

Abstract
Epidermolysis bullosa is a group of inherited disorders that can be both systemic and life-threatening. Standard treatments for the most severe forms of this disorder, typically limited to palliative care, are ineffective in reducing the morbidity and mortality due to complications of the disease. Emerging therapies—such as the use of allogeneic cellular therapy, gene therapy, and protein therapy—have all shown promise, but it is likely that several approaches will need to be combined to realize a cure. For recessive dystrophic epidermolysis bullosa, each particular therapeutic approach has added to our understanding of type VII collagen (C7) function and the basic biology surrounding the disease. The efficacy of these therapies and the mechanisms by which they function also give us insight into developing future strategies for treating this and other extracellular matrix disorders.
“The outcome of any serious research can only be to make two questions grow where only one grew before.”
Thorstein Veblen, The Evolution of the Scientific Point of View, 1908
Born to blister
Recessive dystrophic epidermolysis bullosa (RDEB) is a severe inherited skin disorder characterized by chronic skin blistering, diminished wound healing, joint contractures, esophageal strictures, pseudosyndactyly, corneal abrasions, and a shortened life span [1-3]. Affected individuals suffer through intense pain throughout their lives, with few or no effective treatments available to reduce the severity of their symptoms. Along with the life-threatening infectious complications associated with this disorder, many individuals will develop an aggressive form of squamous cell carcinoma [4,5].
RDEB is caused by mutations in COL7A1, the gene that encodes for C7 [6,7]. One of the most severe types of epidermolysis bullosa, RDEB is typically inherited in an autosomal-recessive fashion. It results from transfer of the mutated COL7A1 copies from both parents, who carry the mutation, to the affected offspring [8]. C7 is the main component of anchoring fibrils, structures that attach the dermis to the epidermis at the dermal-epidermal junction [9-11]. The inability of these anchoring fibrils to form and function properly causes the epidermis to not adhere to the underlying dermis [12]. This loss of structural integrity causes the skin to become susceptible to even slight trauma and also hinders the skin from healing productively [13,14]. It is likely that the constant cellular stress from the skin trying to heal itself, along with the resulting chronic inflammation, is the main reason for the increased risk of squamous cell carcinoma in individuals with RDEB [5,15-17].
Owing to its nature and severity, RDEB presents unique challenges for developing successful therapies that simultaneously alleviate the plethora of complications while having a significant impact on survival and quality of life. Recent approaches such as allogeneic cellular therapy, gene therapy, and protein therapy [18-23] show promise. Beyond the potential translational benefit of these studies, they have also significantly advanced our understanding of the biological properties of skin. Armed with this information and the recent technical advances, we believe the collective ability of multiple teams around the globe to both understand and treat RDEB is approaching a pivotal point in achieving effective, sustainable treatment options.
Allogeneic cellular therapies: from bench to bedside
Initial studies using allogeneic cells for the in situ treatment of epidermolysis bullosa included allogeneic fibroblasts [24-27] and mesenchymal stromal cells [28] and gene-corrected autologous epidermal stem cells [29]. These early studies using donor cells for local skin repair were crucial in demonstrating the capacity of allogeneic cells to correct this extracellular matrix disorder, but the benefits were limited to the site of application. Although the pathology of severe generalized RDEB is most apparent in the skin, its effects are numerous and systemic, and any therapy to treat the systemic manifestations requires broad delivery of C7 throughout the body. The prototype of cell therapy for genetic disorders is hematopoietic cell transplantation (HCT), which allows systemic and long-term distribution of donor cells in the recipient [30,31]. There is a growing amount of evidence describing the participation of cells with hematopoietic origin that are responsible for orchestrating and contributing to productive wound healing [32-35]. The process of wound healing in injured skin is complex, and a wide variety of cells from the bone marrow are recruited and participate in regulating inflammation, re-epithelialization, and extracellular matrix production [35].
Initial studies investigating the potential for bone marrow cells to treat extracellular matrix disorders confirmed this potential [36,37]. In a mouse transplantation model of RDEB, purified populations from the wildtype bone marrow were shown to home to injured skin and secrete C7 [37]. In turn, this improved the blistering phenotype and increased survival rates in treated mice. This approach was also shown to be effective in treating other forms of epidermolysis bullosa [38]. These studies provided the proof of principle needed for the first clinical trial using HCT to treat RDEB. The results from the initial patients enrolled in the clinical trial demonstrated the efficacy of HCTs and also revealed new information about how the bone marrow contributes to wound healing [18]. The patients treated with HCT not only displayed an increase in C7 deposition (Figure 1) but also showed a substantial level of donor chimerism in the skin following transplant. Exactly which cell types are responsible for homing to the skin, producing C7, and contributing to high levels of donor chimerism is still being determined, but several studies of this phenomenon have uncovered potentially relevant mechanisms. For example, a recent study described a particular subset of bone marrow cells expressing the surface marker platelet-derived growth factor receptor alpha that respond to a homing signal in injured skin, high-mobility group box (HMGB1) [39]. This subset was shown to produce C7 in the transplanted mouse model of RDEB. Other studies have demonstrated that certain subsets of bone marrow or cord blood cells were capable of producing C7 and that production increased in the context of wound healing [35,40]. Although it remains to be seen whether this subset of cells can be enriched prior to transplant or whether particular homing signals can be manipulated in order to improve transplant efficacy, these findings improve our understanding of how HCT can treat extracellular matrix disorders [41-43].
Figure 1. Increase in type VII collagen (C7) deposition and improvement of clinical symptoms after hematopoietic cell transplantation (HCT) treatment for recessive dystrophic epidermolysis bullosa (RDEB).
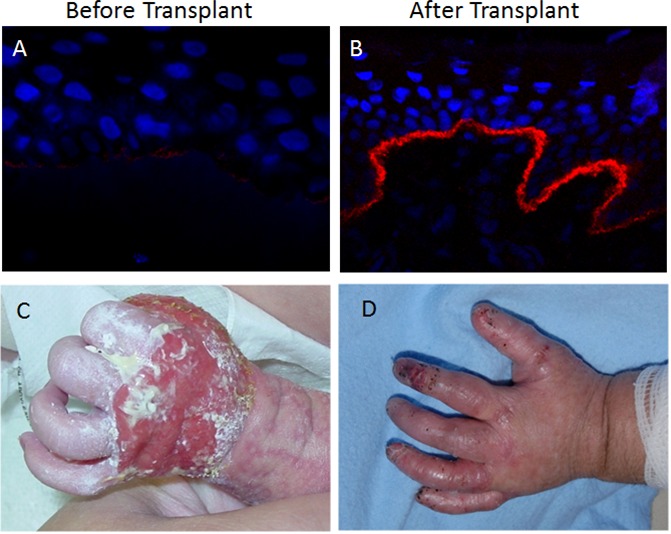
Immunofluorescent stain of C7 (red) and 4ʹ-6-diamidino-2-phenylindole (DAPI) (blue) visualizing the dermal-epidermal junction (A) before transplant and (B) 2.5 years after transplant. Photos of an RDEB patient presenting wounds over the back of the hand (C) before transplant and (D) the improvement after transplant.
Induced pluripotent stem cells: evidence-based approaches
Along with HCT, another option for future therapies in RDEB would be the use of cells derived from personalized induced pluripotent stem (iPS) cells [44-46]. In principle, iPS cells offer an inexhaustible supply of cells capable of differentiating into almost all cell types of the body. They have already been used in ex vivo modelling of many genetic diseases [47-49]. Skin cells isolated from both patients with RDEB and patients who suffer from the closely related disorder, junctional epidermolysis bullosa (JEB), can be reprogrammed into iPS cells that can be used to investigate the mechanisms of mucocutaneous destruction and wound healing in disorders with deficiencies in the protein complexes that support structural integrity of the epidermis and extracellular matrix of the dermis [50,51]. Furthermore, keratinocytes isolated from a healthy patch of skin from a patient with RDEB were reprogrammed into iPS cells [52]. The healthy patch of skin was determined to be a result of somatic mosaicism [53-55], and iPS cells derived from this healthy patch produced functional, biologically relevant levels of C7. These cells, and similar cells derived from mosaic patches in JEB individuals [56-58], represent a serendipitous opportunity for therapeutic use and a spearhead for the future of autologous cellular therapy [59]. RDEB iPS cells can also be differentiated into keratinocytes and fibroblasts, the two cell types that produce C7 in the skin, and can be used to construct full-thickness three-dimensional skin equivalents [60-62]. Fibroblasts, keratinocytes, and skin equivalents produced from iPS cells could be used therapeutically to treat localized, topical wounds. In addition to differentiation into skin cells and reconstruction of epidermis and dermis, recent studies showed that RDEB iPS cells can generate cells with surface markers similar to those expressed by human hematopoietic cells [63]. Intense efforts are under way to derive transplantable human iPS cell-derived hematopoietic stem cells that can be used for HCT [64-70]. These advances, along with the allogeneic HCT being used today, are the first steps needed in developing a more comprehensive therapy for RDEB. Other simultaneous advances in genome engineering should eventually allow a patient's own cells to be gene-corrected and then reprogrammed into iPS cells for use in autologous therapy.
Gene therapy: both inside and outside of the COL7A1 locus
Although allogeneic HCT is the most effective and widespread cellular therapy of genetic disorders to date, it requires a human leucocyte antigen-matched donor, and the HCT process itself can be life-threatening [71-75]. Autologous transplant would be a preferred option. Multiple approaches have been used for correcting COL7A1, including retroviral vectors, self-inactivating retroviral or lentiviral vectors, and retroviral vectors encoding a 3′ pre-trans-splicing molecule [19,76-78]. These approaches demonstrated that transduced cells were capable of producing functional and biologically significant levels of C7. Although viral-mediated transgenesis is an efficient way of correcting a genetic defect in patients' cells [79-83], correcting the endogenous mutation in situ in the genome could offer benefits over the use of viral vectors. Endogenous correction ensures physiological transcriptional control of COL7A1 and expression at biologically appropriate levels and—because the transgene is designed to not integrate in the host genome—reduces off-target, potentially oncogenic events caused by random insertional mutagenesis. Recently, genome-editing strategies using zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) have demonstrated the ability to target specific sites in the human genome and correct endogenous mutations [84-87]. TALENs have been used successfully in combination with homology-directed repair to correct the COL7A1 mutation in human fibroblasts from patients with RDEB (Figure 2) [87]. These corrected fibroblasts were capable of producing wildtype C7 with minimal off-target genomic effects. Moreover, these cells could be reprogrammed into iPS cells and, when xenotransplanted into immunodeficient mice, generated human skin-like structures with apparently normal C7 deposition. These data support the possibility that in situ correction of the COL7A1 locus leads to physiological C7 production and could offer therapeutic benefit to individuals with RDEB. Although TALEN correction appears to be a superior option to previous gene therapy methods, TALEN construction must be tailored to the particular COL7A1 loci that harbor the specific RDEB mutations, which can be both costly and labor-intensive [88]. As there are hundreds of causative mutations for RDEB characterized to date, using this approach on a larger scale may be challenging [89]. With the advent of clustered regulatory interspaced short palindromic repeats and associated proteins (CRISPR/Cas), the ability to correct multiple genetic mutations in human cells might have become considerably easier [90-92], although their off-target profile needs to be carefully analyzed [93].
Figure 2. Using transcription activator-like effector nucleases (TALENs) to genetically correct mutation in COL7A1 gene leads to phenotypic correction.
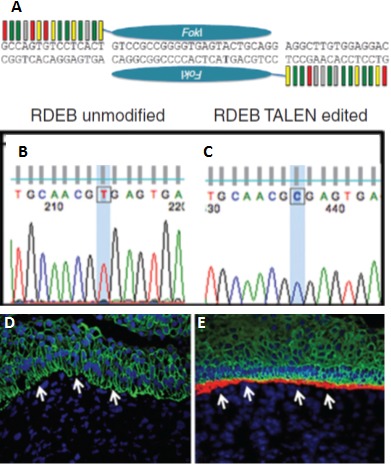
(A) Diagram of TALEN targeting COL7A1 mutation g.1837 C>T, which leads to a premature stop codon. Sequence analysis of base pair 1837 from (B) recessive dystrophic epidermolysis bullosa (RDEB) fibroblasts and (C) the corresponding TALEN-corrected fibroblasts. Immunofluorescent staining of type VII collagen in (D) skin-like structures formed from RDEB-induced pluripotent stem (RDEB-iPS) cells and (E) the corresponding TALEN-corrected iPS cells.
Protein therapy: translation of basic scientific insights
Protein therapy has been used for other inherited disorders of enzyme production due to the inherent capacity of affected cells to take up the missing enzyme [94,95]. Using protein therapy to treat inherited defects of structural protein production has been limited in comparison, but recent studies have demonstrated exciting results, specifically in using C7 protein therapy in pre-clinical models of RDEB [96,97]. Intradermal injections of C7 resulted in the stable incorporation of recombinant C7 into the basement membrane zone and corrected the phenotype in a murine model of RDEB. Intravenous injection of recombinant C7 into RDEB mice resulted in systemic biodistribution and deposition of C7 in wounded skin, but not in unaffected skin sites and internal organs [20]. It is likely that the soluble nature of C7 (unlike other collagens that aggregate and collect in the bloodstream) underlies both the safety and efficacy of systemic C7 infusion [98]. In addition, topical C7 application not only improves the phenotype in an RDEB murine model but may accelerate wound healing in skin that produces functional C7 as well [21]. Thus, the ability of C7 to promote healing in normal skin highlights its importance in coordinating cell migration and extracellular matrix organization in skin repair [13]. The necessary dosing levels and repeated applications of using recombinant C7 for RDEB patients has yet to be determined, although the initial pre-clinical studies have shown promising results with levels that should be attainable for clinical settings.
Future: finding a cure for the incurable
The future of medicine, including the quest to decrease suffering in individuals with RDEB, will involve a nuanced understanding of mechanisms underlying patient-specific therapies and combinatorial approaches to achieve the best possible outcomes. Although cellular therapies have been effective in ameliorating the severe generalized phenotype of RDEB, additional modifications, including local application of recombinant homing signals (such as HMGB1) or topical C7 therapy to remaining wounds, will likely complement the use of systemic cellular therapy. Also, inclusion of multiple cell types, such as hematopoietic stem/progenitor cells, mesenchymal stromal cells, fibroblasts, or keratinocytes, alone or after HCT, may speed and enhance wound healing. Local administration will be required in sites where systemic cell therapy offers little benefit, such as in the eyes, where limbal cell transplantation has been shown to be effective in treating other types of corneal disorders or trauma [3,99-101].
Certain aspects about why particular therapies are effective at treating RDEB are unknown, but findings from one therapy can give clues to questions that remain about another. For instance, the finding that intravenous injection of recombinant C7 results in C7 deposition at the dermal-epidermal junction of injured skin may have implications regarding the mechanisms of HCT for treating RDEB. It is conceivable that cells are not required to be in close proximity to the dermal-epidermal junction in order to produce the C7 that is deposited there. Rather, owing to the soluble nature of C7, cells from the graft could produce C7 in another site (such as the bone marrow), which is then taken up by the bloodstream and distributed systemically to injured skin. Thus, the beneficial effects of donor cells that are present near the dermal-epidermal junction following HCT can be amplified by these distant C7-producing cells. It has also been hypothesized that donor cells, such as those used in allogeneic fibroblast therapies, may not only be producing their own functional C7 but inducing recipient keratinocytes and fibroblasts to produce increased levels of mutant C7 as well, through induction via heparin-binding epidermal growth factor-like growth factor signaling [102]. Investigating such possibilities may help discover or define new roles for cell types or signals that were not previously known to be important for wound healing or extracellular matrix production in RDEB, other genodermatoses, and acquired skin disorders and injuries.
Along with new discoveries, critical information will become available following the treatment of these patients. Whether the new approaches are deemed successful will not only be evaluated by the long-term improvement of their daily lives but also by the reduction of the associated risks of RDEB, including squamous cell carcinoma and systemic infections. Integrating treatments for these complications will be necessary moving forward, as will expanding the use of novel therapies to more complicated cases. It also remains to be seen whether patients treated with cellular, genetic, or protein therapies develop an acquired immune response to antigens derived from the newly synthesized C7 that was not present before therapy, similar to the related autoimmune disorder epidermolysis bullosa acquisita [103-105]. No anti-C7 antibodies were detected initially in patients who received bone marrow transplant, but the long-term results remain to be determined [18]. In the case of HCT, reduced intensity conditioning and using alternative sources of hematopoietic cells may help improve survival rates and lessen the associated risks, such as graft-versus-host disease and infections. Further improvements and adjustments to these novel approaches will hopefully be made.
Acknowledgments
We apologize to the authors whose work could not be quoted because of space constraints. Jakub Tolar is supported by grants from the National Institutes of Health (R01 AR063070 and R01 AR059947), the Department of Defense (USAMRAA/DOD Department of the Army W81XWH-12-1-0609 and USAMRAA/DOD W81XWH-10-1-0874), DebRA International, the Jackson Gabriel Silver Fund, the Epidermolysis Bullosa Medical Research Fund, and the Children's Cancer Research Fund, Minnesota. Skin biopsies and photographs were collected after obtaining written informed patient consent as approved by the Institutional Review Board of the Human Subjects Committee at the University of Minnesota. All procedures adhered to the Helsinki Guidelines.
Abbreviations
- C7
type VII collagen
- HCT
hematopoietic cell transplantation
- iPS
induced pluripotent stem (cell)
- JEB
junctional epidermolysis bullosa
- RDEB
recessive dystrophic epidermolysis bullosa
- TALEN
transcription activator-like effector nuclease
Disclosures
The authors declare that they have no disclosures.
The electronic version of this article is the complete one and can be found at: http://f1000.com/prime/reports/m/6/35
References
- 1.Quality of life in epidermolysis bullosa. Clin Exp Dermatol. 2002;27:707–10. doi: 10.1046/j.1365-2230.2002.01121.x. [DOI] [PubMed] [Google Scholar]
- 2.Uitto J, Pulkkinen L, McLean WH. Epidermolysis bullosa: a spectrum of clinical phenotypes explained by molecular heterogeneity. Mol Med Today. 1997;3:457–65. doi: 10.1016/S1357-4310(97)01112-X. [DOI] [PubMed] [Google Scholar]
- 3.Fine J, Johnson LB, Weiner M, Stein A, Cash S, Deleoz J, Devries DT, Suchindran C. Eye involvement in inherited epidermolysis bullosa: experience of the National Epidermolysis Bullosa Registry. Am J Ophthalmol. 2004;138:254–62. doi: 10.1016/j.ajo.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 4.South AP, O'Toole EA. Understanding the pathogenesis of recessive dystrophic epidermolysis bullosa squamous cell carcinoma. Dermatol Clin. 2010;28:171–8. doi: 10.1016/j.det.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 5.Pourreyron C, Cox G, Mao X, Volz A, Baksh N, Wong T, Fassihi H, Arita K, O'Toole EA, Ocampo-Candiani J, Chen M, Hart IR, Bruckner-Tuderman L, Salas-Alanis JC, McGrath JA, Leigh IM, South AP. Patients with recessive dystrophic epidermolysis bullosa develop squamous-cell carcinoma regardless of type VII collagen expression. J Invest Dermatol. 2007;127:2438–44. doi: 10.1038/sj.jid.5700878. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1088981
- 6.Kern JS, Grüninger G, Imsak R, Müller ML, Schumann H, Kiritsi D, Emmert S, Borozdin W, Kohlhase J, Bruckner-Tuderman L, Has C. Forty-two novel COL7A1 mutations and the role of a frequent single nucleotide polymorphism in the MMP1 promoter in modulation of disease severity in a large European dystrophic epidermolysis bullosa cohort. Br J Dermatol. 2009;161:1089–97. doi: 10.1111/j.1365-2133.2009.09333.x. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1168314
- 7.Hovnanian A, Duquesnoy P, Blanchet-Bardon C, Knowlton RG, Amselem S, Lathrop M, Dubertret L, Uitto J, Goossens M. Genetic linkage of recessive dystrophic epidermolysis bullosa to the type VII collagen gene. J Clin Invest. 1992;90:1032–6. doi: 10.1172/JCI115916. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718345160
- 8.Fine J, Eady RAJ, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, Hintner H, Hovnanian A, Jonkman MF, Leigh I, McGrath JA, Mellerio JE, Murrell DF, Shimizu H, Uitto J, Vahlquist A, Woodley D, Zambruno G. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931–50. doi: 10.1016/j.jaad.2008.02.004. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1115711
- 9.Sakai LY, Keene DR, Morris NP, Burgeson RE. Type VII collagen is a major structural component of anchoring fibrils. J Cell Biol. 1986;103:1577–86. doi: 10.1083/jcb.103.4.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wegener H, Leineweber S, Seeger K. The vWFA2 domain of type VII collagen is responsible for collagen binding. Biochem Biophys Res Commun. 2013;430:449–53. doi: 10.1016/j.bbrc.2012.11.119. [DOI] [PubMed] [Google Scholar]
- 11.Rousselle P, Keene DR, Ruggiero F, Champliaud MF, Rest M, Burgeson RE. Laminin 5 binds the NC-1 domain of type VII collagen. J Cell Biol. 1997;138:719–28. doi: 10.1083/jcb.138.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruckner-Tuderman L, Has C. Disorders of the cutaneous basement membrane zone-The paradigm of epidermolysis bullosa. Matrix Biol. 2013 doi: 10.1016/j.matbio.2013.07.007. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718060536
- 13.Nyström A, Velati D, Mittapalli VR, Fritsch A, Kern JS, Bruckner-Tuderman L. Collagen VII plays a dual role in wound healing. J Clin Invest. 2013;123:3498–509. doi: 10.1172/JCI68127. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718056659
- 14.Küttner V, Mack C, Rigbolt KTG, Kern JS, Schilling O, Busch H, Bruckner-Tuderman L, Dengjel J. Global remodelling of cellular microenvironment due to loss of collagen VII. Mol Syst Biol. 2013;9(657) doi: 10.1038/msb.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718345161
- 15.Kita Y, Mimori K, Tanaka F, Matsumoto T, Haraguchi N, Ishikawa K, Matsuzaki S, Fukuyoshi Y, Inoue H, Natsugoe S, Aikou T, Mori M. Clinical significance of LAMB3 and COL7A1 mRNA in esophageal squamous cell carcinoma. Eur J Surg Oncol. 2009;35:52–8. doi: 10.1016/j.ejso.2008.01.025. [DOI] [PubMed] [Google Scholar]
- 16.Ortiz-Urda S, Garcia J, Green CL, Chen L, Lin Q, Veitch DP, Sakai LY, Lee H, Marinkovich MP, Khavari PA. Type VII collagen is required for Ras-driven human epidermal tumorigenesis. Science. 2005;307:1773–6. doi: 10.1126/science.1106209. [DOI] [PubMed] [Google Scholar]
- 17.Ng Y, Pourreyron C, Salas-Alanis JC, Dayal JHS, Cepeda-Valdes R, Yan W, Wright S, Chen M, Fine J, Hogg FJ, McGrath JA, Murrell DF, Leigh IM, Lane EB, South AP. Fibroblast-derived dermal matrix drives development of aggressive cutaneous squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa. Cancer Res. 2012;72:3522–34. doi: 10.1158/0008-5472.CAN-11-2996. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/717952969
- 18.Wagner JE, Ishida-Yamamoto A, McGrath JA, Hordinsky M, Keene DR, Woodley DT, Chen M, Riddle MJ, Osborn MJ, Lund T, Dolan M, Blazar BR, Tolar J. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med. 2010;363:629–39. doi: 10.1056/NEJMoa0910501. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/4901967
- 19.Titeux M, Pendaries V, Zanta-Boussif MA, Décha A, Pironon N, Tonasso L, Mejia JE, Brice A, Danos O, Hovnanian A. SIN retroviral vectors expressing COL7A1 under human promoters for ex vivo gene therapy of recessive dystrophic epidermolysis bullosa. Mol Ther. 2010;18:1509–18. doi: 10.1038/mt.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718345163
- 20.Woodley DT, Wang X, Amir M, Hwang B, Remington J, Hou Y, Uitto J, Keene D, Chen M. Intravenously injected recombinant human type VII collagen homes to skin wounds and restores skin integrity of dystrophic epidermolysis bullosa. J Invest Dermatol. 2013;133:1910–3. doi: 10.1038/jid.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718345164
- 21.Wang X, Ghasri P, Amir M, Hwang B, Hou Y, Khalili M, Khilili M, Lin A, Keene D, Uitto J, Woodley DT, Chen M. Topical application of recombinant type VII collagen incorporates into the dermal-epidermal junction and promotes wound closure. Mol Ther. 2013;21:1335–44. doi: 10.1038/mt.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718010068
- 22.Nyström A, Bruckner-Tuderman L, Kern JS. Cell- and protein-based therapy approaches for epidermolysis bullosa. Methods Mol Biol. 2013;961:425–40. doi: 10.1007/978-1-62703-227-8_29. [DOI] [PubMed] [Google Scholar]
- 23.Uitto J, Has C, Bruckner-Tuderman L. Cell-based therapies for epidermolysis bullosa - from bench to bedside. J Dtsch Dermatol Ges. 2012;10:803–7. doi: 10.1111/j.1610-0387.2012.08035.x. [DOI] [PubMed] [Google Scholar]
- 24.Wong T, Gammon L, Liu L, Mellerio JE, Dopping-Hepenstal PJC, Pacy J, Elia G, Jeffery R, Leigh IM, Navsaria H, McGrath JA. Potential of fibroblast cell therapy for recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2008;128:2179–89. doi: 10.1038/jid.2008.78. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1105013
- 25.Kern JS, Loeckermann S, Fritsch A, Hausser I, Roth W, Magin TM, Mack C, Müller ML, Paul O, Ruther P, Bruckner-Tuderman L. Mechanisms of fibroblast cell therapy for dystrophic epidermolysis bullosa: high stability of collagen VII favors long-term skin integrity. Mol Ther. 2009;17:1605–15. doi: 10.1038/mt.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1162581
- 26.Venugopal SS, Yan W, Frew JW, Cohn HI, Rhodes LM, Tran K, Melbourne W, Nelson JA, Sturm M, Fogarty J, Marinkovich MP, Igawa S, Ishida-Yamamoto A, Murrell DF. A phase II randomized vehicle-controlled trial of intradermal allogeneic fibroblasts for recessive dystrophic epidermolysis bullosa. J Am Acad Dermatol. 2013;69:898–908. e7. doi: 10.1016/j.jaad.2013.08.014. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718122968
- 27.Petrof G, Martinez-Queipo M, Mellerio JE, Kemp P, McGrath JA. Fibroblast cell therapy enhances initial healing in recessive dystrophic epidermolysis bullosa wounds: results of a randomized, vehicle-controlled trial. Br J Dermatol. 2013;169:1025–33. doi: 10.1111/bjd.12599. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718117495
- 28.Conget P, Rodriguez F, Kramer S, Allers C, Simon V, Palisson F, Gonzalez S, Yubero MJ. Replenishment of type VII collagen and re-epithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy. 2010;12:429–31. doi: 10.3109/14653241003587637. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/4066956
- 29.Mavilio F, Pellegrini G, Ferrari S, Di Nunzio F, Di Iorio E, Recchia A, Maruggi G, Ferrari G, Provasi E, Bonini C, Capurro S, Conti A, Magnoni C, Giannetti A, Luca M de. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 2006;12:1397–402. doi: 10.1038/nm1504. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1067075
- 30.Wynn R. Stem cell transplantation in inherited metabolic disorders. Hematology Am Soc Hematol Educ Program. 2011;2011:285–91. doi: 10.1182/asheducation-2011.1.285. [DOI] [PubMed] [Google Scholar]
- 31.Prasad VK, Kurtzberg J. Cord blood and bone marrow transplantation in inherited metabolic diseases: scientific basis, current status and future directions. Br J Haematol. 2010;148:356–72. doi: 10.1111/j.1365-2141.2009.07974.x. [DOI] [PubMed] [Google Scholar]
- 32.Arwert EN, Hoste E, Watt FM. Epithelial stem cells, wound healing and cancer. Nat Rev Cancer. 2012;12:170–80. doi: 10.1038/nrc3217. [DOI] [PubMed] [Google Scholar]
- 33.Wu Y, Zhao RCH, Tredget EE. Concise review: bone marrow-derived stem/progenitor cells in cutaneous repair and regeneration. Stem Cells. 2010;28:905–15. doi: 10.1002/stem.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sasaki M, Abe R, Fujita Y, Ando S, Inokuma D, Shimizu H. Mesenchymal stem cells are recruited into wounded skin and contribute to wound repair by transdifferentiation into multiple skin cell type. J Immunol. 2008;180:2581–7. doi: 10.4049/jimmunol.180.4.2581. [DOI] [PubMed] [Google Scholar]
- 35.Alexeev V, Uitto J, Igoucheva O. Gene expression signatures of mouse bone marrow-derived mesenchymal stem cells in the cutaneous environment and therapeutic implications for blistering skin disorder. Cytotherapy. 2011;13:30–45. doi: 10.3109/14653249.2010.518609. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718345165
- 36.Chino T, Tamai K, Yamazaki T, Otsuru S, Kikuchi Y, Nimura K, Endo M, Nagai M, Uitto J, Kitajima Y, Kaneda Y. Bone marrow cell transfer into fetal circulation can ameliorate genetic skin diseases by providing fibroblasts to the skin and inducing immune tolerance. Am J Pathol. 2008;173:803–14. doi: 10.2353/ajpath.2008.070977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tolar J, Ishida-Yamamoto A, Riddle M, McElmurry RT, Osborn M, Xia L, Lund T, Slattery C, Uitto J, Christiano AM, Wagner JE, Blazar BR. Amelioration of epidermolysis bullosa by transfer of wild-type bone marrow cells. Blood. 2009;113:1167–74. doi: 10.1182/blood-2008-06-161299. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1127147
- 38.Fujita Y, Abe R, Inokuma D, Sasaki M, Hoshina D, Natsuga K, Nishie W, McMillan JR, Nakamura H, Shimizu T, Akiyama M, Sawamura D, Shimizu H. Bone marrow transplantation restores epidermal basement membrane protein expression and rescues epidermolysis bullosa model mice. Proc Natl Acad Sci USA. 2010;107:14345–50. doi: 10.1073/pnas.1000044107. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718345166
- 39.Tamai K, Yamazaki T, Chino T, Ishii M, Otsuru S, Kikuchi Y, Iinuma S, Saga K, Nimura K, Shimbo T, Umegaki N, Katayama I, Miyazaki J, Takeda J, McGrath JA, Uitto J, Kaneda Y. PDGFRalpha-positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proc Natl Acad Sci USA. 2011;108:6609–14. doi: 10.1073/pnas.1016753108. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/9607956
- 40.Liao Y, Itoh M, Yang A, Zhu H, Roberts S, Highet AM, Latshaw S, Mitchell K, van de Ven C, Christiano A, Cairo MS. Human Cord Blood Derived Unrestricted Somatic Stem Cells Promote Wound Healing and Have Therapeutic Potential for Patients With Recessive Dystrophic Epidermolysis Bullosa. Cell Transplant. 2013 doi: 10.3727/096368913X663569. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718345167
- 41.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, Marchis F de, Liu J, Antonelli A, Preti A, Raeli L, Shams SS, Yang H, Varani L, Andersson U, Tracey KJ, Bachi A, Uguccioni M, Bianchi ME. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209:1519–28. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717953753
- 42.Alexeev V, Donahue A, Uitto J, Igoucheva O. Analysis of chemotactic molecules in bone marrow-derived mesenchymal stem cells and the skin: Ccl27-Ccr10 axis as a basis for targeting to cutaneous tissues. Cytotherapy. 2013;15:171–184. e1. doi: 10.1016/j.jcyt.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petrof G, Abdul-Wahab A, Proudfoot L, Pramanik R, Mellerio JE, McGrath JA. Serum levels of high mobility group box 1 correlate with disease severity in recessive dystrophic epidermolysis bullosa. Exp Dermatol. 2013;22:433–5. doi: 10.1111/exd.12152. [DOI] [PubMed] [Google Scholar]
- 44.Wu SM, Hochedlinger K. Harnessing the potential of induced pluripotent stem cells for regenerative medicine. Nat Cell Biol. 2011;13:497–505. doi: 10.1038/ncb0611-734b. doi: 10.1038/ncb0611-734b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1096022
- 46.Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481:295–305. doi: 10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu P, Paschon DE, Miranda E, Ordóñez A, Hannan NRF, Rouhani FJ, Darche S, Alexander G, Marciniak SJ, Fusaki N, Hasegawa M, Holmes MC, Di Santo JP, Lomas DA, Bradley A, Vallier L. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–4. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/13357095
- 48.Thatava T, Armstrong AS, Lamo JG de, Edukulla R, Khan YK, Sakuma T, Ohmine S, Sundsbak JL, Harris PC, Kudva YC, Ikeda Y. Successful disease-specific induced pluripotent stem cell generation from patients with kidney transplantation. Stem Cell Res Ther. 2011;2(48) doi: 10.1186/scrt89. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717968776
- 49.Hanna J, Wernig M, Markoulaki S, Sun C, Meissner A, Cassady JP, Beard C, Brambrink T, Wu L, Townes TM, Jaenisch R. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920–3. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1098499
- 50.Tolar J, Xia L, Riddle MJ, Lees CJ, Eide CR, McElmurry RT, Titeux M, Osborn MJ, Lund TC, Hovnanian A, Wagner JE, Blazar BR. Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2011;131:848–56. doi: 10.1038/jid.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tolar J, Xia L, Lees CJ, Riddle M, McElroy A, Keene DR, Lund TC, Osborn MJ, Marinkovich MP, Blazar BR, Wagner JE. Keratinocytes from induced pluripotent stem cells in junctional epidermolysis bullosa. J Invest Dermatol. 2013;133:562–5. doi: 10.1038/jid.2012.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tolar J, McGrath JA, Xia L, Riddle MJ, Lees CJ, Eide C, Keene DR, Liu L, Osborn MJ, Lund TC, Blazar BR, Wagner JE. Patient-Specific Naturally Gene-Reverted Induced Pluripotent Stem Cells in Recessive Dystrophic Epidermolysis Bullosa. J Invest Dermatol. 2013 doi: 10.1038/jid.2012.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pasmooij AMG, Garcia M, Escamez MJ, Nijenhuis AM, Azon A, Cuadrado-Corrales N, Jonkman MF, Del Rio M. Revertant mosaicism due to a second-site mutation in COL7A1 in a patient with recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2010;130:2407–11. doi: 10.1038/jid.2010.163. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/5642956
- 54.Almaani N, Nagy N, Liu L, Dopping-Hepenstal PJC, Lai-Cheong JE, Clements SE, Techanukul T, Tanaka A, Mellerio JE, McGrath JA. Revertant mosaicism in recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2010;130:1937–40. doi: 10.1038/jid.2010.64. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718345168
- 55.Lai-Cheong JE, Moss C, Parsons M, Almaani N, McGrath JA. Revertant mosaicism in Kindler syndrome. J Invest Dermatol. 2012;132:730–2. doi: 10.1038/jid.2011.352. [DOI] [PubMed] [Google Scholar]
- 56.Gostyński A, Pasmooij AMG, Jonkman MF. Successful therapeutic transplantation of revertant skin in epidermolysis bullosa. J Am Acad Dermatol. 2014;70:98–101. doi: 10.1016/j.jaad.2013.08.052. [DOI] [PubMed] [Google Scholar]
- 57.Gostyński A, Llames S, García M, Escamez MJ, Martinez-Santamaria L, Nijenhuis M, Meana A, Pas HH, Larcher F, Pasmooij AMG, Jonkman MF, Del Rio M. Long-Term Survival of Type XVII Collagen Revertant Cells in an Animal Model of Revertant Cell Therapy. J Invest Dermatol. 2014;134:571–4. doi: 10.1038/jid.2013.308. [DOI] [PubMed] [Google Scholar]
- 58.Pasmooij AMG, Jonkman MF, Uitto J. Revertant mosaicism in heritable skin diseases: mechanisms of natural gene therapy. Discov Med. 2012;14:167–79. [PubMed] [Google Scholar]
- 59.Lai-Cheong JE, McGrath JA, Uitto J. Revertant mosaicism in skin: natural gene therapy. Trends Mol Med. 2011;17:140–8. doi: 10.1016/j.molmed.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Itoh M, Kiuru M, Cairo MS, Christiano AM. Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc Natl Acad Sci USA. 2011;108:8797–802. doi: 10.1073/pnas.1100332108. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/11001956
- 61.Uitto J, Christiano AM, McLean WHI, McGrath JA. Novel molecular therapies for heritable skin disorders. J Invest Dermatol. 2012;132:820–8. doi: 10.1038/jid.2011.389. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718345169
- 62.Itoh M, Umegaki-Arao N, Guo Z, Liu L, Higgins CA, Christiano AM. Generation of 3D skin equivalents fully reconstituted from human induced pluripotent stem cells (iPSCs) PLoS ONE. 2013;8:e77673. doi: 10.1371/journal.pone.0077673. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718151082
- 63.Amabile G, Welner RS, Nombela-Arrieta C, D'Alise AM, Di Ruscio A, Ebralidze AK, Kraytsberg Y, Ye M, Kocher O, Neuberg DS, Khrapko K, Silberstein LE, Tenen DG. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood. 2013;121:1255–64. doi: 10.1182/blood-2012-06-434407. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717968294
- 64.Kelley JM, Daley GQ. Hematopoietic defects and iPSC disease modeling: lessons learned. Immunol Lett. 2013;155:18–20. doi: 10.1016/j.imlet.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Irion S, Clarke RL, Luche H, Kim I, Morrison SJ, Fehling H, Keller GM. Temporal specification of blood progenitors from mouse embryonic stem cells and induced pluripotent stem cells. Development. 2010;137:2829–39. doi: 10.1242/dev.042119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Szabo E, Rampalli S, Risueño RM, Schnerch A, Mitchell R, Fiebig-Comyn A, Levadoux-Martin M, Bhatia M. Direct conversion of human fibroblasts to multilineage blood progenitors. Nature. 2010;468:521–6. doi: 10.1038/nature09591. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/6305956
- 67.Grigoriadis AE, Kennedy M, Bozec A, Brunton F, Stenbeck G, Park I, Wagner EF, Keller GM. Directed differentiation of hematopoietic precursors and functional osteoclasts from human ES and iPS cells. Blood. 2010;115:2769–76. doi: 10.1182/blood-2009-07-234690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Raya A, Rodríguez-Pizà I, Guenechea G, Vassena R, Navarro S, Barrero MJ, Consiglio A, Castellà M, Río P, Sleep E, González F, Tiscornia G, Garreta E, Aasen T, Veiga A, Verma IM, Surrallés J, Bueren J, Izpisúa Belmonte JC. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature. 2009;460:53–9. doi: 10.1038/nature08129. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1161131
- 69.Lee JB, Werbowetski-Ogilvie TE, Lee J, McIntyre BAS, Schnerch A, Hong S, Park I, Daley GQ, Bernstein ID, Bhatia M. Notch-HES1 signaling axis controls hemato-endothelial fate decisions of human embryonic and induced pluripotent stem cells. Blood. 2013;122:1162–73. doi: 10.1182/blood-2012-12-471649. [DOI] [PubMed] [Google Scholar]
- 70.Risueño RM, Sachlos E, Lee J, Lee JB, Hong S, Szabo E, Bhatia M. Inability of human induced pluripotent stem cell-hematopoietic derivatives to downregulate microRNAs in vivo reveals a block in xenograft hematopoietic regeneration. Stem Cells. 2012;30:131–9. doi: 10.1002/stem.1684. [DOI] [PubMed] [Google Scholar]
- 71.La Nasa G, Caocci G, Efficace F, Dessì C, Vacca A, Piras E, Sanna M, Marcias M, Littera R, Carcassi C, Lucarelli G. Long-term health-related quality of life evaluated more than 20 years after hematopoietic stem cell transplantation for thalassemia. Blood. 2013;122:2262–70. doi: 10.1182/blood-2013-05-502658. [DOI] [PubMed] [Google Scholar]
- 72.Peffault de Latour R, Porcher R, Dalle J, Aljurf M, Korthof ET, Svahn J, Willemze R, Barrenetxea C, Mialou V, Soulier J, Ayas M, Oneto R, Bacigalupo A, Marsh JCW, Peters C, Socie G, Dufour C. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood. 2013;122:4279–86. doi: 10.1182/blood-2013-01-479733. [DOI] [PubMed] [Google Scholar]
- 73.MacMillan ML, Wagner JE. Haematopoeitic cell transplantation for Fanconi anaemia - when and how? Br J Haematol. 2010;149:14–21. doi: 10.1111/j.1365-2141.2010.08078.x. [DOI] [PubMed] [Google Scholar]
- 74.Boelens JJ, Aldenhoven M, Purtill D, Ruggeri A, Defor T, Wynn R, Wraith E, Cavazzana-Calvo M, Rovelli A, Fischer A, Tolar J, Prasad VK, Escolar M, Gluckman E, O'Meara A, Orchard PJ, Veys P, Eapen M, Kurtzberg J, Rocha V. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood. 2013;121:3981–7. doi: 10.1182/blood-2012-09-455238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guffon N, Bertrand Y, Forest I, Fouilhoux A, Froissart R. Bone marrow transplantation in children with Hunter syndrome: outcome after 7 to 17 years. J Pediatr. 2009;154:733–7. doi: 10.1016/j.jpeds.2008.11.041. [DOI] [PubMed] [Google Scholar]
- 76.Murauer EM, Gache Y, Gratz IK, Klausegger A, Muss W, Gruber C, Meneguzzi G, Hintner H, Bauer JW. Functional correction of type VII collagen expression in dystrophic epidermolysis bullosa. J Invest Dermatol. 2011;131:74–83. doi: 10.1038/jid.2010.249. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718345170
- 77.Pendaries V, Gasc G, Titeux M, Tonasso L, Mejía JE, Hovnanian A. siRNA-mediated allele-specific inhibition of mutant type VII collagen in dominant dystrophic epidermolysis bullosa. J Invest Dermatol. 2012;132:1741–3. doi: 10.1038/jid.2012.11. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/13940956
- 78.Ortiz-Urda S, Lin Q, Yant SR, Keene D, Kay MA, Khavari PA. Sustainable correction of junctional epidermolysis bullosa via transposon-mediated nonviral gene transfer. Gene Ther. 2003;10:1099–104. doi: 10.1038/sj.gt.3301978. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718345171
- 79.Thrasher AJ, Gaspar HB, Baum C, Modlich U, Schambach A, Candotti F, Otsu M, Sorrentino B, Scobie L, Cameron E, Blyth K, Neil J, Abina SH, Cavazzana-Calvo M, Fischer A. Gene therapy: X-SCID transgene leukaemogenicity. Nature. 2006;443:E5–6. doi: 10.1038/nature05219. discussion E6-7. [DOI] [PubMed] [Google Scholar]
- 80.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, Vidaud M, Abel U, Dal-Cortivo L, Caccavelli L, Mahlaoui N, Kiermer V, Mittelstaedt D, Bellesme C, Lahlou N, Lefrère F, Blanche S, Audit M, Payen E, Leboulch P, l'Homme B, Bougnères P, Kalle C von, Fischer A, Cavazzana-Calvo M, Aubourg P. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–23. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1255967
- 81.Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, Baldoli C, Martino S, Calabria A, Canale S, Benedicenti F, Vallanti G, Biasco L, Leo S, Kabbara N, Zanetti G, Rizzo WB, Mehta NAL, Cicalese MP, Casiraghi M, Boelens JJ, Del Carro U, Dow DJ, Schmidt M, Assanelli A, Neduva V, Di Serio C, Stupka E, Gardner J, Kalle C von, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341(1233158) doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718029893
- 82.Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC, Bosticardo M, Evangelio C, Assanelli A, Casiraghi M, Di Nunzio S, Callegaro L, Benati C, Rizzardi P, Pellin D, Di Serio C, Schmidt M, Kalle C von, Gardner J, Mehta N, Neduva V, Dow DJ, Galy A, Miniero R, Finocchi A, Metin A, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341(1233151) doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718084720
- 83.Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, Scaramuzza S, Andolfi G, Mirolo M, Brigida I, Tabucchi A, Carlucci F, Eibl M, Aker M, Slavin S, Al-Mousa H, Al Ghonaium A, Ferster A, Duppenthaler A, Notarangelo L, Wintergerst U, Buckley RH, Bregni M, Marktel S, Valsecchi MG, Rossi P, Ciceri F, Miniero R, Bordignon C, Roncarolo M. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447–58. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1148845
- 84.Ramirez CL, Certo MT, Mussolino C, Goodwin MJ, Cradick TJ, McCaffrey AP, Cathomen T, Scharenberg AM, Joung JK. Engineered zinc finger nickases induce homology-directed repair with reduced mutagenic effects. Nucleic Acids Res. 2012;40:5560–8. doi: 10.1093/nar/gks179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Overlack N, Goldmann T, Wolfrum U, Nagel-Wolfrum K. Gene repair of an Usher syndrome causing mutation by zinc-finger nuclease mediated homologous recombination. Invest Ophthalmol Vis Sci. 2012;53:4140–6. doi: 10.1167/iovs.12-9812. [DOI] [PubMed] [Google Scholar]
- 86.Chang C, Bouhassira EE. Zinc-finger nuclease-mediated correction of α-thalassemia in iPS cells. Blood. 2012;120:3906–14. doi: 10.1182/blood-2012-03-420703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Osborn MJ, Starker CG, McElroy AN, Webber BR, Riddle MJ, Xia L, DeFeo AP, Gabriel R, Schmidt M, Kalle C von, Carlson DF, Maeder ML, Joung JK, Wagner JE, Voytas DF, Blazar BR, Tolar J. TALEN-based gene correction for epidermolysis bullosa. Mol Ther. 2013;21:1151–9. doi: 10.1038/mt.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van den Akker PC, Jonkman MF, Rengaw T, Bruckner-Tuderman L, Has C, Bauer JW, Klausegger A, Zambruno G, Castiglia D, Mellerio JE, McGrath JA, van Essen AJ, Hofstra RMW, Swertz MA. The international dystrophic epidermolysis bullosa patient registry: an online database of dystrophic epidermolysis bullosa patients and their COL7A1 mutations. Hum Mutat. 2011;32:1100–7. doi: 10.1002/humu.21551. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718345172
- 90.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–8. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718007692
- 91.Le Cong, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717969062
- 92.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717969003
- 93.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–6. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Du H, Levine M, Ganesa C, Witte DP, Cole ES, Grabowski GA. The role of mannosylated enzyme and the mannose receptor in enzyme replacement therapy. Am J Hum Genet. 2005;77:1061–74. doi: 10.1086/498652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Matsuoka K, Tamura T, Tsuji D, Dohzono Y, Kitakaze K, Ohno K, Saito S, Sakuraba H, Itoh K. Therapeutic potential of intracerebroventricular replacement of modified human β-hexosaminidase B for GM2 gangliosidosis. Mol Ther. 2011;19:1017–24. doi: 10.1038/mt.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Remington J, Wang X, Hou Y, Zhou H, Burnett J, Muirhead T, Uitto J, Keene DR, Woodley DT, Chen M. Injection of recombinant human type VII collagen corrects the disease phenotype in a murine model of dystrophic epidermolysis bullosa. Mol Ther. 2009;17:26–33. doi: 10.1038/mt.2008.234. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718345173
- 97.Woodley DT, Keene DR, Atha T, Huang Y, Lipman K, Li W, Chen M. Injection of recombinant human type VII collagen restores collagen function in dystrophic epidermolysis bullosa. Nat Med. 2004;10:693–5. doi: 10.1038/nm1063. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718345174
- 98.Saelman EU, Nieuwenhuis HK, Hese KM, Groot PG de, Heijnen HF, Sage EH, Williams S, McKeown L, Gralnick HR, Sixma JJ. Platelet adhesion to collagen types I through VIII under conditions of stasis and flow is mediated by GPIa/IIa (alpha 2 beta 1-integrin) Blood. 1994;83:1244–50. [PubMed] [Google Scholar]
- 99.Rama P, Matuska S, Paganoni G, Spinelli A, Luca M de, Pellegrini G. Limbal stem-cell therapy and long-term corneal regeneration. N Engl J Med. 2010;363:147–55. doi: 10.1056/NEJMoa0905955. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/3779956
- 100.Li W, Hayashida Y, Chen Y, Tseng SCG. Niche regulation of corneal epithelial stem cells at the limbus. Cell Res. 2007;17:26–36. doi: 10.1038/sj.cr.7310137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pellegrini G, Rama P, Mavilio F, Luca M de. Epithelial stem cells in corneal regeneration and epidermal gene therapy. J Pathol. 2009;217:217–28. doi: 10.1002/path.2441. [DOI] [PubMed] [Google Scholar]
- 102.Nagy N, Almaani N, Tanaka A, Lai-Cheong JE, Techanukul T, Mellerio JE, McGrath JA. HB-EGF induces COL7A1 expression in keratinocytes and fibroblasts: possible mechanism underlying allogeneic fibroblast therapy in recessive dystrophic epidermolysis Bullosa. J Invest Dermatol. 2011;131:1771–4. doi: 10.1038/jid.2011.85. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718345175
- 103.Woodley DT, Cogan J, Wang X, Hou Y, Haghighian C, Kudo G, Keene DR, Chen M. De Novo Anti-Type VII Collagen Antibodies in Patients with Recessive Dystrophic Epidermolysis Bullosa. J Invest Dermatol. 2013 doi: 10.1038/jid.2013.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Iwata H, Bieber K, Tiburzy B, Chrobok N, Kalies K, Shimizu A, Leineweber S, Ishiko A, Vorobyev A, Zillikens D, Köhl J, Westermann J, Seeger K, Manz R, Ludwig RJ. B cells, dendritic cells, and macrophages are required to induce an autoreactive CD4 helper T cell response in experimental epidermolysis bullosa acquisita. J Immunol. 2013;191:2978–88. doi: 10.4049/jimmunol.1300310. [DOI] [PubMed] [Google Scholar]
- 105.Samavedam UKSRL, Kalies K, Scheller J, Sadeghi H, Gupta Y, Jonkman MF, Schmidt E, Westermann J, Zillikens D, Rose-John S, Ludwig RJ. Recombinant IL-6 treatment protects mice from organ specific autoimmune disease by IL-6 classical signalling-dependent IL-1ra induction. J Autoimmun. 2013;40:74–85. doi: 10.1016/j.jaut.2012.08.002. [DOI] [PubMed] [Google Scholar]