

Abstract
Inflammatory airway diseases such as asthma and chronic obstructive pulmonary disease (COPD) exhibit stereotyped traits that are variably expressed in each person. In experimental mouse models of chronic lung disease, these individual disease traits can be genetically segregated and thereby linked to distinct determinants. Functional genomic analysis indicates that at least one of these traits, mucous cell metaplasia, depends on members of the calcium-activated chloride channel (CLCA) gene family. Here we review advances in the biochemistry of the CLCA family and the evidence of a role for CLCA family members in the development of mucous cell metaplasia and possibly airway hyperreactivity in experimental models and in humans. Based on this information, we develop the model that CLCA proteins are not integral membrane proteins with ion channel function, but instead are secreted signaling molecules that specifically regulate airway target cells in healthy and disease conditions.
Keywords: airway epithelial cell, asthma, chloride channel calcium-activated, mucous cell metaplasia, chronic obstructive pulmonary disease (COPD)
INTRODUCTION
The calcium-activated chloride channel (CLCA) proteins were first isolated in 1991 and have since grown to a complex family that is preserved throughout the animal, plant, and microbial kingdoms. Despite uncertainty over the biological and pathological function of the CLCA family of proteins, they remain as an intriguing target for therapeutic intervention. This practical interest in CLCA proteins was largely confined to the cancer field until new lines of research recognized a connection between CLCA gene expression and the development of inflammatory airway disease both in animal models and in humans with asthma, chronic obstructive pulmonary disease (COPD), and cystic fibrosis. This link between CLCA gene expression and airway disease is particularly interesting because each of these diseases is associated with excess production of airway mucus, and at least some members of the CLCA family are expressed selectively in mucous cells (1–3). Thus, it is possible that the development of mucus hypersecretion in complex airway diseases or even after acute respiratory infection can be marked or perhaps driven by CLCA proteins. The possibility that CLCA controls mucus production is especially relevant to clinical aspects of airway diseases, since mucus hypersecretion is responsible for much of the morbidity and mortality associated with these conditions (4–11). Furthermore, there is limited understanding of the pathobiology underlying the development of hypersecretory diseases, and there are no effective and specific therapeutic strategies to inhibit mucus production or secretion in acute or chronic respiratory disease.
In this review, we provide the background biochemistry that defines the CLCA family, focusing on the genomic organization of the family, and the structure-function relationships of the CLCA proteins. Previous reviews are also available on this topic (12–16), but we will update this information to include more recent genome sequencing and identification of additional CLCA family members. We will then summarize our knowledge of the functional biology of CLCA proteins, focusing on the role of specific CLCA proteins in causing disease. Significant research effort has aimed at the possible activity of CLCA proteins in the control of cell death and the development and spread of cancer, and we will summarize this issue. However, we will concentrate mainly on the role of CLCA proteins in the development of inflammatory airway disease. We devote specific attention to the association of CLCA family members with the inflammatory process that leads to overproduction of airway mucus and the often concomitant development of airway hyperreactivity, since these two disease traits are characteristic of airway disease and are likely responsible for much of the morbidity associated with this type of disease. We review the evidence for CLCA function in animal models of airway disease and in humans with chronic obstructive lung disease in separate sections. We conclude with a perspective on CLCA biology, and a set of questions that need to be addressed for future research on CLCA proteins, since this field is just now poised for definitive understanding and critical therapeutic development.
BACKGROUND BIOCHEMISTRY
In this section, we review the genomic organization of the CLCA gene locus and provide a nomenclature to define all of the members of the CLCA gene family in humans and related animal species. We also review how this genomic information serves as a basis to define the structure of CLCA proteins, and how this structure may relate to function. The information is aimed at providing a background for the subsequent sections on the biological role of CLCA proteins in healthy and diseased tissue.
Genomic organization and gene expression
The initial members of the CLCA family were identified independently in two laboratories based on protein purification and were designated as bovine tracheal calcium-activated chloride channel (CaCC) and bovine lung endothelial cell adhesion molecule 1 (Lu-ECAM-1) (17–19). Subsequent molecular cloning indicated that the two gene products were homologous (13, 20). New nomenclature was developed, and CaCC was named bClca1 and Lu-ECAM-1 was designated bClca2. Since that time, additional Clca gene homologues were discovered in multiple species. In addition, putative orthologues are being discovered in other species as genome sequences are completed. For example, sequencing of the genome for the sea squirt (Ciona intestinalis) led to the description of seven putative CLCA homologues in that species (14). At present, homologues or putative homologues of CLCA family members exist in at least 30 species (Aedes aegypti, Anopheles gambiae, Bos taurus, Canis familiaris, Cavia porcellus, Ciona intestinalis, Ciona savignyi, Danio rerio, Dasypus novemcinctus, Echinops telfairi, Equus caballus, Erinaceus europaeus, Gallus gallus, Loxodonta africana, Macaca mulatta, Microcebus murinus, Mus musculus, Myotis lucifugus, Ochotona princeps, Ornithorhynchus anatinus, Oryctolagus cuniculus, Otolemur garnettii, Pan troglodytes, Pongo pygmaeus, Rattus norvegicus, Sorex araneus, Spermophilus tridecemlineatus, Strongylocentrotus purpuratus, Tupaia belangeri, and Xenopus tropicalis) based on Ensembl GeneTreeView or NCBI Gene database query (21, 22). In many species, the genomic organization of the CLCA locus is highly conserved, with the CLCA genes contained in a single block that preserves the same ordering of CLCA family members (Figure 1). However, it is not yet certain that exon homologies are routinely preserved for noncoding promoter sequences that regulate gene expression.
Figure 1.

A consequence of sequencing multiple species during the discovery of the CLCA gene family is an inconsistent nomenclature for the CLCA genes. A more unified nomenclature scheme was proposed to resolve some of these inconsistencies (12), however, additional homologues and renaming have occurred since that time. Two comprehensive reviews of CLCA cloning and sequencing have been recently published (14, 16). Here we have updated the nomenclature for selected species (human, mouse, bovine, rat, and horse) that are especially relevant to airway disease (Table 1). This nomenclature is based on structural homology for coding sequences (Figure 2). We have preserved the CLCA designation for this gene family, although this approach may have to be revised in the future if CLCA proteins prove to function as signaling molecules instead of ion channels.
Table 1.
Unified nomenclature for CLCA family members. Gene assignments were based on analysis of human, mouse, cow, rat, and horse coding sequences with respect to the closest mouse homologue.
| CLCA Name | Original Name(s) | Reference | GenBank Accession (22) |
|---|---|---|---|
| hCLCA1 | hCaCC-1 | (28, 129) | NM_0001285 |
| hCLCA2 | hCaCC-3, CaCC | (38, 129) | NM_006536 |
| hCLCA3 | (38) | NM_004921 | |
| hCLCA4 | hCaCC-2 | (129) | NM_012128 |
| mClca1 | (34, 130) | NM_009899 | |
| mClca2 | (131) | NM_030601 | |
| mClca3 | gob-5 | (132) | NM_017474 |
| mClca4 | (35) | NM_139148 | |
| mClca5 | (88) | NM_178697 | |
| mClca6 | (88) | NM_207208 | |
| mClca7 | AI747448 | (23) | NM_0001033199 |
| mClca8 | EG622139, A730041H10Rik | (23) | NM_001039222 |
| bClca1 | LOC507643 | (22, 133) | XM_865947 |
| bClca2 | LOC784768 | (20) | XM_001252288, U36445 |
| bClca3 | CaCC, bClca2 | (134) | AF001261-AF001264, NM_181018 |
| bClca4 | (22, 133) | NM_001034300 | |
| rClca1 | Rbclca2 | (135) | AB256513, XM_001063517 |
| rClca2 | Rbclca1 | (26, 136) | NM_001077356 |
| rClca3 | rClca3_Pred, rClca1† | (22, 127) | NM_001107449 |
| rClca4 | rClca6, Prp3 | (22, 127) | NM_201419 |
| rClca5 | rClca2_Pred, rClca2† | (22, 127) | NM_001107450 |
| eClca1 | (25) | NM_001081799 | |
| eClca2 | (22, 137) | XM_001496218 | |
| eClca3 | LOC100052500 | (22, 118, 119, 137) | XM_001495998 |
| eClca4 | (22, 137) | XM_001494684 |
Figure 2.

The human CLCA (hCLCA) locus consists of four genes located on the short arm of chromosome 1 (1p31-p22), a region containing no other known genes (14). These hCLCA genes were identified using homology screens for bClca1 and bClca2 sequences (14). Similarly, there are four Clca genes in the cow and horse genomes, and five Clca genes in rats. In contrast to these species, the mouse Clca (mClca) locus consists of eight genes located on chromosome 3 (23). The basis for the extra Clca genes in the mouse is uncertain, but the situation appears to lead to at least some degree of functional redundancy (24). One member of the human and mouse CLCA gene family (hCLCA3 and mClca8, respectively) may represent pseudogenes since they contain premature stop codons and may not be expressed at detectable levels. However, there are conflicting views on the level of expression for these two genes, so it is still possible that they exert some type of biological function under some conditions.
The pattern of expression of the CLCA genes in different tissues was determined soon after gene identification and was perhaps the first insight into the functional biology of CLCA proteins. Each hCLCA gene has a distinct pattern of expression based on mRNA detection in healthy human tissue (Table 2). Similar to the case for human CLCA genes, the mouse Clca family members are expressed differentially across tissues (Table 3). Of the four horse Clca (eClca) genes, only eClca1 has been characterized to any extent, and it is expressed in the nasal epithelium, trachea, subtracheal glands, mucous (goblet) cells of the airway epithelium, sweat glands, kidney, small intestine, and colon (25). The pattern of CLCA expression, particularly at the protein level still needs to be fully determined. Similarly, there is limited information on the types of cells that express CLCA proteins. However, as further developed in the next section on inflammatory airway disease, the initial analysis indicates that specific CLCA family members (e.g., hCLCA1 in humans and mClca3 in mice) exhibit increased expression in airway mucous cells and are therefore associated with conditions that manifest mucus production.
Table 2.
Tissue distribution of hCLCA gene expression. Levels of expression were determined by detection of mRNA species for the corresponding hCLCA gene.
| Gene | Tissue | Reference |
|---|---|---|
| hCLCA1 | Intestinal goblet cells and basal crypt epithelial cells | (34) |
| airway mucous cells | (58) | |
| Uterus, stomach, testis, kidney, fetal spleen | (129) | |
| Brain | (138) | |
| hCLCA2 | Trachea, breast | (47) |
| Trachea, uterus, prostate, testis, kidney | (129) | |
| Conjunctival and corneal epithelium | (139) | |
| Nasal epithelium | (140) | |
| Cornea, skin, vagina, esophagus, larynx | (141, 142) | |
| hCLCA3 | Lung, trachea, breast, spleen, thymus | (38) |
| hCLCA4 | Brain, colon, bladder, uterus, prostate, stomach, testis, salivary gland, breast, small intestine, appendix, trachea | (129) |
| Nasal epithelium | (140) |
Table 3.
Tissue distribution of mClca gene expression. Levels of expression were determined by detection of mRNA species for the corresponding mClca gene.
| Gene | Tissue | Reference |
|---|---|---|
| mClca1 | Lung, aorta, spleen, bone marrow, lymph nodes, brain | (143) |
| Kidney, skin, liver, spleen | (130) | |
| Intestine, cecum, brain, dorsal root ganglion | (23) | |
| Breast | (48) | |
| mClca2 | Breast | (48, 131) |
| Thymus, colon, small intestine, bladder, epididymis, vesicular gland, skin, breast | (143) | |
| Intestine, cecum, dorsal root ganglion | (23) | |
| mClca3 | Stomach, small intestine, colon, uterus, trachea; limited to mucous cells | (132) |
| Lung, stomach, small intestine, colon, uterus, trachea; limited to mucous cells | (59) | |
| airway mucous cells | (52) | |
| mClca4 | Colon, small intestine, bladder, stomach, esophagus, uterus, skeletal muscle, heart, aorta, lung; endothelial, smooth muscle, and epithelial cells | (35) |
| mClca5 | Eye, spleen, heart, intestine, lung, skeletal muscle, stomach, testis | (88) |
| Dorsal root ganglion | (23) | |
| mClca6 | Intestine, stomach, eye, spleen | (88) |
| Small and large intestine; Non-goblet cell enterocytes | (33) | |
| mClca7 | Intestine and cecum | (23) |
| mClca8 | No transcripts detected | (23) |
Protein structure and function
The proposed structural organization of CLCA proteins has undergone considerable revision in recent years. The initial structural models were based on the observation that the bClca1 protein appeared to regulate calcium-dependent chloride conductance in tracheal epithelial cells (20). The proposal that CLCA proteins may therefore function as ion channels was further supported by experiments in which transfection of 293T cells with several different CLCA isoforms from various species (human, mouse, rat, pig, and cow) were all found to produce a chloride current in response to calcium ionophores or calcium release from the endoplasmic reticulum (16, 26), a process involving calcium/calmodulin-dependent protein kinase activity (27). Subsequent theoretical and experimental studies using hydropathy analysis and epitope insertion in concert with immunohistochemistry produced several different models of transmembrane topology with as many as five transmembrane spanning segments (16). However, these models predicted a transmembrane pass within the common von Willebrand factor type A (VWA) domain, a structurally conserved soluble domain. In addition, some of the epitope insertion experiments likely resulted in misfolding of the protein since the hallmark proteolytic cleavage was blocked (28). Thus, even at this early stage of analysis, there were serious inconsistencies in the proposed model for CLCA proteins as transmembrane ion channels.
Recent experimental results and improved sequence analysis tools (including hidden Markov models for transmembrane segments and protein fold prediction) have led to a new model for CLCA structure. A revised scheme for the domain structure for mouse and human CLCA proteins is provided in Figure 3, and an annotated analysis of amino acid sequence for these proteins is provided in Figure 4. This updated analysis implies that the CLCA proteins are soluble secreted molecules (29–31) with the exception of a subset of CLCA proteins that contain a C-terminal membrane-anchoring region (32, 33). This anchoring region takes the form of a transmembrane alpha-helix or GPI anchor that lacks the structural requirements to function as an ion channel itself (15, 29). A feature common to the CLCA family of proteins is the presence of a proteolytic cleavage site that is located approximately 240 amino acids from the C-terminus. To date, all of the CLCA isoforms that have been tested in mammalian cell culture appear to be processed similarly (29–37). In each case, a precursor 120-kDa glycoprotein is cleaved to produce approximately 85-kDa N-terminal and 35-kDa C-terminal products, both of which contain numerous N-linked glycosylation sites (30, 33, 38). For hCLCA1, mClca3, and mClca4 (which have no identifiable transmembrane region), both the N- and C-terminal products are secreted into the media when expressed in human cell lines (29–31). In similar experiments with hCLCA2 (which has a transmembrane region), the N-terminal product is released into the media while the C-terminal product remains cell surface associated (32). A recent report indicates that mClca4 contains luminal motifs that control its trafficking out of the endoplasmic reticulum (31). Mutation of the forward-trafficking motifs led to trapping of the full-length protein in the endoplasmic reticulum, implying that cleavage occurs after exit from this site. This sequence of events is consistent with the observation that full-length hCLCA1 (as well as the N- and C-terminal cleavage products) is found secreted into the media of hCLCA1-expressing HEK293 cells (29), implying that cleavage might occur outside the cell.
Figure 3.

Figure 4.

One proposal for the nature of CLCA cleavage rests on an analysis of protein sequence. The N-terminal 280 amino acids comprise a cysteine-rich domain conserved only in the CLCA family. The function of this domain is unknown, however a recent bioinformatics analysis suggested that this region contains a zinc metalloprotease domain (39). This finding raised the possibility that the CLCA proteins may self-cleave as part of normal processing. Furthermore, expression of an hCLCA1 mutant in which a predicted critical active site glutamate was changed to glutamine totally abolished cleavage. However, in these experiments only the cell lysates (and not supernatants) were analyzed, so the mutant protein may have been misfolded. Therefore, the precise nature of the process for cleavage of CLCA proteins still needs to be defined. The function of the CLCA protein cleavage remains uncertain, particularly since a specific cleavage site has not yet been precisely identified and characterized. Defining the site and mechanism for CLCA cleavage is critical to defining the function of the mature CLCA peptides.
Directly adjacent to the nCLCA domain is a VWA domain, a widely distributed protein-protein interaction domain (40). The VWA domains commonly engage proteins by coordinating a divalent cation at the binding interface using a highly conserved set of five residues known as a metal-ion dependent adhesion site (MIDAS). All of the CLCA VWA domains contain an intact MIDAS with the exception of hCLCA2 and the homologous mClca5 (Figure 4). Ironically, the VWA domain of hCLCA2 was the first CLCA domain for which a binding partner was identified and it was found to be a beta4-integrin (41). This binding does not depend on MIDAS, but instead involves the consensus sequence F(S/N)R(I/L/V)(S/T)S found in some CLCA proteins and the specificity-determining loop (SDL) of beta4-integrin (41–43). The hCLCA2, mCLCA1, and mCLCA2 proteins contain two of these beta4-binding motifs (Figure 4). This interaction has been observed to promote adhesion of hCLCA2 expressing cell lines to tumorigenic cell lines with upregulated beta4-integrin expression and may be of importance in certain cancer metastases and colonizations. Furthermore, it has been hypothesized that the binding of hCLCA2 to beta4-integrin initiates a novel signaling cascade that promotes tumor invasion.
In addition to cleavage sites and VWA interaction domains, we find that the remaining region C-terminal to the VWA domain of CLCA proteins is rich in beta-sheet structure as determined by a secondary structure prediction using JPRED 3 software (44). This region includes an easily detectable fibronectin type-III (FnIII) domain. FnIII domains are protein-protein interaction modules of the immunoglobulin (Ig) superfamily. No function or binding partners have been identified for this portion of any CLCA family member. Taken together, this updated sequence analysis and recent experimental data indicate that the CLCA proteins cannot act in solo as ion channels and instead imply the revised view that they interact with other proteins in a possible signaling capacity.
MODELS OF INFLAMMATORY AIRWAY DISEASE
In this section, we review cell and animal models that were studied to define the role of CLCA proteins in the development of inflammatory airway diseases. However, before work on the function of CLCA in the lung was even begun, several reports suggested that CLCA proteins might have a role as tumor suppressors. For example, studies of melanoma in mice indicated that the pattern of metastasis correlated with sites of Clca expression, and that anti-Clca antibodies blocked metastasis to the lung (19, 45). Furthermore, mice immunized with purified bClca2 developed resistance to metastasis (46). Other studies showed that human breast cancer cells lacked the usual expression of hCLCA2 expression and overexpression of hCLCA2 decreased the capacity of these cells for invasion and tumor generation in nude mice (47). Similarly, mouse breast cancer cells expressed decreased amounts of mClca1, mClca2, and mClca5, and re-expression or overexpression of these proteins decreased cell growth and increased sensitivity to apoptosis (36, 48). Related work indicated that interaction between hCLCA2 on endothelial cells and its beta-4 integrin ligand on breast cancer cells might mediate more aggressive tumor behavior (41, 43, 49). Similarly, CLCA family members with functional beta-4 integrin binding domains may use this mechanism to activate focal adhesion kinase signaling to extracellular signal-regulated kinase (ERK) with the potential to regulate cell survival (42, 43). Decreased hCLCA2 expression is also observed in breast cancer specimens, and this decrease appears to be due to DNA methylation (50). Recent work also shows that the hCLCA2 gene may be deleted in mantle cell lymphoma specimens (51). These studies highlight the possible role of hCLCA2 as a tumor suppressor, and set the stage for studies of hCLCA1 in inflammatory airway disease. A connection between the activities of CLCA proteins as tumor suppressors and immune modifiers has not been examined.
Isolated cell models
Studies of isolated cells were perhaps the first to develop a role for CLCA proteins in immunity. In particular, overexpression of mClca3 or hCLCA1 in a human mucoepidermoid cell line NCI-H292 causes a significant increase in mucin production (52, 53). These findings infer that CLCA proteins might regulate the level of mucus production and the consequent efficiency of mucociliary clearance of particulate and microbial pathogens. Using this cell model, investigators explored the possibility that CLCA proteins worked as ion channels to somehow influence mucus production. For example, investigators used niflumic acid (a drug that was purported to be a specific inhibitor of chloride channel activity) to block the increase in mucin production found in NCI-H292 cells that expressed hCLCA1 (53). In related work, another group showed that niflumic acid suppressed ATP-mediated exocytosis of mucin in HT29-Cl.16E cells (54). Similarly, McNamara and colleagues found that adenosine induction of mucin biosynthesis depended on transactivation of EGF receptor, and that niflumic acid inhibited this type of transactivation (55). Other work with the niflumic acid derivative MSI-2216 also demonstrated inhibition of mucus production (56, 57). These studies inferred a role for CLCA proteins in the signaling pathway leading to mucin synthesis and secretion, but provided no direct evidence for how CLCA proteins might be involved in this process. Since structural analysis now indicates that CLCA proteins cannot be membrane ion channels themselves, there is a need for more detailed study for how CLCA proteins might control the development of mucous cells.
Although some progress has been made in establishing a role of hCLCA1 in mucous cell metaplasia in isolated cell models, there is little information on CLCA function in airway smooth muscle cultures. It is possible that CLCA proteins may influence airway smooth muscle behavior, since antisense oligonucleotides that knock down mClca expression appeared to inhibit airway hyperreactivity in ovalbumin-challenged mice (52). The available reports indicate that hCLCA1 (and the homologous mClca3) are localized to mucin granules of mucous cells in the lung, gastrointestinal tract, and uterus and are not expressed in airway smooth muscle (28, 34, 58, 59). However, mClca4 is expressed at significant levels in airway smooth muscle in mice (35). In addition, calcium-activated chloride flux is found in smooth muscle of the vasculature, urethra, and lymphatics (60–62). Because at least some CLCA/Clca proteins may depolarize cellular membranes (15), it is possible that CLCA expression in airway smooth muscle could lead to bronchoconstriction. Similar to the case for mucous cells, CLCA function in smooth muscle cells still needs to be defined.
Animal models
Perhaps the best evidence for a biologic role of Clca proteins has been developed in animal models of inflammatory airway disease. In this case, a series of studies have established a link between the expression of Clca proteins in the lung and the development of inflammatory airway disease. One of the initial reports came from a study of a mouse model of allergic asthma that depends on sensitization and subsequent inhalation challenge with ovalbumin. Using this model, it was reported that mClca3 appeared to regulate mucous cell metaplasia and airway hyperreactivity (52). Under these conditions, the development of mucous cell metaplasia and airway hyperreactivity was inhibited by administration of a full-length mClca3 antisense oligonucleotide delivered with an adenoviral vector system. These investigators did not recognize that this full-length construct might also interact with shared sequence homologies in other mClca family members. Nonetheless, expression of mClca3 in the airway epithelium was sufficient to cause mucous cell metaplasia and airway hyperreactivity in these animals. This finding was consistent with the evidence that mClca3 expression was sufficient to drive mucin production in NCI-H292 cells (52, 53).
In addition to these studies of allergic asthma in mice, another group studied the role of Clca proteins in the development of an allergic asthma-like illness in horses. This species develops a disease of episodic recurrent airway obstruction (also known as heaves) that also features mucous cell metaplasia and airway hyperreactivity (63–65). The pathogenesis of this equine disease also involves Th2 cytokine production (especially IL-4 and IL-13) (66). Furthermore, like the airway disease that develops in mice and humans, Muc5ac is the predominant airway mucin that is expressed during the development of mucous cell metaplasia (67). Thus, the disease in horses manifests the characteristic features of human asthma and COPD, as well as the allergen challenge and viral infection mouse models of chronic airway disease (68). Therefore, it appeared likely that equine Clca proteins would also be involved in the pathobiology of mucous cell metaplasia of the asthma-like disease that develops in horses. Indeed, investigators report a significant increase in eClca1 expression in the airway epithelium of horses with equine recurrent airway obstruction (25, 69).
In a distinct approach to understanding the basis for inflammatory airway disease, our lab also came to evaluate the role of the Clca gene family (24). Our group aimed to genetically segregate mucous cell metaplasia from airway hyperreactivity in a mouse model, and thereby identify a molecular pathway that might be associated with a single disease trait. Others have also used inbred mouse strains to define a genetic influence on airway disease, but this work has generally concentrated on the development of airway hyperreactivity (70–78). In the usual case, these previous studies were directed at defining a genetic locus for a single quantitative trait rather than segregating one trait from another in a complex phenotype. Moreover, the usual allergen challenge model of airway disease did not often account for the chronic nature of the phenotype. In view of the well-described role of respiratory syncytial virus (RSV) as well as other viruses in chronic airway disease (79–84), we developed a mouse model of viral bronchiolitis. The infectious agent used in this model is Sendai virus (SeV), which is a mouse parainfluenza virus that is similar to other paramyxoviruses that more commonly infect humans. However, mice are relatively resistant to infection with human pathogens such as RSV. By contrast, SeV replicates at high efficiency in the mouse lung, and SeV infection causes injury and inflammation of the small airways (i.e., bronchiolitis) that is indistinguishable from the comparable condition in humans. This acute response is followed by a delayed but permanent switch to chronic airway disease that is characterized by mucus production (mucous cell metaplasia) and increased airway reactivity to inhaled methacholine (airway hyperreactivity) (24, 85, 86). We came to recognize that both of these traits are inducible on a long-term basis after viral bronchiolitis in the C57BL/6J strain of inbred mice (86). In contrast, we found that the Balb/cJ strain responded similarly during the acute infection but then failed to develop any chronic airway disease (24).
We took advantage of this difference in genetic susceptibility to develop an F2 intercross population with phenotypic extremes that exhibit one or the other disease trait, and analyzed these extremes for gene expression using oligonucleotide microarrays (24). This combined genetic and genomic strategy provided evidence of a selective association between expression of a member of the mClca gene family, i.e., mClca3, with the development of mucous cell metaplasia but not airway hyperreactivity. In support of the relationship between mClca3 expression and mucous cell metaplasia, we also found that transfer of the mClca3 gene into the mouse airway (using an adeno-associated viral vector) was sufficient to produce mucous cell metaplasia, but did not cause airway hyperreactivity (24). We next generated a mouse that was homozygous for a targeted null mutation of the mClca3 gene (mClca3−/−). However, mClca3−/− mice developed the same degree of mucous cell metaplasia (and airway hyperreactivity) as wild-type mice, both in viral and allergen-challenge models of chronic airway disease (24).
Since mClca3 gene expression was sufficient but not necessary for the development of mucous cell metaplasia, we reasoned that another mechanism must also be capable of mediating the chronic change in airway behavior. We specifically questioned whether another mClca gene might be responsible for such an effect. Accordingly, we used the BLAST (87) program to search for homologues of the mClca3 gene and consequently uncovered four additional mClca genes flanking mClca3 on mouse chromosome 3. We tentatively named these genes mClca5, mClca6, mClca7, and mClca8; and subsequently, these genes were given the same designations (23, 88), with homology to human CLCA family members as shown in Figure 2. Because other mClca family members are also expressed in airway tissue (88, 89), they may compensate for the loss of mClca3 and thereby allow for the development of mucous cell metaplasia in the setting of mClca3 deficiency (24). Indeed, we recently showed that mClca family members may exhibit functional redundancy in the development of inflammatory airway disease. In particular, mClca5 expression was also increased in concert with mucous cell metaplasia and airway hyperreactivity after viral infection (24). In addition, mClca5 gene transfer to the airway epithelium was sufficient to cause mucous cell metaplasia but not airway hyperreactivity (24). Preliminary studies indicate that mClca6 expression in the airway epithelium is also sufficient for mucous cell metaplasia but not airway hyperreactivity (90).
Another group also recognized that mClca3 deficiency alone does not influence the development of mucous cell metaplasia after allergen challenge, but their analysis of genetic compensation was limited to expression levels of mClca1, mClca2, and mClca4 (91). In fact, our work indicates that mClca5, mClca6, and mClca7 are more suitable candidates for compensatory function. Given the extensive sequence homology for the mClca family members, it is likely that mClca3 antisense oligonucleotides might bind and inhibit several family members. Under these conditions, mClca gene knockdown may inhibit the induction of mucous cell metaplasia and airway hyperreactivity after viral infection, but this possibility still needs to be formally tested.
The precise signals that regulate mClca gene expression in mouse models of inflammatory airway disease are also under study. Most studies show that Th2 cytokines (IL-4, IL-9, and IL-13) will drive increased expression of CLCA proteins in cell culture (53, 92–94). However, at least one group does not find this relationship (94, 95), suggesting that the state of the cultured cells may influence the responsiveness to cytokine. For example, the degree of cell differentiation could influence the expression of cytokine receptor or receptor signaling function. Indeed, IL-13 receptor expression and signaling function may be a critical determinant of mucous cell metaplasia after viral infection (96). In that regard, the Stat6 transcription factor that transmits the signal from IL-4 and IL-13 receptors is also required to upregulate CLCA gene expression (93). Similarly, increased Clca expression develops in concert with increased IL-13 expression in virus- and allergen-induced mouse models of airway diseases (24, 52, 91, 96, 97). Moreover, increased Clca expression develops after IL-13 instillation into the lung and in IL-13 transgenic mice (93, 94, 98, 99). Thus, Th2 cytokine signals serve to regulate CLCA gene expression under a variety of conditions. This stimulatory effect of IL-13 may depend on Stat6-binding sites in the CLCA gene promoter, but the precise biochemical mechanism still needs to be defined.
INFLAMMATORY AIRWAY DISEASE IN HUMANS
In this section, we review the evidence of a role for CLCA proteins in inflammatory airway disease in humans. We concentrate on the two most common types of chronic airway disease, asthma and COPD, but we include reports of CLCA action in other diseases that also manifest mucus hypersecretion and airway hyperreactivity.
CLCA Polymorphism as Genetic Susceptibility Factor
Several lines of evidence have linked CLCA gene variation to the development of inflammatory airway disease in humans. In particular, two separate studies have used quantitative trait locus (QTL) linkage analyses of large cohorts of COPD patients to track the influence of CLCA genes on airway disease. Each of these studies found that lung function (marked by the degree of abnormality in FEV1/FVC ratio) is linked to genetic change in a region of human chromosome 1 containing the CLCA family locus (100, 101). In addition, specific variations in the hCLCA1 gene have been linked to susceptibility to airway disease. Thus, a haplotype analysis of single nucleotide polymorphisms (SNPs) in the hCLCA1 gene observed linkage to childhood and adult asthma as well as COPD in a Japanese population (102, 103). Analysis of patients with mild forms of cystic fibrosis (that maintains residual gastrointestinal chloride current but still manifests increased gastrointestinal and airway mucus accumulations) also detected linkage with genetic variations in the CLCA gene locus (104). Other investigators have shown that intestinal mucous secretion is improved when mClca3 expression is restored in mouse models of cystic fibrosis (37, 105), suggesting that a physiologic level of Clca function is necessary for normal intestinal as well as airway mucous cell function.
CLCA Expression as a Biomarker
In studies of human tissue, several studies have shown that the levels of hCLCA1 mRNA and protein are significantly increased in the airway epithelium of asthmatic patients (58, 106–108). Preliminary studies suggest that increased levels of hCLCA1 protein can also be detected in bronchoalveolar lavage (BAL) fluid as cleaved N-terminal and C-terminal polypeptides in these types of patients (109). Upregulation of hCLCA1 has also been observed in airways of cystic fibrosis patients with high levels of mucus production (110, 111). Furthermore, in patients with COPD, lung samples may contain increased levels of hCLCA1 as well as hCLCA2 and hCLCA4 (108, 112, 113). Thus, hCLCA1 and perhaps hCLCA2 and hCLCA4 expression appear to be useful biomarkers of inflammatory airway disease. Because of the difficulties in accurately monitoring mucus production or mucous cell metaplasia in vivo, it is therefore possible that measurements of CLCA mRNA or protein may serve as a useful biomarker for quantifying this disease trait in the setting of airway inflammation. Indeed, one study of asthma patients already suggests that hCLCA1 mRNA levels may decrease after treatment with inhaled glucorticoids (107), and a preliminary report suggests that hCLCA1 levels may track with mucous cell metaplasia following withdrawal of glucocorticoid treatment (109). However, additional studies are needed to validate the use of CLCA mRNA or protein levels as a diagnostic or prognostic marker of activity of inflammatory airway disease.
CLCA Proteins as Therapeutic Targets
In addition to use as a biomarker, CLCA proteins (and particularly hCLCA1) may also be useful as specific therapeutic targets in the treatment of inflammatory airway disease, or even in general for controlling excessive mucous secretions. As noted above, one group has supported this approach by showing that knockdown of mClca gene expression using antisense directed against mClca transcripts will block the development of mucous cell metaplasia and airway hyperreactivity after ovalbumin allergen challenge in mice (52). However, this report has not yet been confirmed in this mouse model or other models, and there are no ongoing efforts to apply this strategy to humans with airway disease.
In addition to genetic knockout and knockdown strategies, other groups have pursued the potential for small molecules to inhibit CLCA function. In particular, niflumic acid and its derivatives were proposed initially as CLCA inhibitors. In support of this possibility, one group has shown that niflumic acid can inhibit mucous cell metaplasia induced by hCLCA1 expression or IL-13 administration (53). These findings led to the pursuit of talniflumate (a niflumic acid derivative) as a potential therapy for overproduction of mucus in airway disease. Preclinical data in a mouse model of cystic fibrosis indicated that talniflumate improved survival by decreasing gastrointestinal obstruction caused by mucus (114). In addition, a phase II study of the drug found no toxicity in patients with cystic fibrosis (115). Talniflumate was also being considered for treatment of asthma and COPD, but it appears that drug development was eventually halted (115, 116). Another niflumic acid derivative, MSI-2216, was also found to inhibit mucous secretion both in airway cell lines as well as primary-cultures of upper airway epithelial cells, but further development of this drug was also stopped (56, 57).
Some of the likely problems for drug development of CLCA inhibitors have been the issues of drug specificity and mechanism of action. In the case of niflumic acid, the drug may block multiple members of the CLCA family rather than a single member (13, 88). This effect was attributed to inhibition of a common ion channel function among family members. However, this mechanism became less likely when it was recognized that CLCA proteins were unlikely to function as calcium-dependent chloride channels. Moreover, subsequent studies suggested that niflumic acid and its derivatives may act by directly or indirectly blocking activation of the Stat6 transcription factor (93). The niflumic acid class could thereby block the actions of IL-13 on mucus production, since at least part of the IL-13 receptor signaling travels through Stat6 to drive downstream expression of mucin genes. In any case, it is reasonable to conclude that more precise definition of the molecular mechanism for CLCA action will be needed to rationally develop specific inhibitors.
CONCLUSION
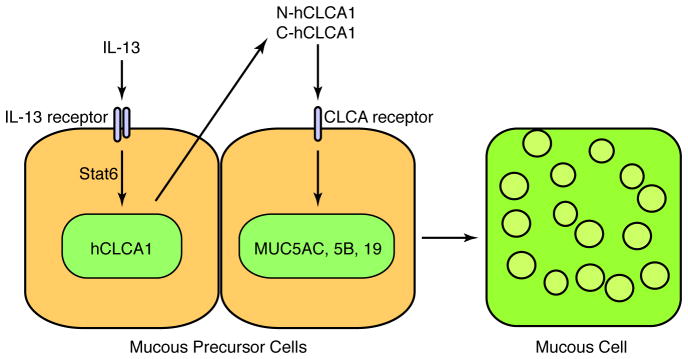
In sum, there is a critical clinical need for better understanding and treatment of inflammatory airway disease in general, and mucous cell metaplasia in particular. Although work is at an early stage, several lines of evidence already suggest that CLCA proteins (particularly hCLCA1) may be master regulators of mucin gene expression in humans. We have gradually developed the proposal that the airway maintains a special program for host defense and that changes in this same program may lead to airway disease. The same argument can be made for mucus secretion. Thus, under normal circumstances a physiologic level of mucus production allows for mucociliary clearance of particulate matter from the airway and aids in defense against respiratory pathogens. However, if mucus secretion is excessive, it leads to airway obstruction and consequent dysfunction. In both normal and abnormal conditions, there appears to be an immune axis that drives cytokine production, and in turn, a downstream mechanism that translates the cytokine signal into mucin gene expression. In humans, this downstream mechanism features hCLCA1, the first member of a newly defined family of “chloride channel calcium-activated” (CLCA) proteins in humans. It is possible that additional CLCA family members may mediate airway hyperreactivity, but this issue requires further study. In either case, the mechanism for how hCLCA1 might signal to cause mucin gene expression and mucous cell metaplasia is a goal of ongoing studies. Initial analysis of CLCA structure and function suggests that these proteins are secreted and cleaved into fragment polypeptides. We are particularly focused on defining the possibility that hCLCA1 is a secreted protein that is cleaved into an active polypeptide that in turn binds to a receptor on the cell surface and signals to activate mucin gene expression (Figure 5). This scheme for hCLCA1 (and orthologous mClca3,5,6) control over epithelial behavior can be tested in isolated airway epithelial cells as well as mouse models and then in patients with mucous cell metaplasia due to asthma or COPD or other inflammatory airway diseases. A full understanding of this pathway will eventually provide multiple targets for therapeutic intervention, including modifications of hCLCA1 expression, processing, and signaling. A particular challenge for therapeutic strategies will be to block excessive mucus prduction without compromising host defense and thereby restore proper balance to the mucociliary system.
Figure 5.
SUMMARY POINTS.
Genomic analysis and gene sequencing has defined a family of CLCA proteins in humans and animals named for the calcium-activated chloride flux associated with purified and recombinant CLCA proteins.
Bioinformatic analysis and experimental data indicates that CLCA proteins are not integral membrane channels but are instead be secreted signaling proteins that are cleaved into polypeptide fragments.
Bioinformatic analysis also indicates that the CLCA proteins exhibit a stereotyped domain architecture that includes VWA and Fn-III domains equipped for protein-protein interaction and possibly signaling function.
Gene expression analysis indicates that CLCA proteins, especially hCLCA1 in humans, are linked to the development of mucous cells in the airway as well as other tissues.
Functional studies combined with genetic and genomic screens indicate that mClca3 (as well as mClca5 and mClca6) are sufficient for the development of mucous cell metaplasia after allergen challenge and viral infection in mouse models.
Clinical studies indicate that CLCA levels (particularly hCLCA1) are linked to the development of inflammatory airway disease, especially mucous cell metaplasia and possibly airway hyperreactivity.
Inflammatory airway diseases such as asthma, COPD, and cystic fibrosis are characterized by mucus obstruction, due to mucin overproduction.
CLCA proteins may serve as useful biomarkers as well as therapeutic targets for the diagnosis and treatment of patients with inflammatory airway disease.
FUTURE ISSUES.
The following questions were raised in this review as areas for future investigation.
What is the basis for redundancy of CLCA/Clca genes? Humans and other species express several CLCA/Clca genes with significant sequence homology. The variable number of CLCA/Clca genes among species suggests the occurrence of gene duplication, but the functional basis for this evolutionary approach still needs to be defined.
What is the pattern of tissue expression for the various CLCA/Clca genes under healthy and disease conditions, and how is this pattern determined? Although we have preliminary analysis of sequence homologies among CLCA family members, we still need to understand what determines cell and tissue specificity for expression and how those patterns translate into function.
How is CLCA/Clca gene expression regulated? In addition to tissue-specific patterns of expression, it appears that CLCA genes are upregulated in disease. For example, CLCA expression is likely induced by Th2 cytokines that are produced during inflammatory airway disease. However, the biochemical basis for how CLCA proteins translate the cytokine signal into mucin gene expression still needs to be defined.
What is the site, mechanism, and functional role of CLCA protein cleavage? CLCA family members appear to uniformly undergo cleavage, but the precise amino acid site for cleavage as well as the protease(s) involved still need to be determined. This information is critical to next explore the structure and function of the mature CLCA peptides.
What is the 3-dimensional structure of CLCA proteins? The understanding of CLCA function as well as the rational design of CLCA inhibitors will be greatly aided by definition of CLCA structure. At present, we have no information on crystal structure for any portion of the CLCA proteins and little understanding of CLCA protein interactions.
Do CLCA proteins function as signaling molecules, and if so, what is the signaling pathway for CLCA/Clca proteins to regulate mucin gene expression? These questions are critical to understanding how CLCA proteins regulate cell function. The development of properly folded recombinant CLCA proteins and the concomitant assay of their functional effects is still needed.
What is the role of CLCA/Clca proteins in airway smooth muscle function? Specific CLCA proteins (notably hCLCA1 and mClca3) are under study for their capacity to regulate mucin gene expression and mucous cell metaplasia. However, study of other CLCA/Clca family members is needed to address their role in controlling smooth muscle cell function in airways and other tissues.
Acknowledgments
The authors gratefully acknowledge their laboratory colleagues for valuable assistance and advice in the course of this work as well as support by grants from the National Institutes of Health (National Heart, Lung, and Blood Institute and National Institute of Allergic and Infectious Diseases), Martin Schaeffer Fund, and Alan A. and Edith L. Wolff Charitable Trust.
Abbreviations and Acronyms
- CLCA
chloride channel calcium-activated
- COPD
Chronic obstructive pulmonary disease
- VWA domain
von Willebrand factor type A domain
- MIDAS
metal-ion dependent adhesion site
- FnIII domain
Fibronectin type III domain
- EGFR
epidermal growth factor receptor
- AHR
airway hyperreactivity
- MCM
mucous cell metaplasia
- AAV
adeno-associated virus
- SeV
Sendai virus
- STAT
signal transducer and activator of transcription
- Th1
T helper type 1
- Th2
T helper type 2
- ERK
extracellular signal-regulated kinase
LITERATURE CITED
- 1.Groneberg DA, Eynott PR, Lim S, Oates T, Wu R, et al. Expression of respiratory mucins in fatal status asthmaticus and mild asthma. Histopathology. 2002;40:367–73. doi: 10.1046/j.1365-2559.2002.01378.x. [DOI] [PubMed] [Google Scholar]
- 2.Kuyper LM, Pare PD, Hogg JC, Lambert RK, Ionescu D, et al. Characterization of airway plugging in fatal asthma. Am J Med. 2003;115:6–11. doi: 10.1016/s0002-9343(03)00241-9. [DOI] [PubMed] [Google Scholar]
- 3.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Eng J Med. 2004;350:2645–53. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 4.Vestbo J, Prescott E, Lange P. Association of chronic mucus hypersecretion with FEV1 decline and chronic obstructive pulmonary disease morbidity. Copenhagen City Heart Study Group. Am J Respir Crit Care Med. 1996;153:1530–5. doi: 10.1164/ajrccm.153.5.8630597. [DOI] [PubMed] [Google Scholar]
- 5.Ekberg-Aronsson M, Pehrsson K, Nilsson JA, Nilsson PM, Lofdahl CG. Mortality in GOLD stages of COPD and its dependence on symptoms of chronic bronchitis. Respir Res. 2005;6:98. doi: 10.1186/1465-9921-6-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ekberg-Aronsson M, Lofdahl K, Nilsson JA, Lofdahl CG, Nilsson PM. Hospital admission rates among men and women with symptoms of chronic bronchitis and airflow limitation corresponding to the GOLD stages of chronic obstructive pulmonary disease--a population-based study. Respir Med. 2008;102:109–20. doi: 10.1016/j.rmed.2007.07.028. [DOI] [PubMed] [Google Scholar]
- 7.Lange P, Nyboe J, Appleyard M, Jensen G, Schnohr P. Relation of ventilatory impairment and of chronic mucus hypersecretion to mortality from obstructive lung disease and from all causes. Thorax. 1990;45:579–85. doi: 10.1136/thx.45.8.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prescott E, Lange P, Vestbo J. Chronic mucus hypersecretion in COPD and death from pulmonary infection. Eur Respir J. 1995;8:1333–8. doi: 10.1183/09031936.95.08081333. [DOI] [PubMed] [Google Scholar]
- 9.Pistelli R, Lange P, Miller DL. Determinants of prognosis of COPD in the elderly: mucus hypersecretion, infections, cardiovascular comorbidity. Eur Respir J Suppl. 2003;40:10s–4s. doi: 10.1183/09031936.03.00403403. [DOI] [PubMed] [Google Scholar]
- 10.Jensen HH, Godtfredsen NS, Lange P, Vestbo J. Potential misclassification of causes of death from COPD. Eur Respir J. 2006;28:781–5. doi: 10.1183/09031936.06.00152205. [DOI] [PubMed] [Google Scholar]
- 11.Hogg JC, Chu FS, Tan WC, Sin DD, Patel SA, et al. Survival after lung volume reduction in chronic obstructive pulmonary disease: insights from small airway pathology. Am J Respir Crit Care Med. 2007;176:454–9. doi: 10.1164/rccm.200612-1772OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gruber AD, Fuller CM, Elble RC, Benos DJ, Pauli BU. The CLCA Gene Family A Novel Family of Putative Chloride Channels. Current Genomics. 2000;1:201. [Google Scholar]
- 13.Fuller CM, Ji HL, Tousson A, Elble RC, Pauli BU, Benos DJ. Ca(2+)-activated Cl(−) channels: a newly emerging anion transport family. Pflugers Arch. 2001;443(Suppl 1):S107–10. doi: 10.1007/s004240100655. [DOI] [PubMed] [Google Scholar]
- 14.Gruber AD, Elble RC, Pauli BU, Catherine Mary F. Current Topics in Membranes. Academic Press; 2002. Discovery and cloning of the CLCA gene family; pp. 367–87. [Google Scholar]
- 15.Hartzell C, Putzier I, Arreola J. Calcium-activated chloride channels. Annu Rev Physiol. 2004;67:719–58. doi: 10.1146/annurev.physiol.67.032003.154341. [DOI] [PubMed] [Google Scholar]
- 16.Loewen ME, Forsyth GW. Structure and function of CLCA proteins. Physiol Rev. 2005;85:1061–92. doi: 10.1152/physrev.00016.2004. [DOI] [PubMed] [Google Scholar]
- 17.Ran S, Benos DJ. Isolation and functional reconstitution of a 38-kDa chloride channel protein from bovine tracheal membranes. J Biol Chem. 1991;266:4782–8. These contemporaneous articles provided for the initial purification and characterization of Clca proteins (bClca1 and bClca2) [PubMed] [Google Scholar]
- 18.Ran S, Benos DJ. Immunopurification and structural analysis of a putative epithelial Cl− channel protein isolated from bovine trachea. J Biol Chem. 1992;267:3618–25. [PubMed] [Google Scholar]
- 19.Zhu D, Cheng CF, Pauli BU. Mediation of lung metastasis of murine melanomas by a lung-specific endothelial cell adhesion molecule. Proc Natl Acad Sci USA. 1991;88:9568–72. doi: 10.1073/pnas.88.21.9568. These contemporaneous articles provided for the initial purification and characterization of Clca proteins (bClca1 and bClca2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cunningham SA, Awayda MS, Bubien JK, Ismailov, Arrate MP, et al. Cloning of an epithelial chloride channel from bovine trachea. J Biol Chem. 1995;270:31016–26. doi: 10.1074/jbc.270.52.31016. [DOI] [PubMed] [Google Scholar]
- 21.Flicek P, Aken BL, Beal K, Ballester B, Caccamo M, et al. Ensembl 2008. Nucleic Acids Res. 2008;36:D707–14. doi: 10.1093/nar/gkm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008;36:D13–21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Jumaily M, Kozlenkov A, Mechaly I, Fichard A, Matha V, et al. Expression of three distinct families of calcium-activated chloride channel genes in the mouse dorsal root ganglion. Neurosci Bull. 2007;23:293–9. doi: 10.1007/s12264-007-0044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel AC, Morton JD, Kim EY, Alevy Y, Swanson S, et al. Genetic segregation of airway disease traits despite redundancy of calcium-activated chloride channel family members. Physiol Genomics. 2006;25:502–13. doi: 10.1152/physiolgenomics.00321.2005. This article demonstrated the activity of Clca family members to regulate mucous cell metaplasia (but not airway hyperreactivity) and to exhibit functional compensation in the mClca3−/− mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anton F, Leverkoehne I, Mundhenk L, Thoreson WB, Gruber AD. Overexpression of eCLCA1 in small airways of horses with recurrent airway obstruction. J Histochem Cytochem. 2005;53:1011–21. doi: 10.1369/jhc.4A6599.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamazaki J, Okamura K, Ishibashi K, Kitamura K. Characterization of CLCA protein expressed in ductal cells of rat salivary glands. Biochim Biophys Acta. 2005;1715:132–44. doi: 10.1016/j.bbamem.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 27.Fuller CM, Ismailov, Keeton DA, Benos DJ. Phosphorylation and activation of a bovine tracheal anion channel by Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1994;269:26642–50. This article provided the first molecular cloning of a Clca gene (bClca1), and provided the first evidence that expression of recombinant Clca protein influenced calcium-dependent chloride flux. [PubMed] [Google Scholar]
- 28.Gruber AD, Elble RC, Ji HL, Schreur KD, Fuller CM, Pauli BU. Genomic cloning, molecular characterization, and functional analysis of human CLCA1, the first human member of the family of Ca2+-activated Cl− channel proteins. Genomics. 1998;54:200–14. doi: 10.1006/geno.1998.5562. These articles were the first to definitively demonstrate that hCLCA1 and mCLCA3 are not ion channels but instead are secreted soluble proteins. [DOI] [PubMed] [Google Scholar]
- 29.Gibson A, Lewis AP, Affleck K, Aitken AJ, Meldrum E, Thompson N. hCLCA1 and mCLCA3 are secreted non-integral membrane proteins and therefore are not ion channels. J Biol Chem. 2005;280:27205–12. doi: 10.1074/jbc.M504654200. These articles were the first to definitively demonstrate that hCLCA1 and mCLCA3 are not ion channels but instead are secreted soluble proteins. [DOI] [PubMed] [Google Scholar]
- 30.Mundhenk L, Alfalah M, Elble RC, Pauli BU, Naim HY, Gruber AD. Both cleavage products of the mCLCA3 protein are secreted soluble proteins. J Biol Chem. 2006;281:30072–80. doi: 10.1074/jbc.M606489200. [DOI] [PubMed] [Google Scholar]
- 31.Huan C, Greene KS, Shi B, Spizz G, Sun H, et al. mCLCA4 processing and secretion requires luminal sorting motifs. Am J Physiol. 2008 doi: 10.1152/ajpcell.00060.2008. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elble RC, Walia V, Cheng HC, Connon CJ, Mundhenk L, et al. The putative chloride channel hCLCA2 has a single C-terminal transmembrane segment. J Biol Chem. 2006;281:29448–54. doi: 10.1074/jbc.M605919200. [DOI] [PubMed] [Google Scholar]
- 33.Bothe MK, Braun J, Mundhenk L, Gruber AD. Murine mCLCA6 is an integral apical membrane protein of non-goblet cell enterocytes and co-localizes with the cystic fibrosis transmembrane conductance regulator. J Histochem Cytochem. 2008;56:495–509. doi: 10.1369/jhc.2008.950592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gandhi R, Elble RC, Gruber AD, Schreur KD, Ji HL, et al. Molecular and functional characterization of a calcium-sensitive chloride channel from mouse lung. J Biol Chem. 1998;273:32096–101. doi: 10.1074/jbc.273.48.32096. [DOI] [PubMed] [Google Scholar]
- 35.Elble RC, Ji G, Nehrke K, DeBiasio J, Kingsley PD, et al. Molecular and functional characterization of a murine calcium-activated chloride channel expressed in smooth muscle. J Biol Chem. 2002;277:18586–91. doi: 10.1074/jbc.M200829200. [DOI] [PubMed] [Google Scholar]
- 36.Beckley JR, Pauli BU, Elble RC. Re-expression of detachment-inducible chloride channel mCLCA5 suppresses growth of metastatic breast cancer cells. J Biol Chem. 2004;279:41634–41. doi: 10.1074/jbc.M408334200. [DOI] [PubMed] [Google Scholar]
- 37.Brouillard F, Bensalem N, Hinzpeter A, Tondelier D, Trudel S, et al. Blue native/SDS-PAGE analysis reveals reduced expression of mClCA3 protein in cystic fibrosis knock-out mice. Mol Cell Proteomics. 2005;4:1762–75. doi: 10.1074/mcp.M500098-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Gruber AD, Pauli BU. Molecular cloning and biochemical characterization of a truncated, secreted member of the human family of Ca2+-activated Cl− channels. Biochim Biophys Acta. 1999;1444:418–23. doi: 10.1016/s0167-4781(99)00008-1. [DOI] [PubMed] [Google Scholar]
- 39.Pawlowski K, Lepisto M, Meinander N, Sivars U, Varga M, Wieslander E. Novel conserved hydrolase domain in the CLCA family of alleged calcium-activated chloride channels. Proteins. 2006;63:424–39. doi: 10.1002/prot.20887. These articles identify and characterize the interaction between hCLCLA2 and beta-4 integrin and highlight the role of this interaction in cancer metastases. [DOI] [PubMed] [Google Scholar]
- 40.Whittaker CA, Hynes RO. Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol Biol Cell. 2002;13:3369–87. doi: 10.1091/mbc.E02-05-0259. These articles identify and characterize the interaction between hCLCLA2 and beta-4 integrin and highlight the role of this interaction in cancer metastases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abdel-Ghany M, Cheng HC, Elble RC, Pauli BU. The breast cancer beta 4 integrin and endothelial human CLCA2 mediate lung metastasis. J Biol Chem. 2001;276:25438–46. doi: 10.1074/jbc.M100478200. These articles identify and characterize the interaction between hCLCLA2 and beta-4 integrin and highlight the role of this interaction in cancer metastases. [DOI] [PubMed] [Google Scholar]
- 42.Abdel-Ghany M, Cheng HC, Elble RC, Pauli BU. Focal adhesion kinase activated by beta(4) integrin ligation to mCLCA1 mediates early metastatic growth. J Biol Chem. 2002;277:34391–400. doi: 10.1074/jbc.M205307200. [DOI] [PubMed] [Google Scholar]
- 43.Abdel-Ghany M, Cheng HC, Elble RC, Lin H, DiBasio J, Pauli BU. The interacting binding domains of the beta(4) integrin and calcium-activated chloride channels (CLCAs) in metastasis. J Biol Chem. 2003;278:49406–16. doi: 10.1074/jbc.M309086200. [DOI] [PubMed] [Google Scholar]
- 44.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008 doi: 10.1093/nar/gkn238. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu D, Pauli BU. Correlation between the lung distribution patterns of Lu-ECAM-1 and melanoma experimental metastases. Int J Cancer. 1993;53:628–33. doi: 10.1002/ijc.2910530417. [DOI] [PubMed] [Google Scholar]
- 46.Zhu D, Cheng CF, Pauli BU. Blocking of lung endothelial cell adhesion molecule-1 (Lu-ECAM-1) inhibits murine melanoma lung metastasis. J Clin Invest. 1992;89:1718–24. doi: 10.1172/JCI115773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gruber AD, Pauli BU. Tumorigenicity of human breast cancer is associated with loss of the Ca2+-activated Cl− channel CLCA2. Cancer Res. 1999;59:5488–91. [PubMed] [Google Scholar]
- 48.Elble RC, Pauli BU. Tumor suppression by a proapoptotic calcium-activated chloride channel in mammary epithelium. J Biol Chem. 2001;276:40510–7. doi: 10.1074/jbc.M104821200. [DOI] [PubMed] [Google Scholar]
- 49.Giancotti FG. Targeting integrin beta4 for cancer and anti-angiogenic therapy. Trends Pharmacol Sci. 2007;28:506–11. doi: 10.1016/j.tips.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 50.Li X, Cowell JK, Sossey-Alaoui K. CLCA2 tumour suppressor gene in 1p31 is epigenetically regulated in breast cancer. Oncogene. 2004;23:1474–80. doi: 10.1038/sj.onc.1207249. [DOI] [PubMed] [Google Scholar]
- 51.Balakrishnan A, von Neuhoff N, Rudolph C, Kamphues K, Schraders M, et al. Quantitative microsatellite analysis to delineate the commonly deleted region 1p22.3 in mantle cell lymphomas. Genes Chromosomes Cancer. 2006;45:883–92. doi: 10.1002/gcc.20352. [DOI] [PubMed] [Google Scholar]
- 52.Nakanishi A, Morita S, Iwashita H, Sagiya Y, Ashida Y, et al. Role of gob-5 in mucus overproduction and airway hyperresponsiveness in asthma. Proc Natl Acad Sci U S A. 2001;98:5175–80. doi: 10.1073/pnas.081510898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou Y, Shapiro M, Dong Q, Louahed J, Weiss C, et al. A calcium-activated chloride channel blocker inhibits goblet cell metaplasia and mucus overproduction. Presented at Novartis Foundation Symposium 248 on Mucus hypersecretion in respiratory disease; 2002. [PubMed] [Google Scholar]
- 54.Bertrand CA, Danahay H, Poll CT, Laboisse C, Hopfer U, Bridges RJ. Niflumic acid inhibits ATP-stimulated exocytosis in a mucin secreting epithelial cell line. Am J Physiol Cell Physiol. 2004;286:C247–C55. doi: 10.1152/ajpcell.00593.2002. [DOI] [PubMed] [Google Scholar]
- 55.McNamara N, Gallup M, Khong A, Sucher A, Maltseva I, et al. Adenosine up-regulation of the mucin gene, MUC2, in asthma. FASEB J. 2004;18:1770–2. doi: 10.1096/fj.04-1964fje. [DOI] [PubMed] [Google Scholar]
- 56.Hauber HP, Daigneault P, Frenkiel S, Lavigne F, Hung HL, et al. Niflumic acid and MSI-2216 reduce TNF-alpha-induced mucin expression in human airway mucosa. J Allergy Clin Immunol. 2005;115:266–71. doi: 10.1016/j.jaci.2004.09.039. [DOI] [PubMed] [Google Scholar]
- 57.Hauber HP, Goldmann T, Vollmer E, Wollenberg B, Hung HL, et al. LPS-induced mucin expression in human sinus mucosa can be attenuated by hCLCA inhibitors. J Endotoxin Res. 2007;13:109–16. doi: 10.1177/0968051907079168. [DOI] [PubMed] [Google Scholar]
- 58.Hoshino M, Morita S, Iwashita H, Sagiya Y, Nagi T, et al. Increased expression of the human Ca2+-activated Cl− channel 1 (CaCC1) gene in the asthmatic airway. Am J Respir Crit Care Med. 2002;165:1132–6. doi: 10.1164/ajrccm.165.8.2107068. [DOI] [PubMed] [Google Scholar]
- 59.Leverkoehne I, Gruber AD. The murine mCLCA3 (alias gob-5) protein is located in the mucin granule membranes of intestinal, respiratory, and uterine goblet cells. J Histochem Cytochem. 2002;50:829–38. doi: 10.1177/002215540205000609. [DOI] [PubMed] [Google Scholar]
- 60.Kirkup AJ, Edwards G, Green ME, Miller M, Walker SD, Weston AH. Modulation of membrane currents and mechanical activity by niflumic acid in rat vascular smooth muscle. Eur J Pharmacol. 1996;317:165–74. doi: 10.1016/s0014-2999(96)00713-3. [DOI] [PubMed] [Google Scholar]
- 61.Sergeant GP, Hollywood MA, McHale NG, Thornbury KD. Spontaneous Ca2+ activated Cl− currents in isolated urethral smooth muscle cells. J Urol. 2001;166:1161–6. [PubMed] [Google Scholar]
- 62.Ledoux J, Greenwood IA, Leblanc N. Dynamics of Ca2+-dependent cl− channel modulation by niflumic Acid in rabbit coronary arterial myocytes. Mol Pharmacol. 2005;67:163–73. doi: 10.1124/mol.104.004168. [DOI] [PubMed] [Google Scholar]
- 63.Lowell FC. Observations on Heaves. An Asthma-Like Syndrome in the Horse. J Allergy Clin Immunol. 1964;35:322–30. doi: 10.1016/0021-8707(64)90095-4. [DOI] [PubMed] [Google Scholar]
- 64.Davis E, Rush BR. Equine recurrent airway obstruction: pathogenesis, diagnosis, and patient management. Vet Clin North Am Equine Pract. 2002;18:453–67. vi. doi: 10.1016/s0749-0739(02)00026-3. [DOI] [PubMed] [Google Scholar]
- 65.Leguillette R. Recurrent airway obstruction--heaves. Vet Clin North Am Equine Pract. 2003;19:63–86. vi. doi: 10.1016/s0749-0739(02)00067-6. [DOI] [PubMed] [Google Scholar]
- 66.Horohov DW, Beadle RE, Mouch S, Pourciau SS. Temporal regulation of cytokine mRNA expression in equine recurrent airway obstruction. Vet Immunol Immunopathol. 2005;108:237–45. doi: 10.1016/j.vetimm.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 67.Gerber V, Robinson NE, Venta RJ, Rawson J, Jefcoat AM, Hotchkiss JA. Mucin genes in horse airways: MUC5AC, but not MUC2, may play a role in recurrent airway obstruction. Equine Vet J. 2003;35:252–7. doi: 10.2746/042516403776148291. [DOI] [PubMed] [Google Scholar]
- 68.Holtzman MJ, Kim EY, Morton JD. Genetic and genomic approaches to complex lung diseases using mouse models. In: Peltz G, editor. Computational Genetics and Genomics: Tools for Understanding Disease. Totawa, NJ: Humana Press Inc; 2005. pp. 99–141. [Google Scholar]
- 69.Range F, Mundhenk L, Gruber AD. A soluble secreted glycoprotein (eCLCA1) is overexpressed due to goblet cell hyperplasia and metaplasia in horses with recurrent airway obstruction. Vet Pathol. 2007;44:901–11. doi: 10.1354/vp.44-6-901. [DOI] [PubMed] [Google Scholar]
- 70.Levitt RC, Mitzner W. Autosomal recessive inheritance of airway hyperreactivity to 5-hydroxytryptamine. J Appl Physiol. 1989;67:1125–32. doi: 10.1152/jappl.1989.67.3.1125. [DOI] [PubMed] [Google Scholar]
- 71.Konno S, Adachi M, Matsuura T, Sunouchi K, Hoshino H, et al. Bronchial reactivity to methacholine and serotonin in six inbred mouse strains. Arerugi. 1993;42:42–7. [PubMed] [Google Scholar]
- 72.Chiba Y, Yanagisawa R, Sagai M. Strain and route differences in airway responsiveness to acetylcholine in mice. Res Commun Mol Pathol Pharmacol. 1995;90:169–72. [PubMed] [Google Scholar]
- 73.Zhang LY, Levitt RC, Kleeberger SR. Differential susceptibility to ozone-induced airways hyperreactivity in inbred strains of mice. Exp Lung Res. 1995;21:503–18. doi: 10.3109/01902149509031755. [DOI] [PubMed] [Google Scholar]
- 74.Miyabara Y, Yanagisawa R, Shimojo N, Takano H, Lim HB, et al. Murine strain differences in airway inflammation caused by diesel exhaust particles. Eur Respir J. 1998;11:291–8. doi: 10.1183/09031936.98.11020291. [DOI] [PubMed] [Google Scholar]
- 75.Brewer JP, Kisselgof AB, Martin TR. Genetic variability in pulmonary physiological, cellular, and antibody responses to antigen in mice. Am J Respir Crit Care Med. 1999;160:1150–6. doi: 10.1164/ajrccm.160.4.9806034. [DOI] [PubMed] [Google Scholar]
- 76.De Sanctis GT, Singer JB, Jiao A, Yandava CN, Lee YH, et al. Quantitative trait locus mapping of airway responsiveness to chromosomes 6 and 7 in inbred mice. Am J Physiol. 1999;277:L1118–L23. doi: 10.1152/ajplung.1999.277.6.L1118. [DOI] [PubMed] [Google Scholar]
- 77.Ewart SL, Kuperman D, Schadt E, Tankersley C, Grupe A, et al. Quantitative trait loci controlling allergen-induced airway hyperresponsiveness in inbred mice. Am J Respir Cell Mol Biol. 2000;23:537–45. doi: 10.1165/ajrcmb.23.4.4199. [DOI] [PubMed] [Google Scholar]
- 78.Van Oosterhout AJ, Jeurink PV, Groot PC, Hofman GA, Nijkamp FP, Demant P. Genetic analysis of antigen-induced airway manifestations of asthma using recombinant congenic mouse strains. Chest. 2002;121:13S. doi: 10.1378/chest.121.3_suppl.13s. [DOI] [PubMed] [Google Scholar]
- 79.Graham BS, Perkins MD, Wright PF, Karzon DT. Primary respiratory syncytial virus infection in mice. J Med Virol. 1988;26:153–62. doi: 10.1002/jmv.1890260207. [DOI] [PubMed] [Google Scholar]
- 80.Seemungal T, Harper-Owen R, Bhowmik A, Moric I, Sanderson G, et al. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164:1618–23. doi: 10.1164/ajrccm.164.9.2105011. [DOI] [PubMed] [Google Scholar]
- 81.Rhode G, Wiethege A, Borg I, Kauth M, Bauer TT, et al. Respiratory viruses in exacerbations of chronic obstructive pulmonary disease requiring hospitalisation: a case-control study. Thorax. 2003;58:37–42. doi: 10.1136/thorax.58.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Borg I, Rohde G, Loseke S, Ittscheidt BJ, Schultze-Werninghaus G, et al. Evaluation of a quantitative real-time PCR for the detection of respiratory syncytial virus in pulmonary diseases. Eur Respir J. 2003;21:944–51. doi: 10.1183/09031936.03.00088102. [DOI] [PubMed] [Google Scholar]
- 83.Falsey A, Hennessey RPA, Formica MA, Cox C, Walsh EE. Respiratory syncytial virus infection in elderly and high-risk adults. New Engl J Med. 2005;352:1749–59. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- 84.Beckham JD, Cadena A, Lin J, Piedra PA, Glezen WP, et al. Respiratory viral infections in patients with chronic obstructive pulmonary disease. J Infect. 2005;50:322–30. doi: 10.1016/j.jinf.2004.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tyner JW, Kim EY, Ide K, Pelletier MR, Roswit WT, et al. Blocking airway mucous cell metaplasia by inhibiting EGFR antiapoptosis and IL-13 transdifferentiation signals. J Clin Invest. 2006;116:309–21. doi: 10.1172/JCI25167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walter MJ, Morton JD, Kajiwara N, Agapov E, Holtzman MJ. Viral induction of a chronic asthma phenotype and genetic segregation from the acute response. J Clin Invest. 2002;110:165–75. doi: 10.1172/JCI14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 88.Evans SR, Thoreson WB, Beck CL. Molecular and functional analyses of two new calcium-activated chloride channel family members from mouse eye and intestine. J Biol Chem. 2004;279:41792–800. doi: 10.1074/jbc.M408354200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gruber AD, Schreur KD, Ji HL, Fuller CM, Pauli BU. Molecular cloning and transmembrane structure of hCLCA2 from human lung, trachea, and mammary gland. Am J Physiol. 2004;276:C1261–C70. doi: 10.1152/ajpcell.1999.276.6.C1261. [DOI] [PubMed] [Google Scholar]
- 90.Patel AC, Battaile JT, Alevy Y, Patterson GA, Swanson S, et al. Homologous mouse Clca3,5,6 and human hCLCA1,2,4 gene clusters control mucous cell metaplasia. Am J Respir Crit Care Med. 2007;175:A499. [Google Scholar]
- 91.Robichaud A, Tuck SA, Kargman S, Tam J, Wong E, et al. Gob-5 is not essential for mucus overproduction in preclinical murine models of allergic asthma. Am J Respir Cell Mol Biol. 2005;33:303–14. doi: 10.1165/rcmb.2004-0372OC. [DOI] [PubMed] [Google Scholar]
- 92.Zhou Y, Dong Q, Louahed J, Dragwa C, Savio D, et al. Characterization of a calcium-activated chloride channel as a shared target of Th2 cytokine pathways and its potential involvement in asthma. Am J Respir Cell Mol Biol. 2001;25:486–91. doi: 10.1165/ajrcmb.25.4.4578. [DOI] [PubMed] [Google Scholar]
- 93.Nakano T, Inoue H, Fukuyama S, Matsumoto K, Matsumura M, et al. Niflumic acid suppresses interleukin-13-induced asthma phenotypes. Am J Respir Crit Care Med. 2006;173:1216–21. doi: 10.1164/rccm.200410-1420OC. [DOI] [PubMed] [Google Scholar]
- 94.Yasuo M, Fujimoto K, Tanabe T, Yaegashi H, Tsushima K, et al. Relationship between calcium-activated chloride channel 1 and MUC5AC in goblet cell hyperplasia induced by interleukin-13 in human bronchial epithelial cells. Respiration. 2006;73:347–59. doi: 10.1159/000091391. [DOI] [PubMed] [Google Scholar]
- 95.Thai P, Chen Y, Dolganov G, Wu R. Differential regulation of MUC5AC/Muc5ac and hCLCA-1/mGob-5 expression in airway epithelium. Am J Respir Cell Mol Biol. 2005;33:523–30. doi: 10.1165/rcmb.2004-0220RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim EY, Battaile JT, Patel AC, You Y, Agapov E, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. 2008;14:633–40. doi: 10.1038/nm1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Holtzman MJ, Battaile JT, Patel AC. Immunogenetic programs for viral induction of mucous cell metaplasia. Am J Respir Cell Mol Biol. 2006;35:29–39. doi: 10.1165/rcmb.2006-0092SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. 2002;8:885–9. doi: 10.1038/nm734. [DOI] [PubMed] [Google Scholar]
- 99.Nath P, Leung SY, Williams AS, Noble A, Xie S, et al. Complete inhibition of allergic airway inflammation and remodelling in quadruple IL-4/5/9/13−/− mice. Clin Exp Allergy. 2007;37:1427–35. doi: 10.1111/j.1365-2222.2007.02789.x. [DOI] [PubMed] [Google Scholar]
- 100.Silverman EK, Palmer LJ, Mosley JD, Barth M, Senter JM, et al. Genomewide linkage analysis of quantitative spirometric phenotypes in severe early-onset chronic obstructive pulmonary disease. Am J Hum Genet. 2002;70:1229–39. doi: 10.1086/340316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Palmer LJ, Celedon JC, Chapman HA, Speizer FE, Weiss ST, Silverman EK. Genome-wide linkage analysis of bronchodilator responsiveness and post-bronchodilator spirometric phenotypes in chronic obstructive pulmonary disease. Hum Mol Genet. 2003;12:1199–210. doi: 10.1093/hmg/ddg125. [DOI] [PubMed] [Google Scholar]
- 102.Kamada F, Suzuki Y, Shao C, Tamari M, Hasegawa K, et al. Association of the hCLCA1 gene with childhood and adult asthma. Genes and Immunity. 2004;5:540–7. doi: 10.1038/sj.gene.6364124. [DOI] [PubMed] [Google Scholar]
- 103.Hegab AE, Sakamoto T, Uchida Y, Nomura A, Ishii Y, et al. CLCA1 gene polymorphisms in chronic obstructive pulmonary disease. J Med Genet. 2004;41:1–7. doi: 10.1136/jmg.2003.012484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ritzka M, Stanke F, Jansen S, Gruber AD, Pusch L, et al. The CLCA gene locus as a modulator of the gastrointestinal basic defect in cystic fibrosis. Hum Genet. 2004;115:483–91. doi: 10.1007/s00439-004-1190-y. [DOI] [PubMed] [Google Scholar]
- 105.Young FD, Newbigging S, Choi C, Keet M, Kent G, Rozmahel RF. Amelioration of cystic fibrosis intestinal mucous disease in mice by restoration of mCLCA3. Gastroenterology. 2007;133:1928–37. doi: 10.1053/j.gastro.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 106.Toda M, Tulic MK, Levitt RC, Hamid Q. A calcium-activated chloride channel (HCLCA1) is strongly related to IL-9 expression and mucus production in bronchial epithelium of patients with asthma. J Allergy Clin Immunol. 2002;109:246–50. doi: 10.1067/mai.2002.121555. [DOI] [PubMed] [Google Scholar]
- 107.Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. 2007;104:15858–63. doi: 10.1073/pnas.0707413104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang K, Wen FQ, Feng YL, Ou XM, Xu D, et al. Increased expression of human calcium-activated chloride channel 1 gene is correlated with mucus overproduction in Chinese asthmatic airway. Cell Biol Int. 2007;31:1388–95. doi: 10.1016/j.cellbi.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 109.Patel AC, Kim EY, Roswit WT, Swanson S, Christie C, et al. Potential utility of CLCA1 as a biomarker: response of hCLCA1/mClca3 to corticosteroid in natural and experimental asthma. Am J Respir Crit Care Med. 2008;177:A75. [Google Scholar]
- 110.Hauber HP, Manoukian JJ, Nguyen LH, Sobol SE, Levitt RC, et al. Increased expression of interleukin-9, interleukin-9 receptor, and the calcium-activated chloride channel hCLCA1 in the upper airways of patients with cystic fibrosis. Laryngoscope. 2003;113:1037–42. doi: 10.1097/00005537-200306000-00022. [DOI] [PubMed] [Google Scholar]
- 111.Hauber HP, Tsicopoulos A, Wallaert B, Griffin S, McElvaney NG, et al. Expression of HCLCA1 in cystic fibrosis lungs is associated with mucus overproduction. Eur Respir J. 2004;23:846–50. doi: 10.1183/09031936.04.00096504. [DOI] [PubMed] [Google Scholar]
- 112.Nakanishi A, Shigeru M. US2004185500 US Patent No Application. 2004
- 113.Szymkowski DE. 7,141,365 B2 US Patent No. 2006
- 114.Walker NM, Simpson JE, Levitt RC, Boyle KT, Clarke LL. Talniflumate increases survival in a cystic fibrosis mouse model of distal intestinal obstructive syndrome. J Pharmacol Exp Ther. 2006;317:275–83. doi: 10.1124/jpet.105.094847. [DOI] [PubMed] [Google Scholar]
- 115.Knight D. Talniflumate (Genaera) Curr Opin Investig Drugs. 2004;5:557–62. [PubMed] [Google Scholar]
- 116.Donnelly LE, Rogers DF. Therapy for chronic obstructive pulmonary disease in the 21st century. Drugs. 2003;63:1973–98. doi: 10.2165/00003495-200363190-00002. [DOI] [PubMed] [Google Scholar]
- 117.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 118.Karolchik D, Kuhn RM, Baertsch R, Barber GP, Clawson H, et al. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res. 2008;36:D773–9. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rimbaut A. FigTree 112. 2008 ( http://tree.bio.ed.ac.uk/software/figtree/)
- 121.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–95. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 122.Bennett-Lovsey RM, Herbert AD, Sternberg MJ, Kelley LA. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins. 2008;70:611–25. doi: 10.1002/prot.21688. [DOI] [PubMed] [Google Scholar]
- 123.Barton GJ. ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng. 1993;6:37–40. doi: 10.1093/protein/6.1.37. [DOI] [PubMed] [Google Scholar]
- 124.Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4:1633–49. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- 125.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 126.Eisenhaber B, Bork P, Eisenhaber F. Prediction of potential GPI-modification sites in proprotein sequences. J Mol Biol. 1999;292:741–58. doi: 10.1006/jmbi.1999.3069. [DOI] [PubMed] [Google Scholar]
- 127.Twigger SN, Pruitt KD, Fernandez-Suarez XM, Karolchik D, Worley KC, et al. What everybody should know about the rat genome and its online resources. Nat Genet. 2008;40:523–7. doi: 10.1038/ng0508-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Petersen G, Stahl F. RGST - Rat Gene Symbol Tracker, a database for defining official rat gene symbols. BMC Genomics. 2008;9:29. doi: 10.1186/1471-2164-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Agnel M, Vermat T, Culouscou JM. Identification of three novel members of the calcium-dependent chloride channel (CaCC) family predominantly expressed in the digestive tract and trachea. FEBS Lett. 1999;455:295–301. doi: 10.1016/s0014-5793(99)00891-1. [DOI] [PubMed] [Google Scholar]
- 130.Romio L, Musante L, Cinti R, Seri M, Moran O, et al. Characterization of a murine gene homologous to the bovine CaCC chloride channel. Gene. 1999;228:181–8. doi: 10.1016/s0378-1119(98)00620-9. [DOI] [PubMed] [Google Scholar]
- 131.Lee D, Ha S, Kho Y, Kim J, Cho K, et al. Induction of mouse Ca(2+)-sensitive chloride channel 2 gene during involution of mammary gland. Biochem Biophys Res Commun. 1999;264:933–7. doi: 10.1006/bbrc.1999.1583. [DOI] [PubMed] [Google Scholar]
- 132.Komiya T, Tanigawa Y, Hirohashi S. Cloning and identification of the gene gob-5, which is expressed in intestinal goblet cells in mice. Biochem Biophys Res Commun. 1999;255:347–51. doi: 10.1006/bbrc.1999.0168. [DOI] [PubMed] [Google Scholar]
- 133.Snelling WM, Chiu R, Schein JE, Hobbs M, Abbey CA, et al. A physical map of the bovine genome. Genome Biol. 2007;8:R165. doi: 10.1186/gb-2007-8-8-r165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Elble RC, Widom J, Gruber AD, Abdel-Ghany M, Levine R, et al. Cloning and characterization of lung-endothelial cell adhesion molecule-1 suggest it is an endothelial chloride channel. J Biol Chem. 1997;272:27853–61. doi: 10.1074/jbc.272.44.27853. [DOI] [PubMed] [Google Scholar]
- 135.Jeong SM, Park HK, Yoon IS, Lee JH, Kim JH, et al. Cloning and expression of Ca2+-activated chloride channel from rat brain. Biochem Biophys Res Commun. 2005;334:569–76. doi: 10.1016/j.bbrc.2005.06.122. [DOI] [PubMed] [Google Scholar]
- 136.Yoon IS, Jeong SM, Lee SN, Lee JH, Kim JH, et al. Cloning and heterologous expression of a Ca2+-activated chloride channel isoform from rat brain. Biol Pharm Bull. 2006;29:2168–73. doi: 10.1248/bpb.29.2168. [DOI] [PubMed] [Google Scholar]
- 137.Chowdhary BP, Raudsepp T. The horse genome derby: racing from map to whole genome sequence. Chromosome Res. 2008;16:109–27. doi: 10.1007/s10577-008-1204-z. [DOI] [PubMed] [Google Scholar]
- 138.Zhang SJ, Steijaert MN, Lau D, Schutz G, Delucinge-Vivier C, et al. Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron. 2007;53:549–62. doi: 10.1016/j.neuron.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 139.Itoh R, Kawamoto S, Miyamoto Y, Kinoshita S, Okubo K. Isolation and characterization of a Ca(2+)-activated chloride channel from human corneal epithelium. Curr Eye Res. 2000;21:918–25. doi: 10.1076/ceyr.21.6.918.6983. [DOI] [PubMed] [Google Scholar]
- 140.Mall M, Gonska T, Thomas J, Schreiber R, Seydewitz HH, et al. Modulation of Ca2+-activated Cl− secretion by basolateral K+ channels in human normal and cystic fibrosis airway epithelia. Pediatr Res. 2003;53:608–18. doi: 10.1203/01.PDR.0000057204.51420.DC. [DOI] [PubMed] [Google Scholar]
- 141.Connon CJ, Kawasaki S, Yamasaki K, Quantock AJ, Kinoshita S. The quantification of hCLCA2 and colocalisation with integrin beta4 in stratified human epithelia. Acta Histochem. 2005;106:421–5. doi: 10.1016/j.acthis.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 142.Connon CJ, Yamasaki K, Kawasaki S, Quantock AJ, Koizumi N, Kinoshita S. Calcium-activated chloride channel-2 in human epithelia. J Histochem Cytochem. 2004;52:415–8. doi: 10.1177/002215540405200313. [DOI] [PubMed] [Google Scholar]
- 143.Leverkoehne I, Horstmeier BA, von Samson-Himmelstjerna G, Scholte BJ, Gruber AD. Real-time RT-PCR quantitation of mCLCA1 and mCLCA2 reveals differentially regulated expression in pre- and postnatal murine tissues. Histochem Cell Biol. 2002;118:11–7. doi: 10.1007/s00418-002-0420-4. [DOI] [PubMed] [Google Scholar]