

Abstract
Chronic Chagas cardiomyopathy (CCC), caused by the obligate intracellular protozoan parasite Trypanosoma cruzi, is a major cause of morbidity and mortality in Latin America. CCC begins when T cruzi enters cardiac cells for intracellular multiplication and differentiation, a process that starts with recognition of host-cell entry receptors. However, the nature of these surface molecules and corresponding parasite counterreceptor(s) is poorly understood. Here we show that antibodies against neurotrophin (NT) receptor TrkC, but not against family members TrkA and TrkB, prevent T cruzi from invading primary cultures of cardiomyocytes and cardiac fibroblasts. Invasion is also selectively blocked by the TrkC ligand NT-3, and by antagonists of Trk autophosphorylation and downstream signaling. Therefore, these results indicate that T cruzi gets inside cardiomyocytes and cardiac fibroblasts by activating TrkC preferentially over TrkA. Accordingly, short hairpin RNA interference of TrkC (shTrkC), but not TrkA, selectively prevents T cruzi from entering cardiac cells. Additionally, T cruzi parasite-derived neurotrophic factor (PDNF)/trans-sialidase, a TrkC-binding protein, but not family member gp85, blocks entry dose-dependently, underscoring the specificity of PDNF as TrkC counterreceptor in cardiac cell invasion. In contrast to invasion, competitive and shRNA inhibition studies demonstrate that T cruzi-PDNF recognition of TrkA, but not TrkC on primary cardiomyocytes and the cardiomyocyte cell line H9c2 protects the cells against oxidative stress. Thus, this study shows that T cruzi via PDNF favors neurotrophin receptor TrkC for cardiac cell entry and TrkA for cardiomyocyte protection against oxidative stress, and suggests a new therapeutic opportunity in PDNF and/or fragments thereof for CCC therapy as entry inhibitors and/or cardioprotection agonists.
Introduction
Chagas disease, caused by infection with the protozoan parasite Trypanosoma cruzi, is a major source of disease burden in the Western Hemisphere, including developed countries (Bern et al., 2009, Rassi et al., 2010, Machado et al., 2012). It starts with an acute phase where robust parasite growth throughout the body coincides with non-descript symptoms such as fever, chills and nausea. Parasitemia typically peaks several weeks after infection, usually following the bite of reduviid insects, and is nearly undetectable after eight weeks, at which point blood and tissue parasitism wane under control from the immune response, giving rise to asymptomatic and pathology-free indeterminate phase that can last many years or a life-time. For reasons that remain unknown, around 30% of Chagas disease patients suffer or will suffer from chronic Chagas cardiomyopathy (CCC) whose symptoms include arrhythmias, heart failure and sudden death, and for which there are no cure except heart transplant (Fiorelli et al., 2011). CCC occurs without a recrudescence of parasitism, and, in spite of the paucity of parasites in chronically infected heart, progression to CCC appears to be driven by the inflammatory response to both residual heart parasitism and to cardiac autoantigens, particularly myosin (Marin-Neto et al., 2007, Machado et al., 2012).
As T cruzi is an obligate intracellular parasite, it must gain access into the host cell cytoplasm for multiplication and differentiation and for avoiding killing by the immune system. Hence, efficient invasion of cardiac cells is critical for replication and differentiation of T cruzi and, thus, for the development of acute heart disease and for parasite persistence in chronic infection, including CCC. Still, mechanisms underlying T cruzi recognition of cardiac cell surface receptors required for the intracellular cycle is poorly understood. Although T cruzi invades both cardiomyocytes and cardiac fibroblasts in vivo (Tafuri, 1970, Wong et al., 1992, Huang et al., 2003b), structural and molecular alterations and immune responses resulting from T cruzi infection of the heart are studied almost exclusively in the context of cardiomyocytes (Machado et al., 2012, Calvet et al., 2012, Araujo-Jorge et al., 2012), even though cardiac fibroblasts are critical for proper cardiomyocyte function either by direct cell-cell interaction or paracrine signaling in physiological conditions and diseases such as myocardial infarction (Souders et al., 2009, Kakkar et al., 2010) and other types of myocarditis (van Nieuwenhoven et al., 2012). Therefore, cardiac fibroblasts likely play an important role in Chagas heart disease and studies designed to understand T cruzi-cardiac fibroblast interactions are sorely needed to better understand Chagas disease progression.
Early findings suggested that T cruzi invasion of primary cultures of embryonic cardiomyocytes may involve mannosyl residues (Soeiro Mde et al., 1999) and recent work indicates that cell entry depends on low-density lipoprotein (LDL) receptor in the cardiomyocyte cell line H9c2 (Nagajyothi et al., 2012). However, the nature of the T cruzi ligand that recognizes mannosyl residues and LDL receptor has not been identified. Given that the protozoan parasite Leishmania, which infects a single cell type (macrophage), takes advantage of at least seven distinct surface receptors for invasion (Cunningham, 2002), and that the relatively simple organism HIV employs three entry receptors (CD4, CCR5 and CXCR4) to invade a single cell type (T-helper cell) (Wilen et al., 2012), then T cruzi should exploit an array of entry receptors in the heart and elsewhere as it is able to enter a wide range of nucleated host cells (Brener, 1973).
We showed earlier that T cruzi binds receptor tyrosine kinases TrkA (Chuenkova et al., 2004) and TrkC (Weinkauf et al., 2009) and that the binding leads to invasion of neurons and glial cells (Weinkauf et al., 2011, de Melo-Jorge et al., 2007). T cruzi recognition of TrkA and TrkC is through its surface parasite derived neurotrophic factor (PDNF), a GPI-linked neuraminidase/transsialidase; however, PDNF interaction with TrkA and TrkC, and with another protein kinase (Akt) (Chuenkova et al., 2009) is independent of sialic acid-binding activity (Chuenkova et al., 2011). TrkA and TrkC are activated primarily by the neurotrophins nerve growth factor (NGF) and neurotrophin-3 (NT-3), respectively, and the activation is essential for development and maintenance of the nervous systems (Huang et al., 2003a). Recently, both TrkC and TrkA have been identified in cardiomyocytes (Kawaguchi-Manabe et al., 2007, Meloni et al., 2010), and very recently in our lab, in neonatal and adult cardiac fibroblasts (Aridgides, 2013), which led to the hypothesis that Trks are responsible, at least in part, for T cruzi homing into the heart.
Here, we report that T cruzi via PDNF invades both primary cardiomyocytes and cardiac fibroblasts preferentially through TrkC compared to TrkA, and that TrkA recognition on cardiomyocytes protects the host cells against oxidative stress. This study is, as far as we know, the first to identify a molecular mechanism underlying T cruzi invasion of cardiac fibroblasts and also the first to show that, prior to homing into the cytosol habitat, direct T cruzi recognition of TrkA on cardiomyocytes protects the cells against oxidative stress.
Results
Preferential use of TrkC for T cruzi entry into cardiomyocytes
To determine whether T cruzi exploits Trk receptors to invade cardiomyocytes, we first assessed the ability of T cruzi strains Colombian (cardiotropic), CL-Brener (skeletal muscle tropic) and Tulahuen (reticulotropic) (Brener, 1973) to invade primary cultures of cardiomyocytes. We find that all three strains readily invade cardiomyocytes dose-dependently, although the Colombian strain is slightly more efficient than the other two (Fig. 1a). We used these strains (as indicated) in subsequent experiments.
Figure 1. Cardiomyocytes are preferentially invaded by T cruzi’s Colombian strain and invasion is specifically inhibited by TrkC antibodies and the TrkC ligand NT-3.
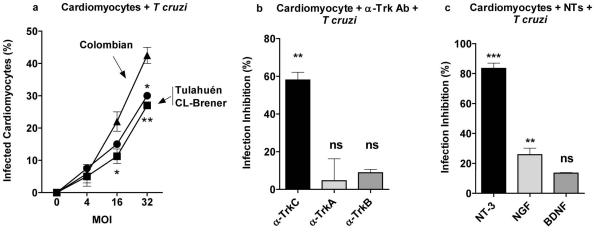
(a) Primary cultures of mouse cardiomyocytes were plated in 96-well plates, and infected in triplicate with T cruzi Colombian, CL-Brener and Tulahuén strains at various multiplicity of infection (MOI). After 2-3 h, parasites that did not invade were washed away and the ones that invaded were allowed to differentiate and multiply for 3 days. Infected cells were counted after Diff-Quik staining18 (average of three experiments).
(b) Primary cardiomyocytes were plated in 96-well plates, pretreated with antibodies (1 μg/ml) against TrkC (a-TrkC), (TrkA (a-TrkA) or TrkB (a-TrkB) followed by T. cruzi Colombian strain as in (a); Standard deviation bars were calculated using Student t test, and the results shown here were similar in three other experiments;*, P<0.05, ***, P<0.001, ns, not significant.
(c) Primary cardiomyocytes were pre-treated with the neurotrophins (NTs) NT-3 (TrkC ligand), NGF (TrkA ligand) and BDNF (TrkB ligand) (10 ng/ml, 30 min), infected with T cruzi and the infection quantitated as in (a). Standard deviation bars were calculated using Student t test, and the results shown here were similar in three other experiments;*, P<0.05, ***, P<0.001.
To determine whether T cruzi uses Trk receptors to invade cardiomyocytes, which express all members of the Trk family (TrkA, TrkB and TrkC) (Kawaguchi-Manabe et al., 2007, Meloni et al., 2010, Cai et al., 2006), we pre-incubated primary cultures of cardiomyocytes with neutralizing antibodies against TrkA (α-TrkA), TrkB (α-TrkB), or TrkC (α-TrkC) followed by T cruzi Colombian strain. We found that infection is inhibited by TrkC antibodies (62% ± 2.3 inhibition at 1 μg/ml) but not by TrkA and TrkB antibodies (Fig. 1b), suggesting that T cruzi invades cardiomyocytes preferentially via TrkC. To further explore this point, we pre-incubated cardiomyocytes with NGF, brain derived neurotrophic factor (BDNF), or NT-3 to block their corresponding primary TrkA, TrkB and TrkC receptors, respectively, and then co-cultured the cardiomyocytes with T cruzi. Similar to the selective inhibition of infection by neutralizing TrkC antibody, the TrkC ligand NT-3 (10 ng/ml) potently blocks infection (80% ± 3.1), whereas NGF is much less effective (30% ± 4.2) and BDNF is without effect (Fig. 1c). Knocking down TrkC in primary cardiomyocytes (five cardiomyocyte clones infected each with distinct lentiviral construct encoding short hairpin against TrkC (shTrkC) prevents T cruzi from efficiently invading cardiomyocytes, whereas knocking down TrkA does not significantly affect T cruzi invasion of cardiomyocytes (Fig. 2a), further establishing TrkC as a preferred invasion receptor for T cruzi invasion of cardiac cells.
Figure 2. T cruzi infection of primary cardiomyocytes is inhibited by TrkC knockdown preferentially over TrkA knockdown and by antagonists of Trk autophosphorylation and Trk downstream signaling.

(a) Primary cardiomyocytes (in quadruplicate wells per point) were plated on 96-well plates, and after one day, were infected with lentivirus expressing small hairpin knockdown plasmids for GFP (shGFP), TrkA (shTrkA, four distinct constructs), or TrkC (shTrkC, five distinct constructs). Six days after lentiviral infection, cells were infected with T cruzi Tulahuen strain (2 × 105 /mL) for three hours, washed to remove unattached parasites, the infection allowed to proceed for two days at 37°C, fixed in 4% paraformaldehyde, and stained with DAPI to visualize host and parasite nuclei. Infected cells (>two parasites/cell) were counted in >200 cells/well, and inhibition of infection was calculated relative to control shGFP-infected cells; * p<0.05 relative to control shGFP-infected cells.
(b) Primary cardiomyocytes were pre-treated (30 min) with AG879 (Trk antagonist), U0126 (Erk signaling antagonist) or IGF1-R (insulin-like growth factor-1 receptor antagonist) followed by T cruzi Tulahuen, as in (a); two experiments, similar results.
In addition, we find that invasion is blocked by the Trk autophosphorylation inhibitor AG879 (Ohmichi et al., 1993) and by U0126, an inhibitor of Erk signaling that is activated downstream of Trks (Huang et al., 2003a), but not by a pharmacological inhibitor of insulin-like growth factor-1 receptor (IGF-1R), which does not mediate T cruzi invasion of neuronal cells (de Melo-Jorge et al., 2007) (Fig. 2b). Thus, T cruzi invasion of cardiomyocytes via TrkC requires TrkC signaling.
Selective inhibition of cardiomyocyte invasion by the T cruzi surface protein parasite-derived neurotrophic factor (PDNF)/ trans-sialidase
Given that the T cruzi surface protein PDNF is the counterreceptor for TrkA (de Melo-Jorge et al., 2007) and TrkC (Weinkauf et al., 2011) in invasion of neuronal cells, PDNF may act likewise during T cruzi entry into cardiomyocytes. We find that a bacterially expressed short version of PDNF (sPDNF) (Chuenkova et al., 1999) lacking C-terminal tandem repeat SAPA (Affranchino et al., 1989) effectively blocks cardiomyocyte invasion by Colombian, CL-Brener and Tulahuen strains (Fig. 3a). PDNF has intrinsic neuraminidase activity (Pereira, 1983, Scudder et al., 1993) but this activity may not be responsible for the ability of sPDNF to competitively inhibit cardiomyocyte invasion because pre-treatment of cardiomyocytes with Vibrio cholera neuraminidase (VCN) (same number of units as sPDNF) does not affect invasion (Fig. 3a). In addition, PDNF family member gp85 (Alves et al., 2008) is also inactive in blocking invasion (Fig. 3b), underscoring the specificity of PDNF/TrkC interaction in triggering cardiomyocyte invasion.
Figure 3. T cruzi infection of cardiomyocytes is blocked by PDNF and not by Vibrio cholerae neuraminidase or PDNF family member gp85.
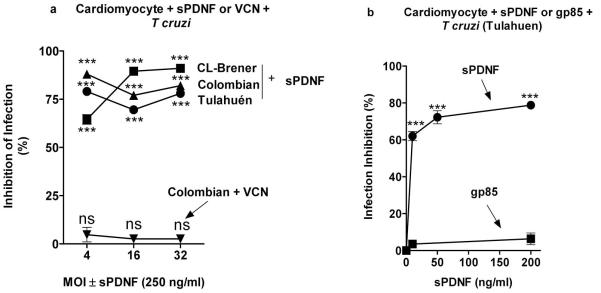
(a) Primary cardiomyocytes (triplicate) were pre-treated with sPDNF (250 ng/ml, 30 min) or Vibrio cholera neuraminidase (VCN, same number of units as PDNF) followed by Colombian, CL-Brener and Tulahuen strains of T cruzi at 4, 16 and 32 MOI for 3 h, washed to remove unattached parasites, and allowed infection to proceed for 2 days. Infection was quantified by light microscopy after Diff-Quik staining, two experiments, similar results.
(b) Primary cardiomyocytes were pre-treated with various doses of sPDNF or gp85 (30 min) followed by T cruzi Tulahuen, as in (a).
Preferential use of surface receptor TrkC for T cruzi via PDNF entry into cardiac fibroblasts
To determine whether T cruzi invades primary cardiac fibroblasts through Trk receptors (Aridgides, 2013) we performed invasion inhibition assays analogous to those described above for cardiomyocytes. As with cardiomyocytes, we find that antibodies against TrkC specifically inhibit T cruzi entry (Fig. 4a), as does the TrkC ligand NT-3 (Fig. 4b), knockdown of TrkC by shRNA as determined with nine distinct lentiviral vectors encoding shTrkC or shTrkA (Fig. 5a), and the Trk autophosphorylation inhibitor K252a (Tapley et al., 1992) (Fig. 5b). These results indicate that T cruzi uses TrkC preferentially over TrkA to invade cardiac fibroblasts, that TrkC must be activated to drive cell entry, and that TrkC-mediated invasion depends on parasite-bound PDNF as judged by the effect of soluble recombinant sPDNF in inhibiting invasion dose-dependently (Fig. 5c).
Figure 4. T cruzi entry into cardiac fibroblasts is specifically inhibited by antibodies against TrkC and by the TrkC ligand neurotrophin-3 (NT-3).
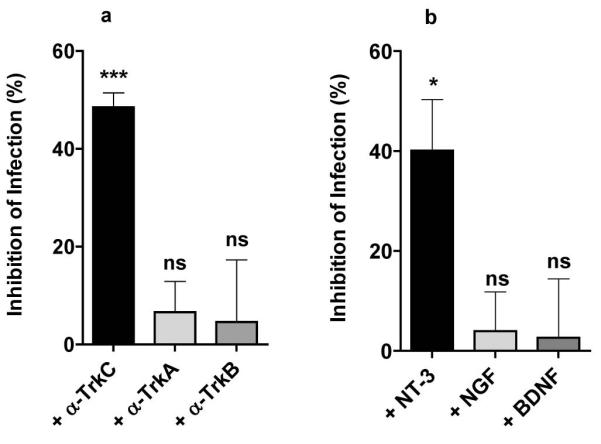
a) Primary cultures of cardiac fibroblasts (in triplicate) were preincubated for 30 min with antibodies against TrkA (α-TrkA), TrkB (α-TrkB), or TrkC (α-TrkC) (1 μg/ml), infected with T cruzi(Tulahuen) for 4 h, washed to remove unattached parasites, allowed to grow for 2 days, and stained with Diff-Quik. Infected cells (more than two parasites per cell) were counted for each well (>200 cells/well); inhibition of infection was computed relative to cells preincubated with medium vehicle; similar results were obtained in three other experiments; **, p<0.001, ns, not significant.
b) Primary cardiac fibroblasts were preincubated (30 min, 37°C) with NGF, BDNF, or NT-3 (50 ng/ml ) prior to T cruzi infection and quantification of infection ascertained as in a); *, p<0.05, ns, not significant.
Figure 5. T cruzi entry into primary cardiac fibroblasts is inhibited by TrkC knockdown, sPDNF and pharmacological inhibitor of Trk autophosphorylation.

(a) Cardiac fibroblasts were infected with lentiviral knockdown vectors against GFP (shGFP), TrkA (shTrkA, four distinct vectors), or TrkC (shTrkC, five distinct vectors) for 7 d before infection, using a protocol otherwise identical to that described in Fig. 2a legend;. *, p<0.05; *** p<0.001; experiments repeated twice with similar results.
b) Primary cultures of cardiac fibroblasts (in triplicate) were preincubated or not with the indicated concentrations of recombinant sPDNF for 30 min prior to T cruzi infection (Tulahuen) for four h. Inhibition of infection was computed relative to cells preincubated with vehicle (PBS); similar results were obtained in three other experiments; ***, p<0.001.
c) Primary cardiac fibroblasts were preincubated (30 min, room temperature) with 250 nM K252a prior to infection; inhibition of infection was calculated relative to infection of fibroblasts pre-incubated with vehicle (DMSO); **, p<0.01.
Recognition of cardiomyocyte-TrkA by T cruzi or recombinant PDNF triggers protection against oxidative stress
As TrkA activation is a bona fide trigger of cell survival and trophic events in neural cells (Huang et al., 2003a) and cardiomyocytes (Caporali et al., 2008), we sought to determine whether T cruzi exploits TrkA for cardiomyocytes protection.
First, to test whether T cruzi infection of cardiomyocytes elicits protective events via PDNF, we pre-incubated T cruzi with the extracellular domain of TrkA (TrkAECD) or (TrkCECD), which, through their neurotrophin-binding sites, bind to and block T cruzi-anchored PDNF (de Melo-Jorge et al., 2007, Weinkauf et al., 2009). T cruzi-bound TrkECD complexes were then co-cultured with cardiomyocytes exposed to a lethal dose of H2O2. This way, and consistently (five experiments, similar results), TrkAECD or TrkCECD prevents cardiomyocyte death as assessed by propidium iodide (PI)/Hoechst 33342 staining (Fig. 6a). In contrast, TrkBECD, which does not bind to PDNF in solution or on the surface T cruzi (Weinkauf et al., 2009), is inactive in preventing H2O2-mediated cardiomyocyte toxicity (Fig. 6a). This indicates that T cruzi infection of cardiomyocytes protects the host cells against oxidative stress through the parasite outer membrane PDNF.
Figure 6. T cruzi infection or sPDNF stimulation of cardiomyocytes confers protection against oxidative stress.

a) T cruzi infection protects cardiomyocytes against H2O2-induced cell death and the protection is blocked by T cruzi interaction with PDNF receptors TrkAECD and TrkCECD but not with PDNF non-receptor TrkBECD. Primary cultures of cardiomyocytes were exposed to 150 μM H2O2 for 4 h following preincubation with vehicle media (H2O2) or with T cruzi (2 × 105/mL) pretreated with vehicle (T cruzi) or with the extracellular domains (ECDs) of TrkA (TrkAECD), TrkC (TrkCECD) or TrkB (TrkBECD) (200 ng/mL, 30 min) to block (TrkAECD and TrkCECD) or not (TrkBECD) GPI-anchored PDNF. Cell death was assessed by counting >200 cells stained with the propidium iodide (PI)/ Hoechst 33342 mixture. Data are combined from five separate experiments after normalization to media alone, each giving results similar to those shown here. Panel on the right shows representative staining from each condition; dead cardiomyocytes are seen a pink dots (the result of the merge of PI and Hoechst staining), while viable cardiomyocytes are stained blue by the vital dye Hoechst 33342 and no PI staining.
b) sPDNF specifically protects primary cardiomyocytes from H2O2-induced cell death. Primary cardiomyocytes were unexposed (Vehicle) or exposed to 150 μM H2O2 for 4 h without (+ H2O2). Prior to H2O2 exposure, cardiomyocytes were preincubated (30 min, room temperature) with sPDNF or with the PDNF/trans-sialidase family member gp85 (150 ng/ml). Prior to H2O2 exposure and sPDNF treatment, cardiomyocytes were additionally preincubated with antibodies against TrkA (α-TrkA IgG) or TrkC (α-TrkC IgG) (1 μg/ mL); **, p<0.001, *, p<0.01; ns, not significant statistically; three experiments, similar results.
c) T cruzi sPDNF protects H9c2 cardiomyocytes from H2O2-induced cell death. H9c2 cardiomyocytes were exposed to 150 μM H2O2 for 4 h with or without preincubation with sPDNF (150 ng/mL); p<0.05, **p<0.01, ***p<0.001; two experiments, similar results.
d) sPDNF protection of cardiomyocytes is selectively abrogated by TrkA knockdown. Primary cardiomyocytes were lentivirally transfected with shRNA to generate four shTrkA clones and five shTrkC clones, and after one week, clones were exposed to 150 ng/mL sPDNF and 500 μM H2O2 for four hours in DMEM/0.1% FCS, and cell death quantified by PI/ Hoechst 33342; *p<0.05; two experiments, similar results.
Second, to directly test the possibility that PDNF triggers cardioprotective events, we sensitized primary cardiomyocytes with recombinant sPDNF (to activate prosurvival Trk signaling) and then challenged the cells with the H2O2 insult. We find that sPDNF treatment rescues cardiomyocytes from H2O2 cell toxicity (Fig. 6b, first three scatter plots) (three experiments, similar results, as it does on the cardiomyocyte cell line H9c2 (Fig. 6c) (two experiments, similar results). sPDNF protection is specific because it does not happen when cardiomyocytes are sensitized with PDNF family member gp85 before exposure to H2O2 (Fig. 6b, first, second, and fourth scatter plots). These findings establish that T cruzi via PDNF protects cardiomyocytes against oxidative stress.
Third, to determine whether T cruzi protects cardiomyocytes against oxidative stress via TrkA, we reacted cardiomyocytes with anti-TrkA IgG or anti-TrkC IgG, and found that only anti-TrkA IgG effectively reverses the protective effect of sPDNF (Fig. 6b, first, second, fifth and sixth scatter plots). This underscores the requirement of cardiomyocyte TrkA for the beneficial effect of sPDNF to take place, the opposite of TrkA usage for cell entry (Fig. 1b). Furthermore, knockdown of TrkA gene expression in primary cardiomyocytes (four clones each infected with lentivirus encoding a distinct short hairpin TrkA construct) annuls sPDNF-induced neutralization of H2O2 cardiotoxicity relative to control cardiomyocytes bearing lentivirus-encoding GFP but without mRNA short hairpins (Fig. 6d). Contrary to TrkA knockdown, interference with TrkC mRNA (five cardiomyocytes clones transfected with different TrkC shRNAs) is much less effective in reversing the protective effect of sPDNF (Fig. 6d).
Discussion
TrkC/NT-3 signaling promotes cardiomyocyte proliferation and ventricular trabeculation in embryogenesis (Lin et al., 2000) and TrkC deletion gives rise to defects in cardiac septation, valvulogenesis, and truncal formation (Tessarollo et al., 1997). However, little is known about the role TrkC signaling plays in cardiomyocyte function in adulthood except that it increases the expression of cardiac hypertrophic markers (skeletal α-actin and atrial natriuretic peptide), cardiomyocytes size, and phenylalanine uptake (Kawaguchi-Manabe et al., 2007). Our results suggest a role for TrkC in Chagas myocarditis as they demonstrate that T cruzi binding to, and activating of, TrkC through its PDNF/trans-sialidase, preferentially over family members TrkA and TrkB, is a prelude to cardiomyocyte invasion, thereby implicating TrkC in T cruzi homing into the heart. Earlier studies showed that activation of transforming growth factor-β (TGF- β) receptor enhances T cruzi entry into cardiomyocyte and other cell types (Ming et al., 1995, Waghabi et al., 2007, Waghabi et al., 2005), as does binding to kinin receptors (Todorov et al., 2003). Although our results suggest that PDNF family member gp85 does not initiate a cascade of events triggering TrkC-dependent cardiomyocyte invasion, independent studies showed that gp85 is important in T cruzi adhesion to laminin and other ECM components (Alves et al., 2008, Mattos et al., 2012).
The idea that T cruzi recognition of TrkC contributes to cardiac homing is further supported by our findings that T cruzi entry into cardiac fibroblast is selectively driven by TrkC-PDNF interaction. Most studies on Chagas heart disease focus on T cruzi recognition of cardiomyocytes and mechanisms underlying T cruzi-cardiac fibroblast interaction are rare to find (Tafuri, 1970, Wong et al., 1992), even though cardiomyocytes require cardiac fibroblasts for proper function either through gap junction-based direct cell-cell interaction or by releasing growth factors and cytokines such as TGF-β, fibroblast growth factor-2, interleukin-6 paracrine signaling (Souders et al., 2009, Kakkar et al., 2010). Cardiac fibroblasts express receptors for growth factors, several in common with cardiomyocytes and some more abundant in the fibroblasts, like angiotensin receptors, which link the renin-angiotensin-aldosterone system to myocardial and extracellular matrix remodeling and which convey paracrine signals to cardiomyocytes via angiotensin secretion (Gray et al., 1998). Results presented here provide a molecular basis for T cruzi entry into cardiac fibroblasts as they show, for the first time, that TrkC is an entry receptor for T cruzi invasion of cardiac fibroblasts, analogous to invasion of cardiomyocytes.
Although TrkA is relatively unimportant for T cruzi entry into cardiomyocytes and cardiac fibroblasts, our results demonstrate that T cruzi interaction with TrkA on cardiomyocytes protects the cells against oxidative stress (Fig. 6). This is in concert with cardiomyocyte-TrkA activation by its natural ligand, NGF, which reduces myocardial infarction (Meloni et al., 2010) and hypoxia/ reoxygenation (Caporali et al., 2008) injury, and with our recent findings that T cruzi-PDNF activation of TrkA in cardiac fibroblasts triggers the secretion of NGF that averts cardiomyocyte damage (Aridgides, 2013).
How might T cruzi-induced cardioprotective events fit in with Chagas heart disease progression? Patients with acute Chagas heart infection present several cardiac dysfunctions including alterations in ventricular repolarization and cardiomegaly due to pericardial effusion (Beltrao Hde et al., 2009, Parada et al., 1997, Pinto et al., 2001, Valente et al., 2009). Paradoxically, acute Chagas myocarditis wanes without sequelae and most patients remain asymptomatic and pathology-free for many years (indeterminate phase) until CCC emerges. But only approximately 30% chagasic patients progress to CCC (Marin-Neto et al., 2007, Rassi et al., 2010, Machado et al., 2012). Thus it may be that the interaction of T cruzi, via PDNF, with cardiac cells in acute infection enhances mutually beneficial host survival mechanisms, as originally suggested in T cruzi recognition of neuronal cells (Chuenkova et al., 2000) and in concert with other beneficial mammalian host/parasite interactions such as suppression of bacterial infection resulting from herpesvirus infection (Barton et al., 2007) and of allergies by worms (Fallon et al., 2007).
Cardiomyocytes are particularly vulnerable to reactive oxygen species due to their high metabolic rate, and oxidative stress is important in Chagas cardiomyopathy (Machado et al., 2012). A T cruzi-driven cardioprotection against oxidative stress such as by direct activation of TrkA on cardiomyocytes (this study) and by paracrine signaling though the stimulation of NGF secretion from cardiac fibroblasts (Aridgides, 2013) fits with the concept of acute heart infection waning without sequelae and with chronic pathology-free infection (indeterminate phase). Consistent with this idea, earlier studies showed that T cruzi can prevent apoptosis in cultured cardiomyocytes via activation of the transcription factor NF-κB (Petersen et al., 2006), an activation that must be distinct from PDNF signaling because NF-κB is not activated by Trks (Huang et al., 2003a).
Given that Maraviroc and other entry receptor antagonists of HIV infection of CD4 lymphocytes are currently used in the treatment of AIDS (Meanwell et al., 2007), and given that sPDNF potently blocks cardiac cell entry of three biologically distinct T cruzi strains, PDNF and/or PDNF fragments may have an opportunity as therapeutics in CCC for which anti-infective therapy in unavailable (Matta Guedes et al., 2012). Furthermore, TrkA-mediated cardioprotective effect should add another dimension for PDNF as therapeutics in CCC.
Experimental procedures
Ethics statement
All mouse work was approved by the Institutional Animal Care and Use Committee of Tufts University School of Medicine and Division of Laboratory Animal Medicine.
Trypanosoma cruzi culture
Parasites were maintained in monolayers of Vero cells. Trypomastigotes, when released from infected cells, were harvested from supernatants by low speed centrifugation (250 xg, 5 min) in a table-top centrifuge to remove any contaminating Vero cells, then at high speed (2500 x g, 10 min) to pellet parasites, which were then resuspended in DMEM/0.1% FCS and counted in a hematocytometer before use.
Cardiomyocyte and cardiac fibroblast preparation
Neonatal (1-3d old) C57BL/6 mice (Jackson Laboratories) were sacrificed by decapitation, hearts dissected, and cardiac cells were prepared as described previously (Aridgides, 2013) according to a procedure modified from Sreejit et al. (Sreejit et al., 2008). Briefly, hearts cut into small cubes and digested with trypsin, separated by differential adhesion to a gelatin-coated substrate, then plated in 20% fetal calf serum and 5% horse serum. Purity of cardiac fibroblasts was assessed by immunofluorescence microscopy using antibodies to vimentin and of cardiomyocytes using antibodies to myosin heavy chain, and used when purity for each cell type was >90%; cardiomyocytes showed a beating phenotype.
In vitro infection assay
Cardiac fibroblasts or cardiomyocytes in 20% FCS/5% horse serum were plated at ~2×104/mL on 1% gelatin-coated 96-well plates and, after incubation for 14 h, medium was changed to DMEM/0.1% FCS. For inhibition of infection assays, cells were pretreated for 30 min with α-Trk antibodies (Abcam), neurotrophins (R&D Systems), Trk chemical inhibitors (Sigma-Aldrich), sPDNF, gp85 (see below), or Vibrio chololera neuraminidase (VCN) (same number of units as sPDNF). Parasites were added at the concentrations indicated in figure legends of figures, and allowed to infect for 3 h, after which non-invaded parasites were washed away twice with PBS and cells were left in DMEM/1% FCS. After two days, cells were fixed with methanol, stained with Diff Quik, and infected cells were counted in each well by microscopy. Inhibition of infection was calculated relative to vehicle-treated cells in each experiment.
Purification of recombinant sPDNF and gp85
sPDNF was cloned into BL21 expression bacteria and purified by Ni-chromatography as previously described (Aridgides, 2013). gp85 (Giordano et al., 1999, Alves et al., 2008) was cloned from CLBrener T cruzi DNA using a nested PCR approach to specifically amplify this single member of a highly conserved gene family (NCBI Red Seq XM_808586). Flanking primers (Forward: 5′GTG GCC TCA CAG TGA TTA AG 3′, Reverse: 5′ACA GGA AGA GTG CGG AAG AA 3′) generated a 3035 base pair product which was then used as a template for PCR to produce the gp85 cDNA product with additional BamHI and XhoI sites (underlined) (Forward: 5′GCA GGA TCC ATG CTC TCA CGT GTT G 3′, Reverse: ATA CTC GAG CGC AGT CGC AAG G 3′). This PCR product was then cloned into the pET-23b vector (Novagen) and transformed into BL21 E. coli for expression. Bacteria were grown in LB medium to an OD600 of 0.7, then induced for 3 h with 200 μM IPTG (isopropyl β-D-1-thiogalactopyranoside). Bacteria were then pelleted, lysed by sonication, and gp85 was purified over a Ni++ column. Purity and concentration were assessed in SDS-PAGE gels after staining with Coomassie brilliant blue.
Lentiviral transfection and shRNA knockdown
Lentiviral vectors (Open Biosystems) encoding shRNA constructs targeting TrkA, TrkC or GFP mRNA were generated from transfected HEK 293 cells according to manufacturer instructions, aliquoted, and frozen at −80°C until use. Lentiviral infection of primary cultures was performed after pretreatment with Polybrene (8μg/mL) (Sigma-Aldrich) then adding lentiviral particles to each well. Optimal concentrations (8 μL vector/100 μL well on 96-well plates) were discerned by preliminary dose-response experiments on cardiac fibroblasts where cells were infected for 7 days, fixed and stained with antibodies specific for TrkA and TrkC (abcam) and assessed by fluorescence microscopy.
Cardiomyocyte survival assay
Primary cardiomyocytes or H9c2 cardiomyocytes (ATCC) were plated on 96-well plates, pre-treated with indicated proteins for 30 min, and hydrogen peroxide or PBS vehicle was added to the cultures for four h. Cells were fixed with 1% paraformaldehyde overnight at 4°C. Cells were stained with Hoechst 33342 (20 μg/mL) and propidium iodide (PI) (10 μL) to visualize live and dead (apoptotic) cells, as we described earlier (Chuenkova et al., 2004). After 5 min, the cells were visualized under ultraviolet irradiation at 340–380 nm with an inverted microscope. DAPI staining was performed as described earlier (Chuenkova et al., 2000). More than 200 cells/well were counted and percent viable cells (Hoechst 33342 positive and PI negative) were calculated based on the percentage of viable cells
Statistics
Statistical analyses were performed using GraphPad Prism software using Student’s t-test or ANOVA with Tukey’s post-test.
Acknowledgements
We thank Dr Ricardo Gazzinelli for the gift of CL-Brener and Colombian strains of T. cruzi. This study was funded by two NIH awards, NS40574 and NS42960.
References
- Affranchino JL, Ibanez CF, Luquetti AO, Rassi A, Reyes MB, Macina RA, et al. Identification of a Trypanosoma cruzi antigen that is shed during the acute phase of Chagas’ disease. Mol Biochem Parasitol. 1989;34:221–228. doi: 10.1016/0166-6851(89)90050-9. [DOI] [PubMed] [Google Scholar]
- Alves MJ, Colli W. Role of the gp85/trans-sialidase superfamily of glycoproteins in the interaction of Trypanosoma cruzi with host structures. Subcell Biochem. 2008;47:58–69. doi: 10.1007/978-0-387-78267-6_4. [DOI] [PubMed] [Google Scholar]
- Araujo-Jorge TC, Waghabi MC, Bailly S, Feige JJ. The TGF-beta Pathway as an Emerging Target for Chagas Disease Therapy. Clinical pharmacology and therapeutics. 2012;92:613–621. doi: 10.1038/clpt.2012.102. [DOI] [PubMed] [Google Scholar]
- Aridgides D, Salvador R, PereiraPerrin M. Preferential use of neurotrophin receptor TrkC for Trypanosoma cruzi entry into cardiomyocytes and cardiac fibroblasts. PLoS One. 2013 (submitting revised manuscript) [Google Scholar]
- Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, et al. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. 2007;447:326–329. doi: 10.1038/nature05762. [DOI] [PubMed] [Google Scholar]
- Beltrao Hde B, Cerroni Mde P, Freitas DR, Pinto AY, Valente Vda C, Valente SA, et al. Investigation of two outbreaks of suspected oral transmission of acute Chagas disease in the Amazon region, Para State, Brazil, in 2007. Tropical doctor. 2009;39:231–232. doi: 10.1258/td.2009.090035. [DOI] [PubMed] [Google Scholar]
- Bern C, Montgomery SP. An estimate of the burden of Chagas disease in the United States. Clin Infect Dis. 2009;49:e52–54. doi: 10.1086/605091. [DOI] [PubMed] [Google Scholar]
- Brener Z. Biology of Trypanosoma cruzi. Annu Rev Microbiol. 1973;27:347–382. doi: 10.1146/annurev.mi.27.100173.002023. [DOI] [PubMed] [Google Scholar]
- Cai D, Holm JM, Duignan IJ, Zheng J, Xaymardan M, Chin A, et al. BDNF-mediated enhancement of inflammation and injury in the aging heart. Physiological genomics. 2006;24:191–197. doi: 10.1152/physiolgenomics.00165.2005. [DOI] [PubMed] [Google Scholar]
- Calvet CM, Melo TG, Garzoni LR, Oliveira FO, Jr., Neto DT, N SLM, et al. Current understanding of the Trypanosoma cruzi-cardiomyocyte interaction. Frontiers in immunology. 2012;3:327. doi: 10.3389/fimmu.2012.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporali A, Sala-Newby GB, Meloni M, Graiani G, Pani E, Cristofaro B, et al. Identification of the prosurvival activity of nerve growth factor on cardiac myocytes. Cell Death Differ. 2008;15:299–311. doi: 10.1038/sj.cdd.4402263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuenkova M, Pereira M, Taylor G. trans-sialidase of Trypanosoma cruzi: location of galactose-binding site(s) Biochem Biophys Res Commun. 1999;262:549–556. doi: 10.1006/bbrc.1999.1154. [DOI] [PubMed] [Google Scholar]
- Chuenkova MV, Pereira MA. A trypanosomal protein synergizes with the cytokines ciliary neurotrophic factor and leukemia inhibitory factor to prevent apoptosis of neuronal cells. Mol Biol Cell. 2000;11:1487–1498. doi: 10.1091/mbc.11.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuenkova MV, PereiraPerrin M. Chagas’ disease parasite promotes neuron survival and differentiation through TrkA nerve growth factor receptor. J Neurochem. 2004;91:385–394. doi: 10.1111/j.1471-4159.2004.02724.x. [DOI] [PubMed] [Google Scholar]
- Chuenkova MV, PereiraPerrin M. Trypanosoma cruzi targets Akt in host cells as an intracellular antiapoptotic strategy. Sci Signal. 2009;2:ra74. doi: 10.1126/scisignal.2000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuenkova MV, PereiraPerrin M. Neurodegeneration and neuroregeneration in Chagas disease. Advances in parasitology. 2011;76:195–233. doi: 10.1016/B978-0-12-385895-5.00009-8. [DOI] [PubMed] [Google Scholar]
- Cunningham AC. Parasitic adaptive mechanisms in infection by leishmania. Exp Mol Pathol. 2002;72:132–141. doi: 10.1006/exmp.2002.2418. [DOI] [PubMed] [Google Scholar]
- de Melo-Jorge M, PereiraPerrin M. The Chagas’ disease parasite Trypanosoma cruzi exploits nerve growth factor receptor TrkA to infect mammalian hosts. Cell host & microbe. 2007;1:251–261. doi: 10.1016/j.chom.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Fallon PG, Mangan NE. Suppression of TH2-type allergic reactions by helminth infection. Nature reviews. Immunology. 2007;7:220–230. doi: 10.1038/nri2039. [DOI] [PubMed] [Google Scholar]
- Fiorelli AI, Santos RH, Oliveira JL, Jr., Lourenco-Filho DD, Dias RR, Oliveira AS, et al. Heart transplantation in 107 cases of Chagas’ disease. Transplant Proc. 2011;43:220–224. doi: 10.1016/j.transproceed.2010.12.046. [DOI] [PubMed] [Google Scholar]
- Giordano R, Fouts DL, Tewari D, Colli W, Manning JE, Alves MJ. Cloning of a surface membrane glycoprotein specific for the infective form of Trypanosoma cruzi having adhesive properties to laminin. J Biol Chem. 1999;274:3461–3468. doi: 10.1074/jbc.274.6.3461. [DOI] [PubMed] [Google Scholar]
- Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovascular research. 1998;40:352–363. doi: 10.1016/s0008-6363(98)00121-7. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003a;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Huang H, Petkova SB, Cohen AW, Bouzahzah B, Chan J, Zhou JN, et al. Activation of transcription factors AP-1 and NF-kappa B in murine Chagasic myocarditis. Infect Immun. 2003b;71:2859–2867. doi: 10.1128/IAI.71.5.2859-2867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakkar R, Lee RT. Intramyocardial fibroblast myocyte communication. Circ Res. 2010;106:47–57. doi: 10.1161/CIRCRESAHA.109.207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi-Manabe H, Ieda M, Kimura K, Manabe T, Miyatake S, Kanazawa H, et al. A novel cardiac hypertrophic factor, neurotrophin-3, is paradoxically downregulated in cardiac hypertrophy. Life Sci. 2007;81:385–392. doi: 10.1016/j.lfs.2007.05.024. [DOI] [PubMed] [Google Scholar]
- Lin MI, Das I, Schwartz GM, Tsoulfas P, Mikawa T, Hempstead BL. Trk C receptor signaling regulates cardiac myocyte proliferation during early heart development in vivo. Dev Biol. 2000;226:180–191. doi: 10.1006/dbio.2000.9850. [DOI] [PubMed] [Google Scholar]
- Machado FS, Dutra WO, Esper L, Gollob KJ, Teixeira MM, Factor SM, et al. Current understanding of immunity to Trypanosoma cruzi infection and pathogenesis of Chagas disease. Seminars in immunopathology. 2012;34:753–770. doi: 10.1007/s00281-012-0351-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Neto JA, Cunha-Neto E, Maciel BC, Simoes MV. Pathogenesis of chronic Chagas heart disease. Circulation. 2007;115:1109–1123. doi: 10.1161/CIRCULATIONAHA.106.624296. [DOI] [PubMed] [Google Scholar]
- Matta Guedes PM, Gutierrez FRS, Nascimento MSL, Do-Valle-Matta MA, Silva JS. Antiparasitical chemotherapy in Chagas’ disease cardiomyopathy: current evidence. Tropical Medicine & International Health. 2012;17:1057–1065. doi: 10.1111/j.1365-3156.2012.03025.x. [DOI] [PubMed] [Google Scholar]
- Mattos EC, Schumacher RI, Colli W, Alves MJ. Adhesion of Trypanosoma cruzi Trypomastigotes to Fibronectin or Laminin Modifies Tubulin and Paraflagellar Rod Protein Phosphorylation. PLoS ONE. 2012;7:e46767. doi: 10.1371/journal.pone.0046767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meanwell NA, Kadow JF. Maraviroc, a chemokine CCR5 receptor antagonist for the treatment of HIV infection and AIDS. Curr Opin Investig Drugs. 2007;8:669–681. [PubMed] [Google Scholar]
- Meloni M, Caporali A, Graiani G, Lagrasta C, Katare R, Van Linthout S, et al. Nerve growth factor promotes cardiac repair following myocardial infarction. Circ Res. 2010;106:1275–1284. doi: 10.1161/CIRCRESAHA.109.210088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming M, Ewen ME, Pereira ME. Trypanosome invasion of mammalian cells requires activation of the TGF beta signaling pathway. Cell. 1995;82:287–296. doi: 10.1016/0092-8674(95)90316-x. [DOI] [PubMed] [Google Scholar]
- Nagajyothi F, Weiss LM, Silver DL, Desruisseaux MS, Scherer PE, Herz J, Tanowitz HB. Trypanosoma cruzi utilizes the host low density lipoprotein receptor in invasion. PLoS Negl Trop Dis. 2012;5:e953. doi: 10.1371/journal.pntd.0000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmichi M, Pang L, Ribon V, Gazit A, Levitzki A, Saltiel AR. The tyrosine kinase inhibitor tyrphostin blocks the cellular actions of nerve growth factor. Biochemistry. 1993;32:4650–4658. doi: 10.1021/bi00068a024. [DOI] [PubMed] [Google Scholar]
- Parada H, Carrasco HA, Anez N, Fuenmayor C, Inglessis I. Cardiac involvement is a constant finding in acute Chagas’ disease: a clinical, parasitological and histopathological study. Int J Cardiol. 1997;60:49–54. doi: 10.1016/s0167-5273(97)02952-5. [DOI] [PubMed] [Google Scholar]
- Pereira ME. A developmentally regulated neuraminidase activity in Trypanosoma cruzi. Science. 1983;219:1444–1446. doi: 10.1126/science.6338592. [DOI] [PubMed] [Google Scholar]
- Petersen CA, Krumholz KA, Carmen J, Sinai AP, Burleigh BA. Trypanosoma cruzi infection and nuclear factor kappa B activation prevent apoptosis in cardiac cells. Infect Immun. 2006;74:1580–1587. doi: 10.1128/IAI.74.3.1580-1587.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto AY, Harada GS, Valente V, Abud JE, Gomes F, Souza GC, Valente SA. [Cardiac attacks in patients with acute Chagas disease in a family micro-outbreak, in Abaetetuba, Brazilian Amazon] Rev Soc Bras Med Trop. 2001;34:413–419. doi: 10.1590/s0037-86822001000500003. [DOI] [PubMed] [Google Scholar]
- Rassi A, Jr., Rassi A, Marin-Neto JA. Chagas disease. Lancet. 2010;375:1388–1402. doi: 10.1016/S0140-6736(10)60061-X. [DOI] [PubMed] [Google Scholar]
- Scudder P, Doom JP, Chuenkova M, Manger ID, Pereira ME. Enzymatic characterization of beta-D-galactoside alpha 2,3-trans-sialidase from Trypanosoma cruzi. J Biol Chem. 1993;268:9886–9891. [PubMed] [Google Scholar]
- Soeiro Mde N, Paiva MM, Barbosa HS, Meirelles Mde N, Araujo-Jorge TC. A cardiomyocyte mannose receptor system is involved in Trypanosoma cruzi invasion and is down-modulated after infection. Cell Struct Funct. 1999;24:139–149. doi: 10.1247/csf.24.139. [DOI] [PubMed] [Google Scholar]
- Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreejit P, Kumar S, Verma RS. An improved protocol for primary culture of cardiomyocyte from neonatal mice. In vitro cellular & developmental biology. Animal. 2008;44:45–50. doi: 10.1007/s11626-007-9079-4. [DOI] [PubMed] [Google Scholar]
- Tafuri WL. Pathogenesis of lesions of the autonomic nervous system of the mouse in experimental acute Chagas’ disease. Light and electron microscope studies. Am J Trop Med Hyg. 1970;19:405–417. doi: 10.4269/ajtmh.1970.19.405. [DOI] [PubMed] [Google Scholar]
- Tapley P, Lamballe F, Barbacid M. K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene. 1992;7:371–381. [PubMed] [Google Scholar]
- Tessarollo L, Tsoulfas P, Donovan MJ, Palko ME, Blair-Flynn J, Hempstead BL, Parada LF. Targeted deletion of all isoforms of the trkC gene suggests the use of alternate receptors by its ligand neurotrophin-3 in neuronal development and implicates trkC in normal cardiogenesis. Proc Natl Acad Sci U S A. 1997;94:14776–14781. doi: 10.1073/pnas.94.26.14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorov AG, Andrade D, Pesquero JB, Araujo Rde C, Bader M, Stewart J, et al. Trypanosoma cruzi induces edematogenic responses in mice and invades cardiomyocytes and endothelial cells in vitro by activating distinct kinin receptor (B1/B2) subtypes. FASEB J. 2003;17:73–75. doi: 10.1096/fj.02-0477fje. [DOI] [PubMed] [Google Scholar]
- Valente SA, da Costa Valente V, das Neves Pinto AY, de Jesus Barbosa Cesar M, dos Santos MP, Miranda CO, et al. Analysis of an acute Chagas disease outbreak in the Brazilian Amazon: human cases, triatomines, reservoir mammals and parasites. Trans R Soc Trop Med Hyg. 2009;103:291–297. doi: 10.1016/j.trstmh.2008.10.047. [DOI] [PubMed] [Google Scholar]
- van Nieuwenhoven FA, Turner NA. The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vascular pharmacology. 2012 doi: 10.1016/j.vph.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Waghabi MC, Keramidas M, Calvet CM, Meuser M, de Nazare CSM, Mendonca-Lima L, et al. SB-431542, a transforming growth factor beta inhibitor, impairs Trypanosoma cruzi infection in cardiomyocytes and parasite cycle completion. Antimicrob Agents Chemother. 2007;51:2905–2910. doi: 10.1128/AAC.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waghabi MC, Keramidas M, Feige JJ, Araujo-Jorge TC, Bailly S. Activation of transforming growth factor beta by Trypanosoma cruzi. Cell Microbiol. 2005;7:511–517. doi: 10.1111/j.1462-5822.2004.00481.x. [DOI] [PubMed] [Google Scholar]
- Weinkauf C, PereiraPerrin M. Trypanosoma cruzi promotes neuronal and glial cell survival through the neurotrophic receptor TrkC. Infect Immun. 2009;77:1368–1375. doi: 10.1128/IAI.01450-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinkauf C, Salvador R, PereiraPerrin M. Neurotrophin receptor TrkC is an entry receptor for Trypanosoma cruzi in neural, glial, and epithelial cells. Infect Immun. 2011;79:4081–4087. doi: 10.1128/IAI.05403-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilen CB, Tilton JC, Doms RW. Molecular mechanisms of HIV entry. Adv Exp Med Biol. 2012;726:223–242. doi: 10.1007/978-1-4614-0980-9_10. [DOI] [PubMed] [Google Scholar]
- Wong WC, Tan CK, Singh M, Yick TY. Ultrastructure of murine cardiac ganglia in experimental Chagas’ disease. Histol Histopathol. 1992;7:371–378. [PubMed] [Google Scholar]