation of 4a to form cyclometalated complex 3a; all energies in kcal·mol–1 at 25 °C.
Figure 10.
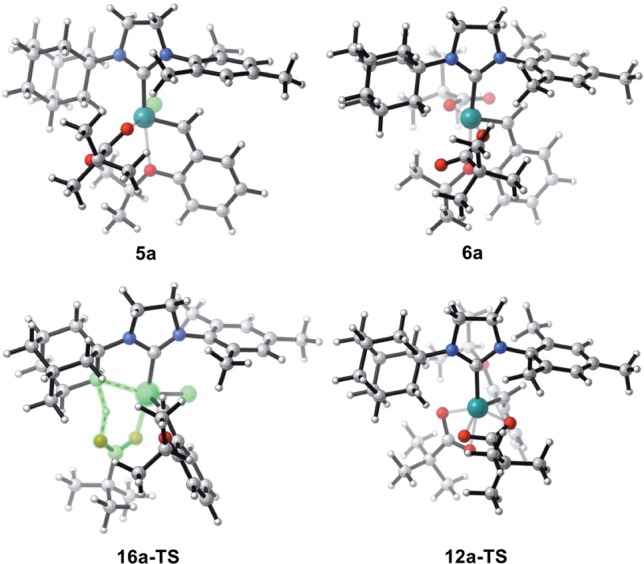
Optimized geometries of the monopivalate complex 5a, dipivalate complex 6a, the monopivalate C–H activation transition state 16a-TS, and the o-isopropoxyphenyl rotation transition state 12a-TS.
Several isomeric C–H activation transition states from the dipivalate complex 6a have been calculated and are all found to be less stable than 14a-TS-A (Figure 11). In 14a-TS-B, the C–H bond is deprotonated by the pivalate bound to the side position (i.e., cis to the NHC). Although this orientation maintains the Ru–O(isopropoxy) chelation, such side-bound C–H activation is 6.9 kcal·mol–1 less favorable than the bottom-bound C–H activation. The four-membered-ring CMD transition state 14a-TS-C, which is also referred to as σ-bond metathesis in the literature,8c is 14 kcal·mol–1 less favorable, as the oxygen coordinated to the Ru is less basic.8a,29 An outer-sphere deprotonation transition state involving an unbound pivalate was also located (14a-TS-D) and is also unfavorable.30 This is consistent with the preference for the six-membered CMD transition states in palladium acetate-catalyzed C–H activations.2e,2i,7d,8
Figure 11.

Four possible transition states for cyclometalation of dipivalate complex 6a.
C–H(D) insertion KIEs were calculated31 using the Bigeleisen–Mayer equation32 with scaled (0.97)33 harmonic frequencies obtained from B3LYP/LANL2DZ-6-31G* ground-state structure 6a and transition structures 14a-TS-A–D (Figure 12). B3LYP/SDD-6-31G(d) and M06/SDD-6-31G(d) models provided comparable KIE estimates for the 14a-TS-A C–H insertion process at 25 °C (6.47 and 6.42, respectively). The computed KIEs decreased with increasing temperature (Figure 12) and were found to originate primarily from zero-point energy contributions.
Figure 12.
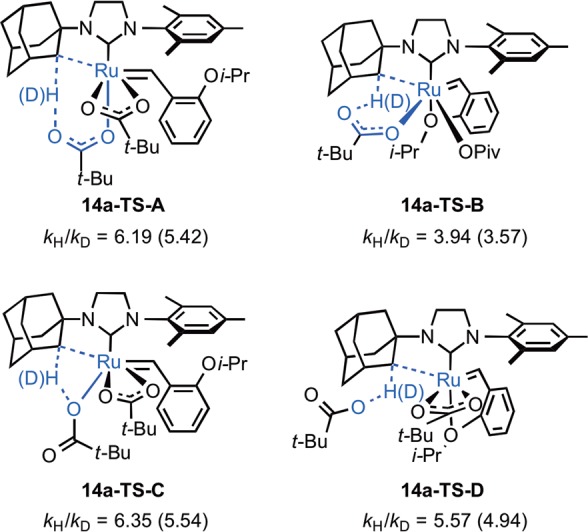
Kinetic isotope effects computed at 25 and 50 °C (in parentheses) for potential C–H(D) activation transition states.31
Discussion
Salt Metathesis Affects the Overall Rate of Cyclometalation
The results outlined above support a general mechanism that requires two salt metathesis steps to preform a ruthenium dicarboxylate species. These salt metathesis events have a profound effect on the observed rate of the cyclometalation reaction when starting from dichloride complexes (Table 2). For instance, the N-mesityl (4a) and N-DIPP (4d) dichloride complexes differ greatly in observed rates of cyclometalation, but the rates for cyclometalation from the dipivalate complexes 6a and 6d are roughly equal (entries 1 and 4). This reduction in the cyclometalation rate of 4d can be attributed to the poor equilibrium concentrations of reactive 6d under the standard conditions. This same effect was also observed for complex 4c, which is computationally expected to undergo cyclometalation at a much faster rate than 4a. Interestingly, complex 4b underwent cycloaddition at a rate similar to that of 4a, although computations predicted the reaction from 6b to be slower than that from 6a.
Table 2. Summary of Rate Information for Ruthenium Complexes 4 and 6a.
| entry | 4 | k from 4 (×10–4 s–1)b | 4:5:6c | k from 6 (×10–3 s–1)b | calcddk from 6 (×10–4 s–1) |
|---|---|---|---|---|---|
| 1 | 4a | 2.7 ± 0.1 | 69:25:6 | 1.6 ± 0.1 | 2.7 |
| 2 | 4b | 1.2 ± 0.1 | 3:38:59 | – | 0.084 |
| 3 | 4c | 2.0 ± 0.1 | 56:43:<1 | – | 1.7 × 106 |
| 4 | 4d | <0.1 | >98:<1:<1 | 1.4 ± 0.2 | 2.6 |
All data from experiments conducted at 40 °C.
Uncertainty reported with 90% confidence intervals.
Relative concentrations of complexes at equilibrium.
See Supporting Information for details.
In general, acceleration of the salt metathesis steps enhanced the overall rate of cyclometalation. For instance, cyclometalation occurred rapidly in the presence of silver pivalate salts as a result of the rapid nature of silver-mediated salt metathesis.13b This hypothesis is further supported by the observation that cyclometalation by exposure of triflate complexes 7 occurs rapidly at low temperature because of the more facile exchange of triflate ligands.
Dicarboxylate Stabilizes the Cyclometalation Transition State
Based on control experiments, only the ruthenium dicarboxylate species 6 is able to undergo C–H activation. This was further supported by computational evidence that the C–H activation transition state of monochloride-monocarboxylate 5a is 8.9 kcal·mol–1 less stable than the corresponding transition state (16a-TS, red, vs 14a-TS, blue, Figure 9). The monopivalate C–H activation also involves a bottom-bound six-membered transition state (16a-TS, Figure 10), similar to 14a-TS-A. The lower energy of the dipivalate TS is in part due to the stronger binding energy of pivalate compared to chloride, as shown in the ground-state structures (6a vs 5a). Additionally, the pivalate is bound to ruthenium in a κ2 fashion in 14a-TS-A and the final product 3a. This binding mode offers additional stabilization compared to the κ1 pivalate species prior to the C–H activation (5a and 6a). In the reaction with monopivalate, there is no such stabilization in the cyclometalation transition state 16a-TS, since the bidentate-bound pivalate is now replaced with chloride.
In addition, the chelating carboxylate ligand allows for further stabilization of the de-chelation of the benzylidene chelate (i.e., 13a). Since the most energetically favorable cyclometalation transition state involves a pseudo-apically oriented carboxylate, the additional carboxylate ligand avoids any unstable 14-electron complexes involved in achieving the required transition-state geometry.
Rate of Cyclometalation Can Be Tuned Electronically
Experimental data reveal some of the electronic characteristics of the C–H activation reaction. The carboxylate ligand acts as a base, as indicated by the negative ρ value for the linear free energy relationship of ruthenium dibenzoates in the C–H activation reaction (Figure 6). Additionally, the stronger correlation to the σ+ value suggests an accumulation of positive charge at the carboxylate carbon that is stabilized by resonance with the aromatic system. These results are supported by computational evidence that more basic carboxylates have lower activation barriers to C–H activation (Table 3).34 Natural population analysis (NPA) calculations of 14a-TS-A indicated polarization of the C–H bond being cleaved: the negative charge at C increases by −0.09, while the positive charge at H increases by +0.23 relative to that of dicarboxylate 6a.35 These electronic characteristics further support the CMD mechanism with the carboxylate acting as a base.
Table 3. Activation Free Energies (at 25 °C) for Cyclometalation with Different Carboxylate Ligands.
| entry | R | dicarboxylate | ΔG⧧ (kcal·mol–1) | pKa |
|---|---|---|---|---|
| 1 | t-Bu | 6a | 23.5 | 5.03 |
| 2 | Me | 18a | 24.0 | 4.76 |
| 3 | Ph | 8a | 26.2 | 4.20 |
The electronic effects of the benzylidene chelate also reveal some characteristics of the ruthenium center during the C–H activation step. The lack of a strong correlation to either the σpara or σmeta values for substituents at the 4-position of the benzylidene chelate indicates a subtle competing effect of the electronics at both the chelating oxygen (σpara values) and the ruthenium benzylidene (σmeta values). Though a stronger correlation to the σmeta values with a positive ρ value supports an increased importance of electron deficiency at ruthenium over the lability of the chelate, definitive conclusions cannot be drawn from the current experimental data. In addition, computations indicate no correlation between the benzylidene de-chelation energy and the C–H activation rate with different benzylidene chelates, 10a–d.36 These conclusions are further muddied by the absence of a correlation between the initiation rates of dichlorides and the rates of cyclometalation of the corresponding dipivalate complexes 10a–d, even though both reactions include de-chelation of the isopropyl ether as a key organizational step.
The importance of increased positive charge at the ruthenium center on the observed rate of the reaction (Figure 7) agrees with the requirement of a dicarboxylate species. The additional electronic stabilization provided by the κ2 pivalate ligand would stabilize the electrophilic character of the ruthenium center. This is further supported by the reduction in positive charge at ruthenium by −0.16 in the transition state, as calculated by natural population analysis. This reduction in positive charge indicates an accumulation of electrons throughout the reaction, a feature that would be more favorable for a more electron-deficient ruthenium center.
Pseudo-Apically Oriented Six-Membered Concerted Metalation–Deprotonation Transition-State Geometry
The results reported above support the reorganization of ligands to a transition-state geometry as depicted in 14a-TS-A (Figure 11). The close agreement between the experimental ΔG⧧298K = 22.2 ± 0.1 kcal·mol–1 and the computed ΔG⧧ = 23.5 kcal·mol–1 lends credence to the computed geometries. Computations indicated that other isomeric C–H activation transition states (14a-TS-B/C/D) are at least 6.9 kcal·mol–1 less favorable than 14a-TS-A. Furthermore, the large, negative ΔS⧧ = −5.2 ± 2.6 eu obtained from the Eyring analysis supports a highly ordered cyclic transition state.
The pseudo-apical orientation of the deprotonating carboxylate in 14a-TS-A is also supported by the KIE (Figure 12). The measured KIE = 8.1 ± 1.7 more closely reflects the computed isotope effects for the two transition states that involve ligand reorganization to place the participating carboxylate in a pseudo-apical position (14a-TS-A and 14a-TS-C). Furthermore, the small and computationally predicted reduction to KIE = 6.4 ± 1.1 at elevated temperatures excludes significant tunneling and lends additional support for this mechanism of C–H insertion.
Site Selectivity of C–H Activation Is Controlled by Steric Effects
In the C–H activation reaction with unsymmetrically substituted NHC complexes such as N-adamantyl-N′-mesityl complex 6a, it is somewhat unexpected that the weaker mesityl C–H bond remains intact while reaction occurs exclusively at the adamantyl C–H bond.2e,2f,37 In addition, the C–H activation in the stereogenic-at-Ru complex is completely stereoselective. Only a single diastereomer of 3a is observed, in which the benzylidene is anti to the C–H bond on the adamantyl (Scheme 5). Computations predicted the same major C–H activation product (3a) as that observed in experiment. Activation of the mesityl C–H bond requires 27.7 kcal·mol–1 (14-TS-F), 4.2 kcal·mol–1 higher than the adamantyl C–H bond activation (14a-TS-A). The transition state in which the benzylidene is syn to the C–H bond on the adamantyl (14a-TS-E) is 2.1 kcal·mol–1 less stable. The major product 3a is also thermodynamically the most stable among the three isomers.
Scheme 5. Activation Free Energies (in kcal·mol–1) of C–H Activation at Different Sites of 6a.
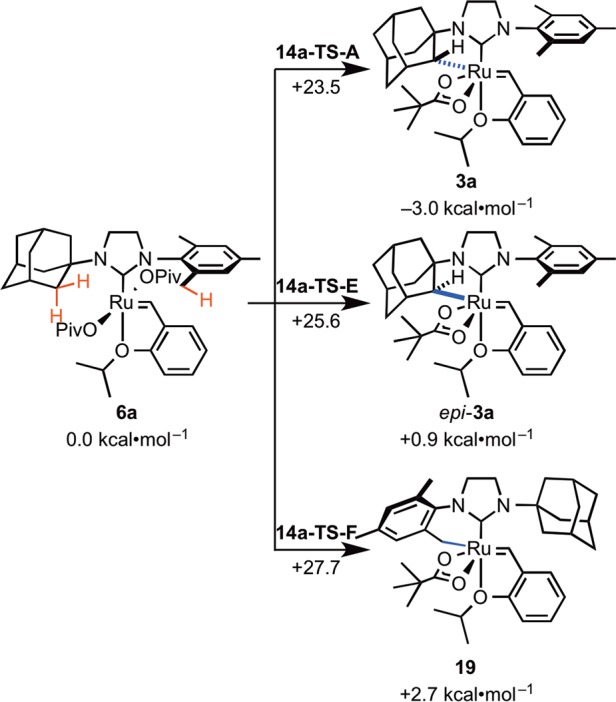
Steric interactions in the transition state disfavor activation of the weaker benzylic C–H bonds of 6a (Figure 13). There is substantial distortion in 14a-TS-F because of the steric repulsion between one of the ortho Me groups on the mesityl and a methylene group on the NHC backbone. This repulsion prevents the other methyl substituent from rotating into proximity of the ruthenium center. In addition, orientation of the bulky N-adamantyl group toward the benzylidene leads to steric interactions with the carbene carbon atom (H···C distance of 2.46 Å).
Figure 13.
Transition-state geometry 14a-TS-F.
In 14a-TS-A and the resulting cyclometalated intermediate 3a, the α-C–H bond on adamantyl is anti to the Ru-benzylidene double bond in a staggered conformation, while complexes 14a-TS-E and epi-3a assume an eclipsed conformation with the α-C–H bond syn to the Ru-benzylidene bond (Figure 14). The staggered anti transition state 14a-TS-A is favored by 2.1 kcal·mol–1. The anti intermediate 3a is also more stable than the syn intermediate epi-3a to a greater extent (3.9 kcal·mol–1).
Figure 14.
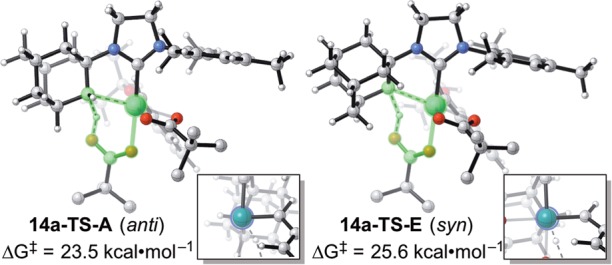
Comparison of the diastereomeric C–H activation transition states 14a-TS-A (anti) and 14a-TS-E (syn). Insets are Newman projections along the forming Ru–C bond. Energies are relative to 6a.
Effects of N-Substituents on the Barriers for C–H Activation
Barriers for C–H activation with N-t-Bu-N′-Mes complex 6c and N-Adm-N′-DIPP complex 6d were computed (Figure 15). Replacing the adamantyl group with the smaller tert-butyl group significantly decreased the barrier to 15.2 kcal·mol–1. There are fewer steric repulsions in the activation of the primary C–H bond in t-Bu than the secondary C–H bond on adamantyl. This reactivity trend agrees with palladium-catalyzed C(sp3)–H activations,1b,2k,38 in which less-hindered primary C–H bonds are preferred. In contrast, iron oxo-catalyzed reactions are governed by a combination of steric and electronic effects.39
Figure 15.

Computed activation barriers at 25 °C for cyclometalation of complexes 6b–d.
On the other hand, replacing the N-mesityl group with N-DIPP has negligible effects on the activation barrier. The N-mesityl and N-DIPP groups both remain perpendicular to the NHC ring in the dipivalate resting state and in the transition state. There are no significant steric repulsions with either the N-mesityl or N-DIPP group in the dipivalate complex or in the transition state.40
Conclusion
Studies in the development of a new family of Z-selective olefin metathesis catalysts revealed an interesting and relevant C–H activation reaction at ruthenium(II). This reaction involves a carboxylate-assisted concerted metalation–deprotonation via an organized six-membered metallacyclic transition state. The resulting ruthenium alkyl complexes are unusually stable, and these benzylidene complexes exhibit high levels of Z-selectivity in olefin metathesis reactions.
Isolation of the dicarboxylate complexes 6 allowed for the direct study of the C–H activation reaction. Kinetic studies revealed first-order reaction kinetics, and thermodynamic results agreed with computed transition states. Furthermore, ΔS⧧ values agree with a highly ordered intramolecular transition state. Computational studies revealed an interesting pseudo-apically oriented carboxylate geometry in the key cyclometalation step and confirmed that a second carboxylate ligand is necessary for transition-state stabilization. Electronic effects of the complex control the rate by modulating either the electron density at the ruthenium center or the overall basicity of the assisting carboxylate.
Experimental and computational results showed that steric effects control the site-selectivity of C–H activation, directing metalation toward typically less-reactive C–H bonds. The computational model was then able to corroborate indirect mechanistic evidence about the relative rates of cyclometalation at complexes that could not be studied directly.
New mechanistic information was developed regarding the inner-sphere activation of methylene C(sp3)–H bonds. It is envisioned that the results of this study will enable the further development of CMD C(sp3)–H activation at ruthenium centers both for the more efficient synthesis of selective ruthenium metathesis catalysts and for functionalization of C–H bonds in general.
Acknowledgments
This work was financially supported by the NIH (NIH R01-GM031332 and NRSA to J.S.C.: F32-GM103002), the NSF (CHE-1212767, CHE-1059084), and the NSF CCI Center for Stereoselective C–H Functionalization (CHE-1205646). NMR spectra were obtained by instruments supported by the NIH (RR027690). Materia, Inc. is acknowledged for the generous donation of complex 4b. D.J.O. thanks Pomona College for supporting a portion of this research. Calculations were performed on the Hoffman2 cluster at UCLA and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF. K. M. Engle, J. Hartung, and Z. K. Wickens are gratefully acknowledged for helpful discussions.
Supporting Information Available
Experimental details, NMR spectra of new compounds, additional kinetics data, optimized Cartesian coordinates and energies, details of computational methods, and complete ref (26). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Lapointe D.; Fagnou K. Chem. Lett. 2010, 39, 1118. [Google Scholar]; b Lyons T. W.; Sanford M. S. Chem. Rev. 2010, 110, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ackermann L. Chem. Rev. 2011, 111, 1315. [DOI] [PubMed] [Google Scholar]; d Yamaguchi J.; Yamaguchi A. D.; Itami K. Angew. Chem., Int. Ed. 2012, 51, 8960. [DOI] [PubMed] [Google Scholar]; e Neufeldt S. R.; Sanford M. S. Acc. Chem. Res. 2012, 45, 936. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Engle K. M.; Mei T.-S.; Wasa M.; Yu J.-Q. Acc. Chem. Res. 2012, 45, 788. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Yeung C. S.; Dong V. M. Chem. Rev. 2011, 111, 1215. [DOI] [PubMed] [Google Scholar]
- a Canty A. J.; Ariafard A.; Sanford M. S.; Yates B. F. Organometallics 2013, 32, 544. [Google Scholar]; b Lyons T. W.; Hull K. L.; Sanford M. S. J. Am. Chem. Soc. 2011, 133, 4455. [DOI] [PubMed] [Google Scholar]; c Baxter R. D.; Sale D.; Engle K. M.; Yu J.-Q.; Blackmond D. G. J. Am. Chem. Soc. 2012, 134, 4600. [DOI] [PubMed] [Google Scholar]; d Rousseaux S.; Gorelsky S. I.; Chung B. K. W.; Fagnou K. J. Am. Chem. Soc. 2010, 132, 10692. [DOI] [PubMed] [Google Scholar]; e Gorelsky S. I.; Lapointe D.; Fagnou K. J. Am. Chem. Soc. 2008, 130, 10848. [DOI] [PubMed] [Google Scholar]; f Gorelsky S. I.; Lapointe D.; Fagnou K. J. Org. Chem. 2012, 77, 658. [DOI] [PubMed] [Google Scholar]; g Sun H.-Y.; Gorelsky S. I.; Stuart D. R.; Campeau L.-C.; Fagnou K. J. Org. Chem. 2010, 75, 8180. [DOI] [PubMed] [Google Scholar]; h Wakioka M.; Nakamura Y.; Hihara Y.; Ozawa F.; Sakaki S. Organometallics 2013, 32, 4423. [Google Scholar]; i García-Cuadrado D.; Braga A. A. C.; Maseras F.; Echavarren A. M. J. Am. Chem. Soc. 2006, 128, 1066. [DOI] [PubMed] [Google Scholar]; j Chen X.; Engle K. M.; Wang D.-H.; Yu J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Daugulis O.; Do H.-Q.; Shabashov D. Acc. Chem. Res. 2009, 42, 1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Davies D. L.; Donald S. M. A.; Al-Duaij O.; Macgregor S. A.; Pölleth M. J. Am. Chem. Soc. 2006, 128, 4210. [DOI] [PubMed] [Google Scholar]; b Li L.; Brennessel W. W.; Jones W. D. Organometallics 2009, 28, 3492. [Google Scholar]; c Hartwig J. F. Acc. Chem. Res. 2012, 45, 864. [DOI] [PubMed] [Google Scholar]; d Mkhalid I. A. I.; Barnard J. H.; Marder T. B.; Murphy J. M.; Hartwig J. F. Chem. Rev. 2010, 110, 890. [DOI] [PubMed] [Google Scholar]
- a Rhinehart J. L.; Manbeck K. A.; Buzak S. K.; Lippa G. M.; Brennessel W. W.; Goldberg K. I.; Jones W. D. Organometallics 2012, 31, 1943. [Google Scholar]; b Davies H. M. L.; Manning J. R. Nature 2008, 451, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Davies H. M. L.; Morton D. Chem. Soc. Rev. 2011, 40, 1857. [DOI] [PubMed] [Google Scholar]; d Colby D. A.; Tsai A. S.; Bergman R. G.; Ellman J. A. Acc. Chem. Res. 2012, 45, 814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Davies D. L.; Al-Duaij O.; Fawcett J.; Giardiello M.; Hilton S. T.; Russell D. R. Dalton Trans. 2003, 4132. [Google Scholar]; b Arockiam P. B.; Bruneau C.; Dixneuf P. H. Chem. Rev. 2012, 112, 5879. [DOI] [PubMed] [Google Scholar]; c Kozhushkov S. I.; Ackermann L. Chem. Sci. 2013, 4, 886. [Google Scholar]; d Ackermann L.; Vicente R.; Potukuchi H. K.; Pirovano V. Org. Lett. 2010, 12, 5032. [DOI] [PubMed] [Google Scholar]; e Hashiguchi B. G.; Young K. J. H.; Yousufuddin M.; Goddard W. A. III; Periana R. A. J. Am. Chem. Soc. 2010, 132, 12542. [DOI] [PubMed] [Google Scholar]; f Gruver B. C.; Adams J. J.; Warner S. J.; Arulsamy N.; Roddick D. M. Organometallics 2011, 30, 5133. [Google Scholar]
- a Gephart R. T. III; Warren T. H. Organometallics 2012, 31, 7728. [Google Scholar]; b Yamaguchi J.; Muto K.; Itami K. Eur. J. Org. Chem. 2013, 19. [Google Scholar]
- a Ryabov A. D.; Sakodinskaya I. K.; Yatsimirsky A. K. J. Chem. Soc., Dalton Trans. 1985, 2629. [Google Scholar]; b Kurzeev S. A.; Kazankov G. M.; Ryabov A. D. Inorg. Chim. Acta 2002, 340, 192. [Google Scholar]; c Maleckis A.; Kampf J. W.; Sanford M. S. J. Am. Chem. Soc. 2013, 135, 6618. [DOI] [PubMed] [Google Scholar]; d Sanhueza I. A.; Wagner A. M.; Sanford M. S.; Schoenebeck F. Chem. Sci. 2013, 4, 2767. [Google Scholar]
- a Davies D. L.; Donald S. M. A.; Macgregor S. A. J. Am. Chem. Soc. 2005, 127, 13754. [DOI] [PubMed] [Google Scholar]; b García-Cuadrado D.; de Mendoza P.; Braga A. A. C.; Maseras F.; Echavarren A. M. J. Am. Chem. Soc. 2007, 129, 6880. [DOI] [PubMed] [Google Scholar]; c Balcells D.; Clot E.; Eisenstein O. Chem. Rev. 2010, 110, 749. [DOI] [PubMed] [Google Scholar]; d Ke Z.; Cundari T. R. Organometallics 2010, 29, 821. [Google Scholar]; e Musaev D. G.; Kaledin A.; Shi B.-F.; Yu J.-Q. J. Am. Chem. Soc. 2012, 134, 1690. [DOI] [PubMed] [Google Scholar]; f Giri R.; Lan Y.; Liu P.; Houk K. N.; Yu J.-Q. J. Am. Chem. Soc. 2012, 134, 14118. [DOI] [PubMed] [Google Scholar]; g Yang Y.-F.; Cheng G.-J.; Liu P.; Leow D.; Sun T.-Y.; Chen P.; Zhang X.; Yu J.-Q.; Wu Y.-D.; Houk K. N. J. Am. Chem. Soc. 2014, 136, 344. [DOI] [PubMed] [Google Scholar]; h Cheng G.-J.; Yang Y.-F.; Liu P.; Chen P.; Sun T.-Y.; Li G.; Zhang X.; Houk K. N.; Yu J.-Q.; Wu Y.-D. J. Am. Chem. Soc. 2014, 136, 894. [DOI] [PubMed] [Google Scholar]; i Sokolov V. I.; Troitskaya L. L.; Reutov O. A. J. Organomet. Chem. 1979, 182, 537. [Google Scholar]
- a Prokopcová H.; Bergman S. D.; Aelvoet K.; Smout V.; Herrebout W.; Van der Veken B.; Meerpoel L.; Maes B. U. W. Chem.—Eur. J. 2010, 16, 13063. [DOI] [PubMed] [Google Scholar]; b Bergman S. D.; Storr T. E.; Prokopcová H.; Aelvoet K.; Diels G.; Meerpoel L.; Maes B. U. W. Chem.—Eur. J. 2012, 18, 10393. [DOI] [PubMed] [Google Scholar]; c Kumar N. Y. P.; Jeyachandran R.; Ackermann L. J. Org. Chem. 2013, 78, 4145. [DOI] [PubMed] [Google Scholar]; d Jun C.-H.; Hwang D.-C.; Na S.-J. Chem. Commun. 1998, 1405. [Google Scholar]; e Dastbaravardeh N.; Kirchner K.; Schnürch M.; Mihovilovic M. D. J. Org. Chem. 2013, 78, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Dastbaravardeh N.; Schnürch M.; Mihovilovic M. D. Org. Lett. 2012, 14, 3792. [DOI] [PubMed] [Google Scholar]; g Chatani N.; Asaumi T.; Yorimitsu S.; Ikeda T.; Kakiuchi F.; Murai S. J. Am. Chem. Soc. 2001, 123, 10935. [DOI] [PubMed] [Google Scholar]; h Weissman H.; Song X.; Milstein D. J. Am. Chem. Soc. 2001, 123, 337. [DOI] [PubMed] [Google Scholar]; i Oi S.; Fukita S.; Hirata N.; Watanuki N.; Miyano S.; Inoue Y. Org. Lett. 2001, 3, 2579. [DOI] [PubMed] [Google Scholar]; j Ozdemir I.; Demir S.; Çetinkaya B.; Gourlaouen C.; Maseras F.; Bruneau C.; Dixneuf P. H. J. Am. Chem. Soc. 2008, 130, 1156. [DOI] [PubMed] [Google Scholar]; k Ackermann L. Org. Lett. 2005, 7, 3123. [DOI] [PubMed] [Google Scholar]; l Padala K.; Jeganmohan M. Org. Lett. 2011, 13, 6144. [DOI] [PubMed] [Google Scholar]; m Burling S.; Paine B. M.; Nama D.; Brown V. S.; Mahon M. F.; Prior T. J.; Pregosin P. S.; Whittlesey M. K.; Williams J. M. J. J. Am. Chem. Soc. 2007, 129, 1987. [DOI] [PubMed] [Google Scholar]; n Häller L. J. L.; Page M. J.; Macgregor S. A.; Mahon M. F.; Whittlesey M. K. J. Am. Chem. Soc. 2009, 131, 4604. [DOI] [PubMed] [Google Scholar]
- Diggle R. A.; Kennedy A. A.; Macgregor S. A.; Whittlesey M. K. Organometallics 2008, 27, 938. [Google Scholar]
- a Liang J.-L.; Yuan S.-X.; Huang J.-S.; Yu W.-Y.; Che C.-M. Angew. Chem., Int. Ed. 2002, 41, 3465. [DOI] [PubMed] [Google Scholar]; b McNeill E.; Du Bois J. J. Am. Chem. Soc. 2010, 132, 10202. [DOI] [PubMed] [Google Scholar]; c Harvey M. E.; Musaev D. G.; Du Bois J. J. Am. Chem. Soc. 2011, 133, 17207. [DOI] [PubMed] [Google Scholar]; d McNeill E.; Du Bois J. Chem. Sci. 2012, 3, 1810. [Google Scholar]
- a Ferrer Flegeau E.; Bruneau C.; Dixneuf P. H.; Jutand A. J. Am. Chem. Soc. 2011, 133, 10161. [DOI] [PubMed] [Google Scholar]; b Fabre I.; von Wolff N.; Le Duc G.; Ferrer Flegeau E.; Bruneau C.; Dixneuf P. H.; Jutand A. Chem.—Eur. J. 2013, 19, 7595. [DOI] [PubMed] [Google Scholar]
- a Keitz B. K.; Endo K.; Patel P. R.; Herbert M. B.; Grubbs R. H. J. Am. Chem. Soc. 2012, 134, 693. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Endo K.; Grubbs R. H. J. Am. Chem. Soc. 2011, 133, 8525. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rosebrugh L. E.; Herbert M. B.; Marx V. M.; Keitz B. K.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 1276. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Rosebrugh L. E.; Marx V. M.; Keitz B. K.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 10032. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Endo K.; Herbert M. B.; Grubbs R. H. Organometallics 2013, 32, 5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx V. M.; Herbert M. B.; Keitz B. K.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Herbert M. B.; Marx V. M.; Pederson R. L.; Grubbs R. H. Angew. Chem., Int. Ed. 2013, 52, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cannon J. S.; Grubbs R. H. Angew. Chem., Int. Ed. 2013, 52, 9001. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Quigley B. L.; Grubbs R. H. Chem. Sci. 2014, 5, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hartung J.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 10183. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hartung J.; Grubbs R. H. Angew. Chem., Int. Ed. 2014, 53, 3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki H.; Herbert M. B.; Liu P.; Dong X.; Xu X.; Keitz B. K.; Ung T.; Mkrtumyan G.; Houk K. N.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Meek S. J.; O’Brien R. V.; Llaveria J.; Schrock R. R.; Hoveyda A. H. Nature 2011, 471, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Flook M. M.; Jiang A. J.; Schrock R. R.; Müller P.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 7962. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Marinescu S. C.; Levine D. S.; Zhao Y.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2011, 133, 11512. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jiang A. J.; Zhao Y.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 16630. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Yu M.; Ibrahem I.; Hasegawa M.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2012, 134, 2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Herbert M. B.; Lan Y.; Keitz B. K.; Liu P.; Endo K.; Day M. W.; Houk K. N.; Grubbs R. H. J. Am. Chem. Soc. 2012, 134, 7861. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hong S. H.; Chlenov A.; Day M. W.; Grubbs R. H. Angew. Chem., Int. Ed. 2007, 46, 5148. [DOI] [PubMed] [Google Scholar]; c Mathew J.; Koga N.; Suresh C. H. Organometallics 2008, 27, 4666. [Google Scholar]; d Poater A.; Cavallo L. J. Mol. Catal. A—Chem. 2010, 324, 75. [Google Scholar]; e Poater A.; Bahri-Laleh N.; Cavallo L. Chem. Commun. 2011, 47, 6674. [DOI] [PubMed] [Google Scholar]; f Leitao E. M.; Dubberley S. R.; Piers W. E.; Wu Q.; McDonald R. Chem.—Eur. J. 2008, 14, 11565. [DOI] [PubMed] [Google Scholar]
- Krause J. O.; Nuyken O.; Wurst K.; Buchmeiser M. R. Chem.—Eur. J. 2004, 10, 777. [DOI] [PubMed] [Google Scholar]
- a Ess D. H.; Nielsen R. J.; Goddard W. A. III; Periana R. A. J. Am. Chem. Soc. 2009, 131, 11686. [DOI] [PubMed] [Google Scholar]; b Ess D. H.; Goddard W. A. III; Periana R. A. Organometallics 2010, 29, 6459. [Google Scholar]
- a Thiel V.; Hendann M.; Wannowius K.-J.; Plenio H. J. Am. Chem. Soc. 2012, 134, 1104. [DOI] [PubMed] [Google Scholar]; b Bujok R.; Bieniek M.; Masnyk M.; Michrowska A.; Sarosiek A.; Stȩpowska H.; Arlt D.; Grela K. J. Org. Chem. 2004, 69, 6894. [DOI] [PubMed] [Google Scholar]; c Van Veldhuizen J. J.; Gillingham D. G.; Garber S. B.; Kataoka O.; Hoveyda A. H. J. Am. Chem. Soc. 2003, 125, 12502. [DOI] [PubMed] [Google Scholar]; d Zaja M.; Connon S. J.; Dunne A. M.; Rivard M.; Buschmann N.; Jiricek J.; Blechert S. Tetrahedron 2003, 59, 6545. [Google Scholar]
- Sanford M. S.; Love J. A.; Grubbs R. H. J. Am. Chem. Soc. 2001, 123, 6543. [DOI] [PubMed] [Google Scholar]
- a Jones W. D. Acc. Chem. Res. 2003, 36, 140. [DOI] [PubMed] [Google Scholar]; b Gómez-Gallego M.; Sierra M. A. Chem. Rev. 2011, 111, 4857. [DOI] [PubMed] [Google Scholar]; c Simmons E. M.; Hartwig J. F. Angew. Chem., Int. Ed. 2012, 51, 3066. [DOI] [PubMed] [Google Scholar]
- Liu P.; Xu X.; Dong X.; Keitz B. K.; Herbert M. B.; Grubbs R. H.; Houk K. N. J. Am. Chem. Soc. 2012, 134, 1464. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, 2009.
- Ragone F.; Poater A.; Cavallo L. J. Am. Chem. Soc. 2010, 132, 4249. [DOI] [PubMed] [Google Scholar]
- Ackermann L.; Vicente R.; Althammer A. Org. Lett. 2008, 10, 2299. [DOI] [PubMed] [Google Scholar]
- a Bischof S. M.; Ess D. H.; Meier S. K.; Oxgaard J.; Nielsen R. J.; Bhalla G.; Goddard W. A. III; Periana R. A. Organometallics 2010, 29, 742. [Google Scholar]; b Ess D. H.; Gunnoe T. B.; Cundari T. R.; Goddard W. A. III; Periana R. A. Organometallics 2010, 29, 6801. [Google Scholar]; c Ess D. H.; Bischof S. M.; Oxgaard J.; Periana R. A.; Goddard W. A. III Organometallics 2008, 27, 6440. [Google Scholar]
- The outer-sphere deprotonation transition state may be stabilized by coordination of solvent molecules with the unbound pivalate. Because of the implicit solvation model used in this study, the energy difference between 14a-TS-D and 14a-TS-A might be overestimated.
- Saunders M.; Laidig K. E.; Wolfsberg M. J. Am. Chem. Soc. 1989, 111, 8989. [Google Scholar]
- a Bigeleisen J.; Mayer M. G. J. Chem. Phys. 1947, 15, 261. [Google Scholar]; b Bigeleisen J.; Wolfsberg M. Adv. Chem. Phys. 1958, 1, 15. [Google Scholar]
- Halls M. D.; Tripp C. P.; Schlegel H. B. Phys. Chem. Chem. Phys. 2001, 3, 2131. [Google Scholar]
- Carboxylates have minimal effects on the de-chelation of the benzylidene chelate. The energy difference between 6a and 13a is 6.7, 6.7, and 6.4 kcal·mol–1 respectively for pivalate, acetate, and benzoate.
- For details, see the Supporting Information.
- The energy differences between the Hoveyda chelate (10a–d) and the C–H coordinated complex (analogue of 13a) are similar (5.3, 5.5, 6.7, and 4.7 kcal·mol–1 respectively for X = OMe, Me, H, and Cl).
- For computational investigations on regioselectivity in palladium carboxylate-catalyzed C–H activations:; a Guihaumé J.; Clot E.; Eisenstein O.; Perutz R. N. Dalton Trans. 2010, 39, 10510. [DOI] [PubMed] [Google Scholar]; b Petit A.; Flygare J.; Miller A. T.; Winkel G.; Ess D. H. Org. Lett. 2012, 14, 3680. [DOI] [PubMed] [Google Scholar]
- a Baudoin O.; Herrbach A.; Guéritte F. Angew. Chem., Int. Ed. 2003, 42, 5736. [DOI] [PubMed] [Google Scholar]; b Giri R.; Maugel N.; Li J.-J.; Wang D.-H.; Breazzano S. P.; Saunders L. B.; Yu J.-Q. J. Am. Chem. Soc. 2007, 129, 3510. [DOI] [PubMed] [Google Scholar]
- a Chen M. S.; White M. C. Science 2010, 327, 566. [DOI] [PubMed] [Google Scholar]; b Hitomi Y.; Arakawa K.; Funabiki T.; Kodera M. Angew. Chem., Int. Ed. 2012, 51, 3448. [DOI] [PubMed] [Google Scholar]; c Bigi M. A.; Liu P.; Zou L.; Houk K. N.; White M. C. Synlett 2012, 23, 2768. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Prat I.; Gómez L.; Canta M.; Ribas X.; Costas M. Chem.—Eur. J. 2013, 19, 1908. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for 3D structures of complexes 14b-TS, 14c-TS, and 14d-TS.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.