

Abstract
Targeting the mammalian target of rapamycin (mTOR) is a promising strategy for cancer therapy. However, the mTOR kinase functions in two complexes, TORC1 and TORC2, neither of which is fully inhibited by the allosteric inhibitor rapamycin or analogs. We compared rapamycin with the active-site TORC1/2 inhibitor PP242, in acute leukemia models harboring the Philadelphia chromosome (Ph) translocation. We demonstrate that PP242, but not rapamycin, causes death of mouse and human leukemia cells. In vivo, PP242 delays leukemia onset and augments the effects of current front-line tyrosine kinase inhibitors, more effectively than rapamycin. Surprisingly, PP242 has much weaker effects than rapamycin on proliferation and function of normal lymphocytes. PI-103, a less selective TORC1/2 inhibitor that also targets phosphoinositide 3-kinase, is more immunosuppressive than PP242. These findings establish that Ph+ transformed cells are more sensitive than normal lymphocytes to selective TORC1/2 inhibitors, and support the development of such inhibitors for leukemia therapy.
The phosphoinositide 3-kinase (PI3K)/AKT/mTOR signaling axis is central to the transformed phenotype of most cancer cells 1. PI3K is a lipid kinase whose products mediate membrane assembly of signaling complexes downstream of activated tyrosine kinases and the small GTPase Ras. AKT serine/threonine kinases are activated in a PI3K-dependent manner to phosphorylate numerous substrates, thereby promoting cell growth, proliferation and survival 2. Most tumor cells bear mutations that increase PI3K/AKT/mTOR signaling output 1,3, and activation of this pathway has been linked to chemoresistance in a variety of clinical settings 4,5. Therefore, a priority has been to develop agents targeting PI3K, AKT, or downstream enzymes such as mTOR 6. However, this signaling network mediates essential physiological functions, and is subject to complex cross-talk and feedback, which has complicated efforts to identify an optimal pharmacological profile to achieve effective and selective killing of cancer cells.
mTOR is present in two cellular complexes, TORC1 and TORC2, with distinct substrates and mechanisms of activation (Fig. 1a) 7,8. The best-known substrates of TORC1 are S6 kinase (S6K) and 4EBP1 (eukaryotic initiation factor-4E (eIF4E)-binding protein); the main substrates of TORC2 are AKT and related kinases. Rapamycin (Sirolimus) and its analogs, such as RAD001 (Everolimus) and CCI-779 (Temsirolimus), suppress mTOR activity through an allosteric mechanism distant to the ATP-catalytic binding site 6,9,10. This class of mTOR inhibitor has profound immunomodulatory activity 11,12 but has achieved limited success as anti-cancer agents 9. Mechanistically, rapamycin has two main drawbacks (Fig. 1a). First, the drug suppresses TORC1-mediated S6K activation, thereby blocking a negative feedback loop, but does not acutely inhibit TORC2. In many cancer cells this leads to elevated PI3K/AKT signaling and promotes cell survival 10. Second, rapamycin is an incomplete inhibitor of TORC1, reducing phosphorylation of 4EBP1 only partially in most cell contexts 13-17.
Figure 1.

PP242 induces apoptosis of p190 BCR-ABL-transformed mouse hematopoietic progenitors and human Ph+ B-ALL cells in vitro. (a) Schematic model of BCR-ABL driven mechanisms of oncogenic survival (top) and models of incomplete mTOR inhibition (bottom, left) versus complete and selective mTOR inhibition (bottom, middle) versus nonselective PI3K/mTOR inhibition (bottom, right) in Ph+ disease. Green = activated; red = inhibited. Selective mTOR inhibitors (middle) may affect PI3K (purple) differently depending on the degree of off-target inhibition. (b) The number of viable mouse p190, human SUP-B15, or human K562 cells following treatment (48 hr) with the indicated inhibitors was determined by the MTS assay (n = 3–9 independent experiments, error bars omitted for clarity). (c) p190 cells were cultured for 24 hr with the inhibitors indicated, then DNA content was measured by flow cytometry. (d–f) Anti-clonogenic effects of PP242 combined with DA in primary Ph+ leukemias. Purified bone marrow (BM) or peripheral blood (PB) from untreated, newly diagnosed individuals with Ph+ B-ALL (n = 8) (d), relapsed or refractory Ph+ B-ALL (n = 6) (e), and imatinib-resistant BC-CML (n = 4 LyBC-CML and n = 1 MyBC-CML) (f), were assessed for colony formation potential in MethoCult cultures with dasatinib (DA; 5 nM) alone or in combination with increasing concentrations (20 or 200 nM) of rapamycin (RAP), PP242, or BEZ235 (*P < 0.05, **P < 0.01, #P < 0.001, RM-ANOVA, measured vs. control except where indicated by brackets). See also Supplementary Table 4 for details of clinical subjects.
A promising approach to overcome these limitations is through ATP-competitive “active-site” mTOR inhibitors. One strategy has been to use small molecule TORC1/2 inhibitors that also inhibit PI3K lipid kinases (Fig. 1a) 6. One such compound, PI-103, is more potent than rapamycin in mouse models of leukemia and in primary human leukemia colony assays 18-21. However, the clinical therapeutic efficacy as well as tolerability of such dual PI3K/mTOR inhibitors remains to be established. Recently, four independent groups reported the discovery and characterization of selective ATP-competitive TORC1/2 inhibitors 14-17. Active-site mTOR inhibitors strongly suppress 4EBP1 phosphorylation and reduce phosphorylation of TORC2 substrates including AKT (Fig. 1a), without strongly inhibiting PI3K. Here we report for the first time a comparison of rapamycin and a selective TORC1/2 inhibitor, PP242, in models of leukemia and normal lymphocyte function. We demonstrate that PP242 has potent and cytotoxic activity against leukemia cells, and enhances the efficacy of the tyrosine kinase inhibitors (TKIs) imatinib and dasatinib in Ph+ acute leukemia models. The effects of PP242 are similar to panPI3K-TORC1/2 inhibitors yet stronger than rapamycin. We also report a surprising reversal of potency in normal lymphocytes, such that rapamycin produces much stronger immunosuppression than PP242 when using a set of in vitro and in vivo assays of adaptive immune function. At doses that show therapeutic effects in leukemia models, the panPI3K-TORC1/2 inhibitor PI-103 is also more immunosuppressive than PP242. Thus, selective TORC1/2 inhibitors might achieve a favorable balance of efficacy and tolerability that is superior to other approaches targeting this pathway in cancer.
Results
Selective TORC1/2 inhibition causes apoptosis in BCR-ABL+ cells
The structure and selectivity of the pyrazolopyrimidine compound PP242 were reported previously 14, and further drug-related information is provided in Supplementary Table 1. We tested the efficacy of PP242 in models of Philadelphia chromosome-positive (Ph)+ B-precursor Acute Lymphoblastic Leukemia (B-ALL), a subtype of leukemia initiated by the BCR-ABL oncogene 22,23. When mouse bone marrow cells are infected with a retrovirus expressing human p190-BCR-ABL, transformed progenitor-B cell lines (termed p190 cells) emerge that initiate aggressive B-ALL upon transfer to recipient mice 19,24. We monitored proliferation and survival of p190 cells treated with mTOR inhibitors in comparison to TKIs, imatinib and dasatinib, presently used in the clinic (Fig. 1b and Supplementary Table 2). Using a colorimetric MTS assay, we observed that both imatinib and dasatinib fully suppressed growth, as expected. Rapamycin displayed anti-proliferative effects (GI50 = 6.5 nM), yet it reached a plateau in efficacy at ∼ 60% inhibition. In contrast, PP242 suppressed growth by > 90%, with low nanomolar potency (GI50 = 12 nM). The selective TORC1/2 inhibitor Ku-0063794 15, which is structurally unrelated to PP242, also fully suppressed growth though with lesser potency (GI50 = 36 nM). We also tested agents that inhibit both class I PI3Ks and TORC1/2 through an ATP-competitive mechanism, PI-103 25 and BEZ-235 26. As we have reported previously 19, panPI3K-TORC1/2 inhibition fully suppressed growth of p190 cells (GI50 = 86 nM for PI-103, 4 nM for BEZ-235). Similar relative GI50 values were obtained when we compared compounds in the human Ph+ B-ALL cell line SUP-B15 and the Ph+ chronic myeloid leukemia (CML) cell line K562 (Fig. 1b and Supplementary Table 2). Cell cycle analysis confirmed that TORC1/2 inhibitors and panPI3K-TORC1/2 inhibitors cause both cell cycle arrest and death, whereas rapamycin was primarily cytostatic (Fig. 1c).
We also observed greater anti-proliferative potency of PP242 relative to rapamycin in a panel of solid tumor cell lines carrying either PI3K gain-of-function or PTEN (phosphatase and tensin homolog) loss-of-function (Supplementary Table 3), indicating a generally stronger anti-cancer effect of TORC1/2 inhibitors, in agreement with a study of TORC1/2 inhibitors in solid tumor lines reported by Wyeth Research Labs 17. We noted that PI-103 was consistently two-fold less potent than PP242 on a molar basis.
PP242 enhances anti-leukemic effects of TKIs in vitro
Imatinib provides overall poor long-term survival in the Ph+ B-ALL setting 27,28. Relapse of both Ph+ B-ALL and blast crisis CML are due to mutations in the BCR-ABL kinase domain, and other genetic anomalies. Some BCR-ABL mutations, particularly T315I, also confer resistance to dasatinib. Targeting downstream of the driving oncogene could overcome resistance 29,30. The observation that PP242 caused apoptosis suggested that TORC1/2 inhibitors might negate resistance and also synergize with current therapies to kill leukemia cells. Indeed, mouse bone marrow transformation with BCR-ABL kinase domain mutants showed similar sensitivity to PP242 (Supplementary Fig. 1a). Furthermore, PP242 strongly synergized with imatinib in p190 cells, similar to the potency achieved using BEZ235. This effect was superior to rapamycin combinations (Supplementary Fig. 1b and Supplementary Table 2). The efficacy of PP242 alone, or combined with ABL inhibition, was further documented with primary human Ph+ B-ALL, including samples representing new diagnoses and drug-resistant relapses (Fig. 1d–e and see Supplementary Table 4 for subject characteristics). For these experiments we used dasatinib to inhibit BCR-ABL, as this agent is emerging as a favored TKI for Ph+ B-ALL treatment, either with chemotherapy or as monotherapy 27,31. PP242 significantly reduced colony formation of newly diagnosed Ph+ B-ALL, and augmented the effect of dasatinib in samples derived from subjects who had relapsed following combination chemotherapy with imatinib. BEZ235 had similar potency as PP242, whereas rapamycin had a lesser effect with these sample groups and did not augment dasatinib efficacy. In five specimens representing CML cells in blast crisis (CML-BC), rapamycin was equally effective as PP242 and BEZ-235 (Fig. 1f).
PP242 and rapamycin have distinct effects on TORC1/2 signaling
In fibroblasts and solid tumor cell lines, active-site inhibitors suppress rapamycin-resistant outputs of TORC1 and TORC2 14-17. Similarly, PP242 suppressed Akt phosphorylation on S473 in p190 cells, whereas rapamycin caused an increase over time in phosphorylation on the TORC2 site S473 and on the phosphoinositide-dependent-kinase-1 site T308 (Fig. 2a–b). Whereas both drugs effectively suppressed phosphorylation of the ribosomal S6 protein (rS6), only PP242 reduced phosphorylation of eukaryotic initiation factor-4E-binding-protein-1 (4EBP1) (Fig. 2a–b). These distinct phosphoprotein signatures of PP242 and rapamycin remained apparent when the drugs were combined with ABL kinase inhibitors (Fig. 2b); thus, p4EBP1 and pAkt were more effectively suppressed by TKIs + PP242 compared to TKIs + rapamycin. It should be emphasized that TKIs alone did not fully suppress TORC1/2 signaling, as observed in other BCR-ABL-driven cell models 32,33. Very similar results were obtained using SUP-B15 and K562 cells (Fig. 2c–f). Cap-dependent mRNA translation is facilitated by TORC1 mediated phosphorylation of 4EBP1, which releases this inhibitory protein from eukaryotic initiation factor-4E (eIF4E) 7,8. PP242 treatment of p190 cells increased binding of 4EBP1 to eIF4G with a concomitant decrease in eIF4E binding, whereas rapamycin had little effect (Supplementary Fig. 2).
Figure 2.
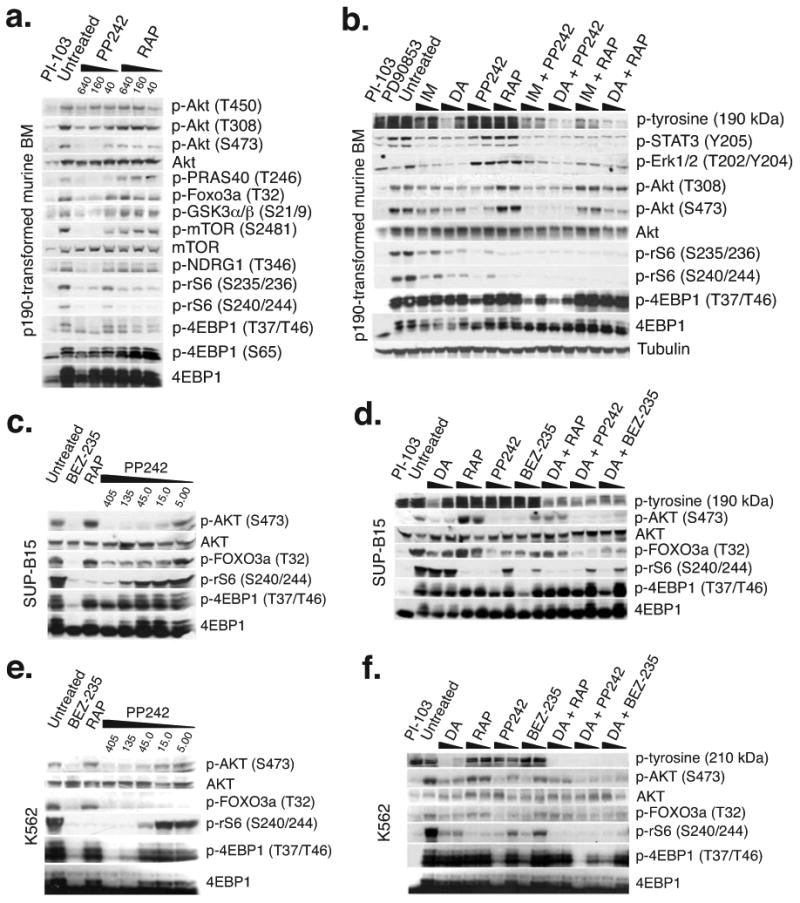
PP242 completely inhibits TORC1 and TORC2 signaling in BCR-ABL+ cells whereas rapamycin partially suppresses TORC1 and drives a PI3K/AKT surge. (a–b) Western blots of p190 cells treated for 1.5 hr (a) or 3 hr (b) with indicated inhibitors. Cells were treated with imatinib (IM; 0.5 and 1.0 μM), DA (5 and 50 nM), PP242 and RAP (50 and 400 nM). Clinically achievable concentrations of IM (1.0 μM) and DA (100 nM) were used for the combination treatments. (c–d) Western blots of SUP-B15 or (e–f) K562 cells treated for 3 hr with indicated inhibitors.
Although PP242 at high concentrations can inhibit PI3K enzymes in vitro 14, the drug did not alter cellular production of the PI3K product phosphatidylinositol-3,4,5-trisphosphate (PIP3) (Fig. 3a). In contrast, rapamycin treatment increased PIP3. It is possible that PP242 counteracts feedback activation of PI3K through weak inhibition of PI3K enzymes.
Figure 3.
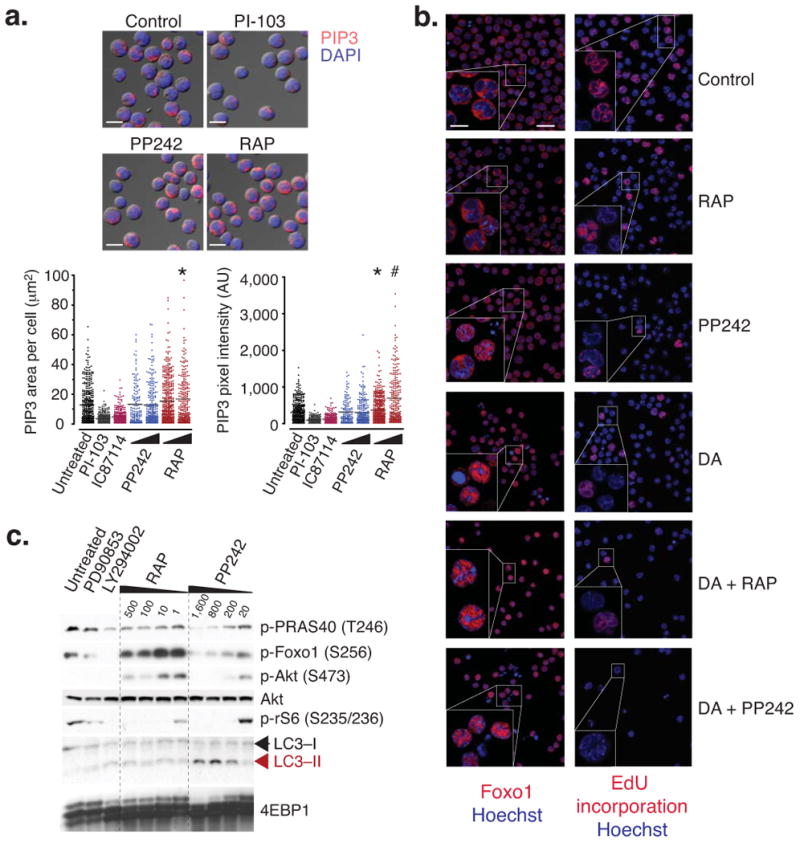
Immunofluorescence analysis shows that PP242 induces Foxo1 nuclear entry without affecting PIP3 levels. (a) Activation of PI3K was quantified in cells by signal pixel intensity and localized area of PIP3 accumulation by confocal microscopy (*P < 0.05, #P < 0.001, ANOVA). Cells were cultured for 4 hr with PI-103 (2 μM), the PI3K inhibitor IC87114 (10 μM), PP242 (20 and 200 nM), or RAP (20 and 200 nM). A minimum of 250 cells was quantified from 2 separate images. Representative images depict PIP3 accumulation, nuclear content (DAPI stain) merged onto DIC images (13.5 μm scale bar). (b) p190 cells were cultured for 8 hr in chamber wells with DA (10 nM), PP242 (250 nM), BEZ-235 (250 nM), RAP (250 nM), and pulsed with EdU 1 hr prior to fixation. Loss of proliferation (EdU accumulation), and distinct localization patterns of Foxo1 were assessed by confocal microscopy (22.5 μm scale bar) and representative cells were magnified for clarity (10 μm scale bar). Hoechst is a nuclear stain. (c) Cells were treated with the Mek inhibitor PD90853 (10,000 nM), LY294002 (1,000 nM), and -indicated concentrations of RAP and PP242 for 24 hr. Lysates were analyzed by western blotting. An increase in LC3-II is characteristic of increased autophagy.
To assess TORC2 function, we examined the phosphorylation and localization of Forkhead Box O (Foxo) transcription factors, which are substrates of both Akt and Sgk (serum and glucocorticoid-induced kinase) downstream of TORC2 34,35. PP242, but not rapamycin, reduced Foxo phosphorylation on Akt consensus sites (Fig. 2a, c–f) and caused nuclear accumulation of Foxo1 correlating with greater inhibition of cell cycling (Fig. 3b). PP242, but not rapamycin, also reduced mTOR autophosphorylation on S2481, phosphorylation of the Akt substrates GSK3 and PRAS40, and of the Sgk substrate NDRG1, all indicators of TORC2 activity 14-17,36,37 (Fig. 2a). Thus, PP242 blocked all TORC1 and TORC2 outputs tested without altering PIP3 levels. PP242 also appeared to increase autophagy more than did rapamycin (Fig. 3c).
PP242 suppresses leukemia better than rapamycin in vivo
To compare the pharmacological characteristics and anti-leukemic efficacy of mTOR inhibitors alone or in combination with TKIs in vivo, we employed three models: (1) a mouse model of p190BCR-ABL pre-B lymphoid leukemia transplanted into syngeneic recipients; (2) a bioluminescent xenograft model of human SUP-B15 cells stably expressing luciferase, transplanted into immunocompromised NSG mice (NOD-SCID-IL2RγKO 38); (3) a xenograft model of four independent leukemia specimens into NSG mice (n = 4–5 recipients per treatment group, totaling ∼ 60 recipients; see also Supplementary Table 4).
The mouse p190 model produces reproducible engraftment and fatal leukemia in recipients that are either irradiated or nonirradiated, with disease resembling the pathologic features of human B-ALL, and is most convenient for both survival measurements and pharmacodynamic monitoring. In this model, short-term oral dosing with PP242 in a dose-dependent manner significantly reduced leukemic burden in the spleen and bone marrow, which correlated with a halt in cell cycle progression and induction of apoptosis (Fig. 4a–d and Supplementary Figs. 3–5). These effects were associated with inhibition of TORC1 and TORC2 substrate phosphorylation and a reduction of leukemic cell size (Fig. 4d and Supplementary Fig. 3). PI-103 (60 mg kg−1 twice per day (b.i.d.)) treatment significantly potentiated the effects of imatinib in this model (Fig. 4e), similar to our previous findings 19. In contrast, mice treated with rapamycin showed no significant reduction in leukemic burden (Fig. 4e and Supplementary Fig. 4). This was not due to poor bioavailability because rapamycin treatment caused strong inhibition of rS6 phosphorylation (Supplementary Fig. 4), and was severely immunosuppressive at this dose (see below). We also detected decreased pAkt-S473 in rapamycin-treated mice, consistent with reports that rapamycin can inhibit assembly of TORC2 after extended exposure to the drug 39.
Figure 4.
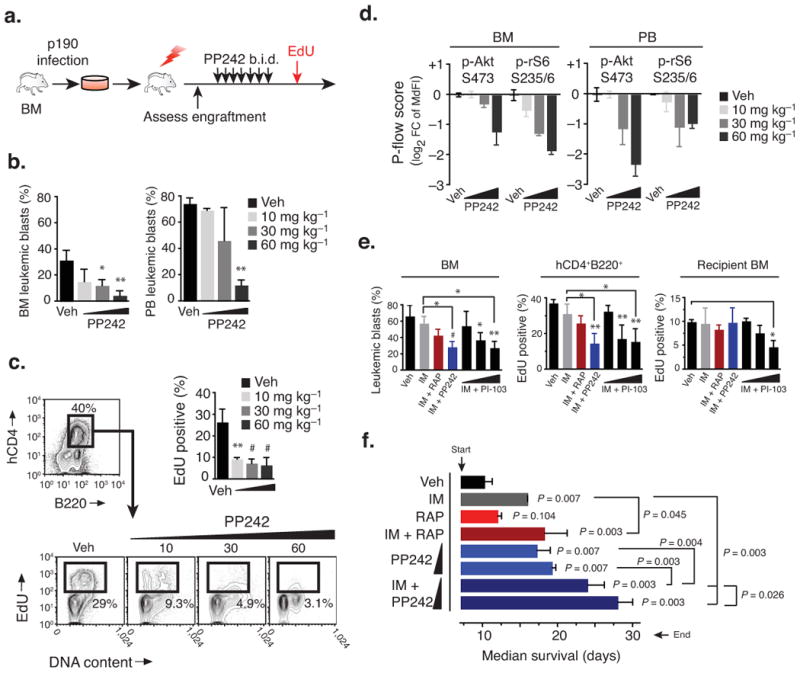
PP242 selectively suppresses leukemic expansion in vivo and extends survival. (a–d) Short-term anti-leukemic efficacy of PP242 in conditioned recipients (450 rad) engrafted with mouse p190 cells. (a) Schematic of treatment design where mice injected i.v. with p190 cells (n = 3) were treated twice daily (p.o., b.i.d.) starting on day 7 for 4 days, with PP242 or vehicle (PEG400). (b) Leukemic burden (mean % ± s.d., ANOVA) was assessed by flow cytometry. (c) Leukemic cells actively cycling (EdU+) was measured by flow cytometry (mean ± s.d., ANOVA). (d) The pharmacodynamic activity of PP242 was assessed using intracellular phospho-staining of leukemic (hCD4+B220+) cells from the bone marrow of recipient mice. (e) Short-term treatment study comparing p190 leukemia burden as in (a), but comparing combinations of IM (150 mg kg−1 i.p., qd) with RAP (7 mg kg−1 i.p., qd), PP242 (60 mg kg−1 p.o., qd), or different doses of PI-103 (i.p., b.i.d.). Graphs show leukemic burden in the bone marrow (left), or percent cycling cells among leukemic hCD4+ (middle) or host hCD4− bone marrow (right). (f) Mice injected with p190 cells were treated daily (qd) starting on day 7 post-transplant with IM (150 mg kg−1, i.p.), RAP (7 mg kg−1, i.p.) and PP242 (30 and 60 mg kg−1, p.o.). Mice were followed daily for survival (n = 5 per group, median ± interquartile range).
In a long-term survival study, oral dosing of PP242 (30 and 60 mg kg−1) significantly delayed the onset of leukemia (Fig. 4f). Rapamycin had no effect on its own, but augmented the protective effect of a suboptimal dose of imatinib (150 mg kg−1) as reported 40. The combination of PP242 with imatinib provided the greatest survival benefit, with 2 of 5 mice displaying minimal disease (2–20% in the bone marrow) even after one month (Fig. 4f and Supplementary Figure 3b).
In the xenograft models, we focused on the ability of PP242 to enhance the efficacy of dasatinib, an emerging standard-of-care for treatment of patients with Ph+ B-ALL. This was motivated in part by the greater efficacy of combination treatments in human Ph+ B-ALL colony assays (Fig. 1), and also by the expectation that novel, active-site mTOR inhibitors will be tested clinically in combination with approved TKIs. The SUP-B15-luciferase xenograft model allowed us to perform temporal quantitation of leukemia cell expansion using sequential bioluminescent imaging, and to correlate these results with signaling differences observed in SUP-B15 cells treated with drug combinations in vitro (Fig. 2c–d). We chose a daily dose of dasatinib (2.5 mg kg−1) reported to be efficacious in K562 xenografts 41. This dose modestly delayed SUP-B15 expansion (Fig. 5a), providing a useful system to model partial TKI resistance. Although rapamycin enhanced the effect of dasatinib, leukemic burden continued to increase with this combination. In contrast, the combination of dasatinib with PP242 caused regression of leukemic disease, and prevented dissemination to the central nervous system (Fig. 5a).
Figure 5.
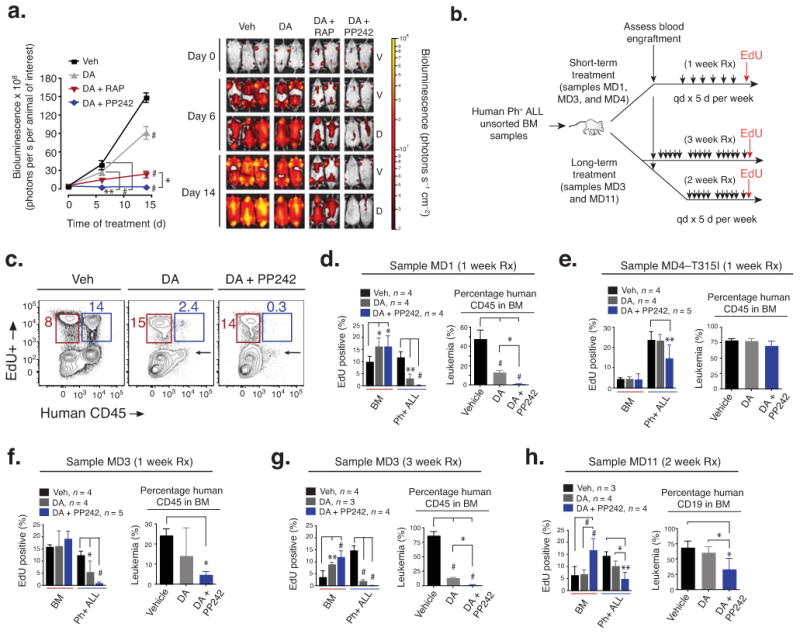
PP242 enhances efficacy of dasatinib and the combination causes regression of human Ph+ B-ALL xenografts. (a) SUP-B15ffLuc cells were injected into NSG mice and leukemia development was monitored by sequential bioluminescent imaging. The scale on the right shows the color scheme for low (red) to high (yellow) photon flux. Both ventral (V) and dorsal (D) images were taken, allowing detection of luminescent cells in the brain and spinal cord. Representative images are shown. The graph on the left shows the bioluminescence (photon flux) detected in each treatment group (mean ± s.d., RM-ANOVA). Vehicle (n = 4), DA (2.5 mg kg−1, n = 5), or DA combined with PP242 (60 mg kg−1, n = 5) or combined with RAP (7.5 mg kg−1). (b) Schematic of treatment design for xenografts of primary human Ph+ B-ALL. NSG mice were treated five days per week (samples MD1, MD3, MD4 for 1 week; MD11 for 2 weeks; and MD3 for 3 weeks) with DA, DA combined with PP242, or vehicle. (c–h) Leukemic burden (mean ± s.d., ANOVA) and cycling cells (mean ± s.d., two-way ANOVA) were assessed by flow cytometry. (c) shows representative cycling ability (EdU+) of normal marrow (red gate) and leukemic (blue gate) populations. Arrow signifies selective regression of leukemia. The total leukemic burden (panels d–h) was determined from %hCD45 or hCD19 in the BM. *P < 0.05, **P < 0.01, #P < 0.001.
In xenograft experiments using primary human leukemia samples, we tested PP242 in combination with a higher dose of dasatinib (5 mg kg−1) that significantly suppressed expansion of some Ph+ B-ALL samples (MD1 and MD3) but not others (MD4 and MD11) (Fig. 5b–h and Supplementary Fig. 6). The combination of PP242 and dasatinib significantly reduced leukemia burden and/or leukemia cell cycling (Fig. 5b–h and Supplementary Fig. 6). Dasatinib combined with PP242 was more effective than dasatinib alone in the short-term (1 week) treatment study of sample MD1, and in a long-term (3 week) treatment study of sample MD3 (Fig. 5c–d, g). Dasatinib plus PP242 caused apoptosis of human CD19+ cells in the bone marrow of xenograft recipients, while supporting recovery of cycling mouse recipient hematopoietic cells (Fig. 5c–d, g–h and Supplementary Fig. 6b). Sample MD4 (T315I mutation in BCR-ABL) showed reduced proliferation in xenografted mice treated with dasatinib plus PP242 (Fig. 5e); although bone marrow leukemic burden was not significantly affected, spleen weight was reduced (Supplementary Fig. 6c). For sample MD11, which displayed dasatinib-resistance in the bone marrow of mice despite no detectable BCR-ABL mutations, 2 week treatment with dasatinib plus PP242 reduced leukemic burden, proliferation, and spleen weight (Fig. 5h and Supplementary Fig. 6c). The greater response of MD11 relative to MD4 might have resulted from a difference in leukemic burden at the start of treatment, different lengths of treatment, or leukemia-specific genetic differences. We did not observe drug-related toxicity in mice at therapeutic doses of PP242 (Supplementary Fig. 6d and data not shown).
TORC1/2 inhibitors are less immunosuppressive than rapamycin
Rapamycin and analogs are immunosuppressive and myelosuppressive 42. In contrast, we observed that in mice treated with PP242, there was a highly selective response of leukemia cells compared to normal hematopoietic cells. PP242 prevented infiltration of the spleen and lymph nodes by p190 cells while largely preserving the normal splenic architecture and relative T cell abundance (Supplementary Figs. 4 and 5). PP242 in combination with dasatinib significantly inhibited proliferation and survival of xenografted human B-ALL cells while allowing hematopoietic expansion of endogenous mouse marrow (Fig. 5c–d, g and Supplementary Fig. 6b). Rapamycin did not fully preserve splenic architecture or T cell counts in the p190 model (Supplementary Figs. 4 and 5b), and was myelosuppressive at much lower concentrations than PP242 in human hematopoietic colony-forming assays (Fig. 6a). The dual PI3K/mTOR inhibitor PI-103 yielded an activity comparable to PP242 in normal human hematopoietic cell colony assays (Fig. 6a). Considering that the GI50 of PI-103 in cancer cell lines was two-fold to ten-fold greater than the GI50 of PP242, it was striking that PI-103 showed much greater hematotoxicity at a dose (2 μM) that was twofold higher than the highest dose tested of PP242 (1 μM). We also observed that p190-transplanted mice treated with imatinib and a therapeutic dose of PI-103 (60 mg kg−1 b.i.d.), but not imatinib plus PP242, had reduced cycling of endogenous bone marrow cells (Fig. 4e).
Figure 6.
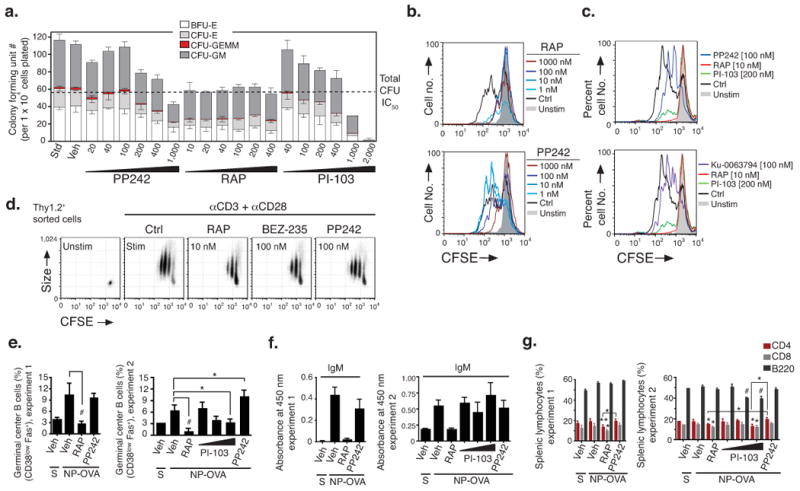
In vitro and in vivo selectivity of PP242 compared to rapamycin. (a) Hematopoietic clonogenic progenitors of bone marrow cells from a healthy human donor were assessed for colony formation in the presence of indicated concentrations of inhibitors. (b–d) Cell division tracking of mouse lymphocytes was done by labeling cells with CFSE before incubation for 3 d with the indicated stimuli. Reduced CFSE fluorescence denotes cell division. The histogram overlays (b–c) show cell division history of CFSE-labeled B cells pretreated with RAP, PP242, PI-103 or Ku-0063794 (15 min) before stimulation with anti-IgM (b) or LPS (c). Dot plots (d) depict CFSE fluorescence versus cell size (forward scatter) of activated T cells pretreated with RAP, PP242 or BEZ-235. Shown are representative examples of 3–5 independent experiments. In (b–c), upper and lower histograms are from same experiment. (e–g) Groups of 3–4 mice were immunized with NP-OVA in alum (i.p.) and immune responses measured 8 days later. Mice were treated with vehicle, RAP (7.5 mg kg−1, i.p., qd), PP242 (60 mg kg−1, p.o., qd), or PI-103 (10, 30 or 60 mg kg−1, i.p., b.i.d.) starting one day before immunization. S = sham immunized. (e) NP-specific IgM in serum were quantified by ELISA (mean ± s.d.); equivalent results were obtained for NP-specific IgG1 (data not shown). (f) Splenic GCB cells (CD38lowFas+, gated on B220+IgD−) and (g) total B cells and T cell subsets were quantified (mean ± s.d.) by flow cytometry.
Remarkably, PP242 showed consistently weaker effects than rapamycin in assays of adaptive immune function. Rapamycin (1–10 nM) strongly suppressed ex vivo B and T cell proliferation (Fig. 6b–d and Supplementary Fig. 7). In contrast, PP242 had little effect at 1–10 nM, with increasing effects at 100 nM and 1000 nM, the latter far exceeding effective concentrations in leukemia cell lines. The reduced anti-proliferative potency of PP242 in primary lymphocytes was not due to altered signaling impact as the compound inhibited phosphorylation of Akt, 4EBP1, and rS6 (Supplementary Fig. 7c), and reduced the size of activated T cells (Fig. 6d) at concentrations similar to those found to be effective in p190 cells (Fig. 2a–b). As observed in human hematopoietic colony assays, PI-103 suppressed normal B cell proliferation much more strongly when tested at concentrations two-fold higher than PP242 (Fig. 6c). Another panPI3K-TORC1/2 inhibitor, BEZ-235, suppressed T cell proliferation more strongly than PP242 when the compounds were compared at equal concentrations (Fig. 6d and Supplementary Fig. 7b). Similar to PP242, the TORC1/2 inhibitor Ku-0063794 had little effect on B cell proliferation when present at 100 nM (Fig. 6c) and partially inhibited the response at 1000 nM (data not shown).
To compare the effects of PP242 and rapamycin on adaptive immunity in vivo, we measured antibody responses to the T cell-dependent antigen nitrophenyl-ovalbumin (NP-OVA). Mice were treated orally with PP242 (60 mg kg−1), a dose that achieved rapid leukemic clearance in other experiments (Fig. 4 and 5), or with rapamycin (7.5 mg kg−1), starting one day before immunization. Whereas rapamycin strongly reduced NP-specific antibody titers, PP242 had little or no effect (Fig. 6e). These patterns were reflected in the percentage of B cells with a germinal center (GC) phenotype (Fig. 6f). Furthermore, rapamycin treatment reduced the percentages of total T and B cells in the spleen whereas PP242 did not (Fig. 6g). In a separate experiment, we tested PI-103 at 10, 30 and 60 mg kg−1 b.i.d. Although PI-103-treated mice developed anti-NP antibody titers that were equivalent to vehicle or PP242-treated mice (Fig. 6e), PI-103 reduced the fraction of B cells with a GC phenotype (Fig. 6f) as well as the percentages of total splenic B and T cells (Fig. 6g). PI-103 also caused dose-dependent displacement of marginal zone B cells (Supplementary Fig. 8), a surrogate readout for PI3K (p110δ) inhibition 43.
Discussion
We have shown that a selective, active-site mTOR kinase inhibitor has potent anti-leukemic effects in vitro and in vivo, while leaving lymphocyte function largely intact. Inhibiting TORC1/2 addresses rapamycin-resistant TORC1 outputs, and prevents feedback AKT activation. TORC1/2 inhibition causes selective apoptosis of leukemic cells, and also synergizes with clinically relevant TKIs in assays of both mouse and human leukemic expansion. A likely mechanism is that the PI3K/mTOR pathway might be sustained by cytokines and serum factors even when BCR-ABL is inhibited. These findings support the importance of TORC1/2 as a druggable target downstream of BCR-ABL in a disease in which resistance to TKIs develops rapidly. TORC1/2 inhibitors might be particularly effective when used with current induction regimens consisting of dasatinib with or without chemotherapy in the Ph+ B-ALL setting.
Whereas PP242 is toxic to leukemia cells, normal hematopoietic cells and mature lymphocytes survive in mice treated with therapeutic doses. Moreover, lymphocyte function exhibits an inverse pattern of drug sensitivity relative to leukemia cells, with rapamycin having more potent suppressive effects than PP242. The finding that pharmacological TORC1/2 inhibition is tolerated by the adaptive immune system is important and surprising, considering that complete deletion of the gene encoding mTOR (Frap1) in T cells has more severe effects on proliferation 44. One possibility is that mTOR has a noncatalytic scaffolding function that is abolished by the full gene deletion but not by the active-site kinase inhibitor. Another possibility is that PP242 does not constitutively ablate mTOR kinase activity, at doses that achieve therapeutic anti-leukemic effects via “oncogenic shock” 45. At higher concentrations of PP242, lymphocyte proliferation was strongly suppressed in vitro. Of note, mice heterozygous for a kinase-dead mTOR exhibit normal lymphocyte proliferation 46, supporting the idea that partial and/or temporary TORC1/2 kinase inhibition is tolerated by normal lymphocytes.
An important issue is whether selective TORC1/2 inhibitors provide advantages over panPI3K-TORC1/2 pathway inhibitors. PI3K has numerous roles in cell survival, differentiation, metabolism and migration, some of which are independent of AKT and mTOR 1,47. Our data suggest that whereas TORC1/2 and panPI3K-TORC1/2 inhibitors both have anti-leukemic efficacy, the panPI3K-TORC1/2 inhibitors cause greater immune suppression. When we tested PI-103 at two-fold higher concentration than PP242, based on the GI50 differences in cancer cell lines, we observed greater suppression of hematopoietic colony formation and B cell proliferation. In vivo treatment of mice with PI-103 at concentrations that had anti-leukemic effects also reduced abundance of key lymphocyte subsets that were unaffected by PP242. A recent report also found that PI-103 reduces lymphocyte numbers in vivo, and showed that PI-103 suppresses immune rejection of melanoma xenografts 48. It remains to be seen whether panPI3K-TORC1/2 inhibitors will provide an acceptable therapeutic window in humans. Our data indicate that selective TORC1/2 inhibition is an attractive alternative that provides equivalent anti-leukemic efficacy while maintaining PI3K activity.
Overall, these findings provide encouragement for further preclinical and clinical studies of selective active-site mTOR inhibitors in cancer. Based on the efficacy of PP242 when combined with dasatinib in Ph+ B-ALL xenografts, incorporating TORC1/2 kinase inhibitors with TKIs that target other driving mutations may improve treatment outcomes in other leukemias and epithelial malignancies.
Methods
Chemical synthesis and inhibitors
We synthesized PP242 as described previously 49. We obtained imatinib, dasatinib, and rapamycin from LC Laboratories. We obtained LY294002 and PD98059 from Sigma-Aldrich. Some compounds were synthesized as described in patents (BEZ235: patent # WO 2006122806; IC87114: patent # WO 2001081346; PI-103: patent # WO 2001083456, TG101348, patent # WO 2007053452; Ku-0063794, patent # WO 2007060404).
Cell culture and viruses
We obtained SUP-B15 human Ph+ B-ALL cells, K562 cells and solid tumor cell lines (SKOV3, PC-3, 786-O, U87) from ATCC. The SUP-B15 cells stably expressing firefly luciferase (SUP-B15ffLuc) were a kind gift from Michael Jensen (City of Hope). We obtained retroviruses by transient transfection of 293T cells with various pMSCV vectors 19. We infected mouse hematopoietic cells using standard procedures, which are detailed in the Supplementary Methods.
In vitro cell viability studies, cap pull-down assay and protein expression analysis
We performed cell viability assays, retroviral transduction, cap pull-down, western blot, flow cytometry assays, and confocal fluorescence microscopy following standard procedures, which are detailed in the Supplementary Methods.
Mice
All mice were kept in specific pathogen-free animal facilities at the University of California, Irvine, and all mouse procedures were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of University of California, Irvine. We used 8-week-old female BALB/cJ (Jackson Laboratory) mice as recipients of mouse p190 BCR-ABL transformed BM as has been previously described 19. We used 6–12-week-old male and female NSG (JAX mouse stock name NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ; Jackson Laboratory) as recipients for human leukemic transplants 38. We used 8-week-old female C57BL/6J (Jackson Laboratory) mice for immunization experiments, and 6–8-week-old Balb/cJ (Jackson Laboratory) mice for ex vivo lymphocyte cultures.
Primary leukemia samples
Cryopreserved peripheral blood samples were provided by one of the authors (M.B. Lilly) while treating the subjects at Loma Linda Medical Center, Loma Linda, California, USA. Heparanized peripheral blood was collected during the course of routine clinical care or as part of a Loma Linda Medical Center Institutional Review Board-approved specimen bank protocol. Their use for this study was approved by the University of California Irvine Institutional Review Board, with the stipulation that the samples lacked personal identifiers and that the donors were either deceased or had initially signed a specimen banking consent. We obtained cryopreserved bone marrow from the University of Texas M.D. Anderson Cancer Center with approval of their Institutional Review Board. All bone marrow samples were collected during routine diagnostic procedures after informed consent was obtained in accordance with the institutions regulations and the Declaration of Helsinki. Brief clinical history, pathological characteristics, and procedures of isolation, purification, and culturing of leukemic samples have been previously described 19, and are further detailed in the Supplementary Methods.
Primary human bone marrow cultures
We used a contract service (Stem Cell Technologies) to compare the effects of inhibitors on human bone marrow colony formation. Details are provided in the Supplementary Methods.
Leukemia transplantation procedures
Transplantation, engraftment procedures, bioluminescent imaging procedures, and flow cytometric evaluation of disease endpoints are further detailed in the Supplementary Methods.
In vivo drug formulation and treatments
Details of formulation of all compounds are provided in the Supplementary Methods. For Balb/cJ recipients engrafted with p190 cells, we treated mice with dosing schedules as indicated in the figure legends. For NSG recipients engrafted with SUP-B15ffLuc cells, we treated mice with daily doses of vehicle, dasatinib (2.5 mg kg−1, p.o.) alone or in combination with rapamycin (7.5 mg kg−1, i.p.) or PP242 (60 mg kg−1, p.o.) throughout the full treatment duration. All recipients received both p.o. and i.p. doses of vehicle or equivalent for fidelity. For NSG recipients engrafted with primary human bone marrow samples, we treated mice with daily doses (5 d per week with a consecutive 2 d rest) of vehicle, dasatinib (5 mg kg−1, p.o.) alone or in combination with PP242 (60 mg kg−1, p.o.). For C57BL/6J mice used for NP-OVA immunizations, we treated animals with dosing schedules as indicated in the figure legends.
Immunizations and measurement of NP-specific immunoglobulin
We immunized mice with NP(15)-ovalbumin (Biosearch Technologies) in Imject Alum (Pierce) at a concentration of 0.5 mg ml−1 (50 μg per mouse) by i.p. injection. Mice were sacrificed 8 days post-immunization for immune assessment. We detected serum Ig by ELISA using standard procedures that are further detailed in the Supplementary Methods.
Statistical analysis
To validate the significance of the observed differences, we analyzed random continuous variables using two-sided t tests, one-way ANOVA, repeated measures ANOVA (RM-ANOVA), and two-way ANOVA. Tukey-Kramer post-hoc analysis was used throughout. We compared differences in survival using the log-rank test. Homogeneity of variances was calculated by Bartlett's test. We used two-tailed Pearson's correlation to find correlation between continuous variables. We used GraphPad Prism (4.0c) software for all statistical analysis.
Supplementary Material
Supplementary Figure 1: TKI combination studies with mouse BM hematopoietic cultures transformed with native and mutant p190 BCR-ABL
Supplementary Figure 2: Distinct effects of PP242 and rapamycin in cap-binding assay
Supplementary Figure 3: PP242 induces leukemia regression in vivo by inhibiting mTORC1 and mTORC2 activation that correlates with loss in cell size
Supplementary Figure 4: Comparison of PP242 and rapamycin in vivo, in short-term p190 leukemia model with non-irradiated immunocompetent recipients
Supplementary Figure 5: Spleen pathology of p190 leukemia model comparing PP242 and rapamycin treatment
Supplementary Figure 6: TKI combination studies with PP242 in primary Ph+ BALL xenograft model
Supplementary Figure 7: Lymphocyte function in the presence of pharmacologic mTOR kinase inhibition
Supplementary Figure 8: Comparison of splenic architecture in NP-OVA immunized mice treated with PP242, rapamycin, or PI-103
Supplementary Table 1: PP242 pharmacologic properties
Supplementary Table 2: Drug GI50 values and synergy studies in p190 cells
Supplementary Table 3: Drug GI50 values in solid tumor cell lines
Supplementary Table 4: Clinical history and characteristics of human samples
Acknowledgments
We thank K. Shokat, T. Wilson, and M. Kharas for support and helpful discussions, and R. Nguyen for constructing BCR-ABL mutants. For access to the LSM710 Zeiss Confocal Microscope we thank E. Gratton's Lab, Department of Biomedical Engineering, University of California, Irvine. Technical assistance was provided through the Optical Biology Core facility of the Developmental Biology Center, a Shared Resource supported in part by the Cancer Center Support Grant (CA-62203) and Center for Complex Biological Systems Support Grant (GM-076516) at the University of California, Irvine. The Lumina IVIS bioluminescent imager was supported by a grant to University of California, Irvine from the California Institute of Regenerative Medicine. This work was supported by NIH training grant T32-CA009054 (to MRJ), NIH MARC grant T34GM069337 (to MAC), a Research Scholar Grant from the American Cancer Society (to DAF), a Discovery Grant from the University of California Industry-University Cooperative Research Program (to DAF), a sponsored research agreement from Intellikine, Inc. (to DAF), and a Bridge award from the Chao Family Comprehensive Cancer Center. MRJ is an awardee of the Jackie Murphy and Carol Malouf Scholar Achievement Rewards for College Scientists.
Footnotes
Author Contribution Statement: MRJ designed and performed experiments, analyzed data, and wrote the manuscript. JJL, LS, JC, and CV designed, performed experiments and analyzed data.
MAC, RJL and SM performed experiments and analyzed data.
MBL, STO and MK provided clinical samples, analyzed data and edited the manuscript.
MBM and PR provided materials and supervised groups performing chemical synthesis and formulation.
YL and CR designed and supervised experiments and edited the manuscript.
DAF designed and supervised experiments and wrote the manuscript.
Competing Interests: MBM, PR, YL and CR are employees of Intellikine, a company developing mTOR inhibitors for therapeutic use. DAF is a member of the Scientific Advisory Board of Intellikine.
References
- 1.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 2.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 4.Martelli AM, et al. Involvement of the phosphoinositide 3-kinase/Akt signaling pathway in the resistance to therapeutic treatments of human leukemias. Histol Histopathol. 2005;20:239–252. doi: 10.14670/HH-20.239. [DOI] [PubMed] [Google Scholar]
- 5.Wee S, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286–4293. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- 6.Yap TA, et al. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8:393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Abraham RT. Regulation of the mTOR signaling pathway: from laboratory bench to bedside and back again. F1000 Biology Reports. 2009;1:a8. doi: 10.3410/B1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 9.Abraham RT, Eng CH. Mammalian target of rapamycin as a therapeutic target in oncology. Expert Opin Ther Targets. 2008;12:209–222. doi: 10.1517/14728222.12.2.209. [DOI] [PubMed] [Google Scholar]
- 10.Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- 11.Janes MR, Fruman DA. Immune regulation by rapamycin: moving beyond T cells. Sci Signal. 2009;2:pe25. doi: 10.1126/scisignal.267pe25. [DOI] [PubMed] [Google Scholar]
- 12.Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. 2009;9:324–337. doi: 10.1038/nri2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feldman ME, et al. Active-Site Inhibitors of mTOR Target Rapamycin-Resistant Outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Martinez JM, et al. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR) Biochem J. 2009 doi: 10.1042/BJ20090489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thoreen CC, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu K, et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009;69:6232–6240. doi: 10.1158/0008-5472.CAN-09-0299. [DOI] [PubMed] [Google Scholar]
- 18.Chiarini F, et al. Dual inhibition of class IA phosphatidylinositol 3-kinase and mammalian target of rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Res. 2009;69:3520–3528. doi: 10.1158/0008-5472.CAN-08-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kharas MG, et al. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. J Clin Invest. 2008;118:3038–3050. doi: 10.1172/JCI33337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kojima K, et al. The dual PI3 kinase/mTOR inhibitor PI-103 prevents p53 induction by Mdm2 inhibition but enhances p53-mediated mitochondrial apoptosis in p53 wild-type AML. Leukemia. 2008 doi: 10.1038/leu.2008.158. [DOI] [PubMed] [Google Scholar]
- 21.Park S, et al. PI-103, a dual inhibitor of Class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia. 2008 doi: 10.1038/leu.2008.144. [DOI] [PubMed] [Google Scholar]
- 22.Burgess MR, Sawyers CL. Treating imatinib-resistant leukemia: the next generation targeted therapies. ScientificWorldJournal. 2006;6:918–930. doi: 10.1100/tsw.2006.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112:4808–4817. doi: 10.1182/blood-2008-07-077958. [DOI] [PubMed] [Google Scholar]
- 24.Li S, Ilaria RL, Jr, Million RP, Daley GQ, Van Etten RA. The P190 P210 P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J Exp Med. 1999;189:1399–1412. doi: 10.1084/jem.189.9.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raynaud FI, et al. Pharmacologic Characterization of a Potent Inhibitor of Class I Phosphatidylinositide 3-Kinases. Cancer Res. 2007;67:5840–5850. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- 26.Maira SM, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 27.Gruber F, Mustjoki S, Porkka K. Impact of tyrosine kinase inhibitors on patient outcomes in Philadelphia chromosome-positive acute lymphoblastic leukaemia. Br J Haematol. 2009;145:581–597. doi: 10.1111/j.1365-2141.2009.07666.x. [DOI] [PubMed] [Google Scholar]
- 28.Talpaz M, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–2541. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 29.Hu Y, et al. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci U S A. 2006;103:16870–16875. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCubrey JA, et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008;22:708–722. doi: 10.1038/leu.2008.27. [DOI] [PubMed] [Google Scholar]
- 31.Foa R, et al. Line Treatment of Adult Ph+ Acute Lymphoblastic Leukemia (ALL) Patients. Final Results of the GIMEMA LAL1205 Study. Blood. 2008;112:A305. [Google Scholar]
- 32.Prabhu S, et al. A novel mechanism for Bcr-Abl action: Bcr-Abl-mediated induction of the eIF4F translation initiation complex and mRNA translation. Oncogene. 2007;26:1188–1200. doi: 10.1038/sj.onc.1209901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang M, et al. Inhibition of polysome assembly enhances imatinib activity against chronic myelogenous leukemia and overcomes imatinib resistance. Mol Cell Biol. 2008;28:6496–6509. doi: 10.1128/MCB.00477-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hedrick SM. The cunning little vixen: Foxo and the cycle of life and death. Nat Immunol. 2009;10:1057–1063. doi: 10.1038/ni.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479–2487. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 36.Copp J, Manning G, Hunter T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009;69:1821–1827. doi: 10.1158/0008-5472.CAN-08-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 38.Kennedy JA, Barabe F. Investigating human leukemogenesis: from cell lines to in vivo models of human leukemia. Leukemia. 2008;22:2029–2040. doi: 10.1038/leu.2008.206. [DOI] [PubMed] [Google Scholar]
- 39.Sarbassov DD, et al. Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 40.Mohi MG, et al. Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc Natl Acad Sci U S A. 2004;101:3130–3135. doi: 10.1073/pnas.0400063101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo FR, et al. Dasatinib (BMS-354825) pharmacokinetics and pharmacodynamic biomarkers in animal models predict optimal clinical exposure. Clin Cancer Res. 2006;12:7180–7186. doi: 10.1158/1078-0432.CCR-06-1112. [DOI] [PubMed] [Google Scholar]
- 42.Sankhala K, et al. The emerging safety profile of mTOR inhibitors, a novel class of anticancer agents. Target Oncol. 2009;4:135–142. doi: 10.1007/s11523-009-0107-z. [DOI] [PubMed] [Google Scholar]
- 43.Durand CA, et al. Phosphoinositide 3-kinase p110delta regulates natural antibody production, marginal zone and B-1 B cell function, and autoantibody responses. J Immunol. 2009;183:5673–5684. doi: 10.4049/jimmunol.0900432. [DOI] [PubMed] [Google Scholar]
- 44.Delgoffe GM, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma SV, Fischbach MA, Haber DA, Settleman J. “Oncogenic shock”: explaining oncogene addiction through differential signal attenuation. Clin Cancer Res. 2006;12:4392s–4395s. doi: 10.1158/1078-0432.CCR-06-0096. [DOI] [PubMed] [Google Scholar]
- 46.Shor B, Cavender D, Harris C. A kinase-dead knock-in mutation in mTOR leads to early embryonic lethality and is dispensable for the immune system in heterozygous mice. BMC Immunol. 2009;10:28. doi: 10.1186/1471-2172-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fruman DA, Bismuth G. Fine tuning the immune response with PI3K. Immunol Rev. 2009;228:253–272. doi: 10.1111/j.1600-065X.2008.00750.x. [DOI] [PubMed] [Google Scholar]
- 48.Lopez-Fauqued M, et al. The dual PI3K/mTOR inhibitor (PI-103) promotes immunosupression, in vivo tumor growth and increases survival of sorafenib treated melanoma cells. Int J Cancer. 2009 doi: 10.1002/ijc.24926. [DOI] [PubMed] [Google Scholar]
- 49.Apsel B, et al. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4:691–699. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: TKI combination studies with mouse BM hematopoietic cultures transformed with native and mutant p190 BCR-ABL
Supplementary Figure 2: Distinct effects of PP242 and rapamycin in cap-binding assay
Supplementary Figure 3: PP242 induces leukemia regression in vivo by inhibiting mTORC1 and mTORC2 activation that correlates with loss in cell size
Supplementary Figure 4: Comparison of PP242 and rapamycin in vivo, in short-term p190 leukemia model with non-irradiated immunocompetent recipients
Supplementary Figure 5: Spleen pathology of p190 leukemia model comparing PP242 and rapamycin treatment
Supplementary Figure 6: TKI combination studies with PP242 in primary Ph+ BALL xenograft model
Supplementary Figure 7: Lymphocyte function in the presence of pharmacologic mTOR kinase inhibition
Supplementary Figure 8: Comparison of splenic architecture in NP-OVA immunized mice treated with PP242, rapamycin, or PI-103
Supplementary Table 1: PP242 pharmacologic properties
Supplementary Table 2: Drug GI50 values and synergy studies in p190 cells
Supplementary Table 3: Drug GI50 values in solid tumor cell lines
Supplementary Table 4: Clinical history and characteristics of human samples