

Abstract
Bacterial RNA polymerase (RNAP) is essential for transcription and is an antibacterial target for small molecule inhibitors. The binding region of myxopyronin B (MyxB), a bacterial RNAP inhibitor, offers the possibility of new inhibitor design. The molecular design program SPROUT has been used in conjunction with the X-ray cocrystal structure of Thermus thermophilus RNAP with MyxB to design novel inhibitors based on a substituted pyridyl-benzamide scaffold. A series of molecules, with molecular masses <350 Da, have been prepared using a simple synthetic approach. A number of these compounds inhibited Escherichia coli RNAP.
Keywords: Bacterial RNA polymerase, structure-based ligand discovery, myxopyronin B, small molecule inhibitors
RNA polymerase (RNAP) catalyzes the synthesis of RNA from a DNA template.1 Because this enzyme is essential for the growth and survival of bacteria, inhibition of bacterial RNAP is an established strategy for antibacterial therapy; for example, the rifamycin antibiotics (known RNAP inhibitors) are used in the frontline treatment of tuberculosis.2 RNAP is considered an attractive antibacterial drug target since it is well conserved across bacterial genera but has distinct structural differences from eukaryotic RNAP.3 This qualifies bacterial RNAP as a target for broad-spectrum antibacterial therapy and permits the design of selective inhibitors.3 Despite the number of known inhibitors of bacterial RNAP,4 the rifamycins are currently the only class of bacterial RNAP inhibitor approved for clinical use,1 highlighting the challenge of RNAP as a drug target.
Combating the rise in bacterial resistance to antibiotics is an important field in drug discovery.4,5 Novel antibacterial agents with new modes of action offer the best possibility to overcome existing resistance mechanisms.6 New opportunities to explore bacterial RNAP as a target have arisen following the 2008 discovery of the myxopyronin B 1 (MyxB) binding site (Figure 1).7,8 MyxB is an antibiotic produced from the myxobacterium Myxococcus fulvus Mxf50, which inhibits bacterial RNAP and the growth of Gram-positive and some Gram-negative bacteria [1: IC50, 0.92 μM; minimum inhibitory concentration (MIC), 1 μg/mL Staphylococcus aureus].9,10 MyxB binds to the switch region of RNAP, where its mode of action has been proposed to involve either inhibition of the opening and closing of the β′ subunit8 or stabilization of the refolding of the β′-subunit switch-2 region, leading to an inactive conformation that cannot bind to template DNA.7 As bacteria have become resistant to the rifamycins, the MyxB binding site presents itself as an excellent opportunity for the design of new RNAP inhibitors as the two antibiotic binding sites are distant from each other.8
Figure 1.
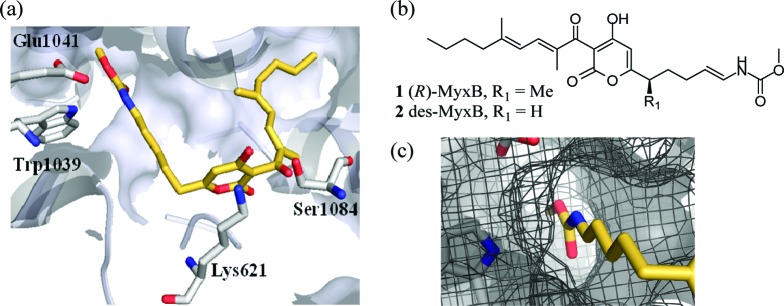
(a) MyxB binding region (PDB ID: 3EQL). des-MyxB 2 is shown in gold with key residues of T. Thermophilus RNAP (gray) labeled. (b) Structure of MyxB. (c) Insert of the enecarbamate side chain and the narrow channel that it fills.
Inspection of the available cocrystal structures7,8 revealed that the MyxB binding site within RNAP is U-shaped and sandwiched between two hydrophobic pockets (Figure 1a). MyxB fills this cavity via the location of lipophilic side chains deep within these pockets. Additionally, the core pyrone ring of MyxB makes key interactions with residues near the cavity entrance including, for example, H-bonds to Lys621 and Ser1084.8 Given the high logP (7.5) of MyxB, it is presumed that most of its binding interactions are hydrophobic,7 although the whole molecule is sensitive to change, as demonstrated in studies of synthetic analogues.10,11 To date, only desmethyl myxopyronin B 2 (des-MyxB) has been found to have comparable biological activity to MyxB (2: IC50, 0.34 μM; MIC, 4 μg/mL S. aureus).10
The premise of structure-based ligand design was introduced in the late 1980s12 and is currently undergoing a revival with the improvement of computer processing power and the increase in the number of high-resolution protein crystal structures that are available.13 New approaches to antibacterial drug discovery are needed since alternative methods, such as high-throughput screening (HTS), have failed to identify useful new antibacterial drug leads, including screening programs against bacterial RNAP.14 SPROUT15 is a powerful software suite for structure-based molecular design and has been shown to be a successful approach in the identification of inhibitors of enzymes derived from infectious agents. This includes penicillin-binding proteins16 and the plasmodia-derived enzyme DHODH,17 as well as in the identification of an RNAP inhibitor targeted at the rifamycin binding site.18 SPROUT utilizes a fragment-based approach and can be used to design putative ligands with novel scaffolds, ranked by predicted binding affinity, ligand efficiency, or synthetic accessibility.
We describe here the application of SPROUT to the development of the first designed small molecule inhibitors targeted at the MyxB binding region of RNAP and report a novel scaffold that shows improved inhibitory activity to that displayed by MyxB in our assay.
Using the cocrystal structure of T. thermophilus RNAP-des-myxopyronin B complex (PDB ID: 3EQL),7 the MyxB binding site was explored using SPROUT. Given the large volume of this cavity, we focused on the region around the enecarbamate side chain, since it was narrow and offered a bound inhibitor the potential for making hydrogen-bonding interactions with residues Trp1039 and Glu1041 (Figure 1c). Using SPROUT, a substituted pyridyl-benzamide scaffold was predicted to fill this volume and make interactions with residues Trp1039, Glu1041, and Gln611 (Figure 2a). Compound 7 was synthesized and tested, along with MyxB, in an in vitro Escherichia coli RNAP assay using a Kool NC-45 RNAP template (Epicenter Biotechnologies).19 This revealed compound 7 to be a weak inhibitor of E. coli RNAP (IC50, 151 μM) and MyxB to have an IC50 of 46.5 ± 5.9 μM. Although in our assay, the measured IC50 value for MyxB is somewhat higher than previously reported,9 we believe that this is due to a difference in assay conditions since Moy et al. recently reported20 an IC50 of 24 μM for MyxB using a similar assay method to that used in the present study.
Figure 2.

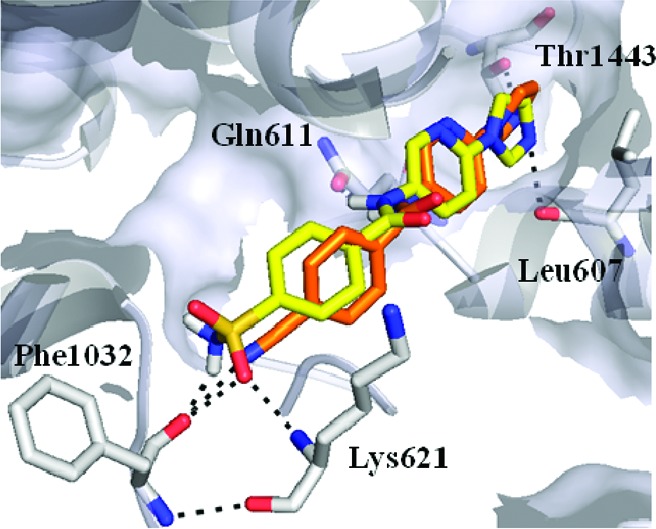
(a) Compound 7 showing predicted interactions with RNAP. (b) AUTODOCK predicted docking poses of compound 7 (blue) within RNAP (gray): lowest energy pose (pink) and highest populated pose (green). (c) AUTODOCK predicted conformation of compounds 8 (yellow) and 10 (orange) within RNAP (gray).
To probe the possible binding conformations of compound 7, the molecular docking program AUTODOCK21 was used to generate and evaluate a range of potential binding poses of 7 within the MyxB binding region of T. thermophilus RNAP. From these studies, the picolyl group of 7 was predicted to be located near to the solvent-exposed entrance of the binding cavity and adopt a range of different conformations of comparable energy (Figure 2b). We reasoned that restricting the degrees of freedom of this moiety could increase the binding affinity of this type of inhibitor. Additionally, we sought groups that could maintain the polarity within this region of the inhibitor due to its exposure to solvent at the entrance to the binding cavity. We therefore prepared compounds 8 and 9 where nitrogen-containing heterocycles were substituted for the picolyl group of 7. As predicted by our modeling, compounds 8 and 9 exhibited a marked increase in potency (Table 1) and were prepared as shown in Scheme 1. Commercially available 2-hydroxy-5-nitropyridine 3 was treated with POCl3 to give 2-chloro-5-nitropyridine 4. Nucleophilic aromatic substitution using imidazole or 1,2,4-triazole, reduction of the nitro group and subsequent amide couplings with 4-carboxybenzenesulfonamide gave compounds 8 and 9 (Scheme 1).
Table 1. Inhibition of E. coli RNAP by MyxB 1 and Pyridyl-Benzamide Compounds 8–13.
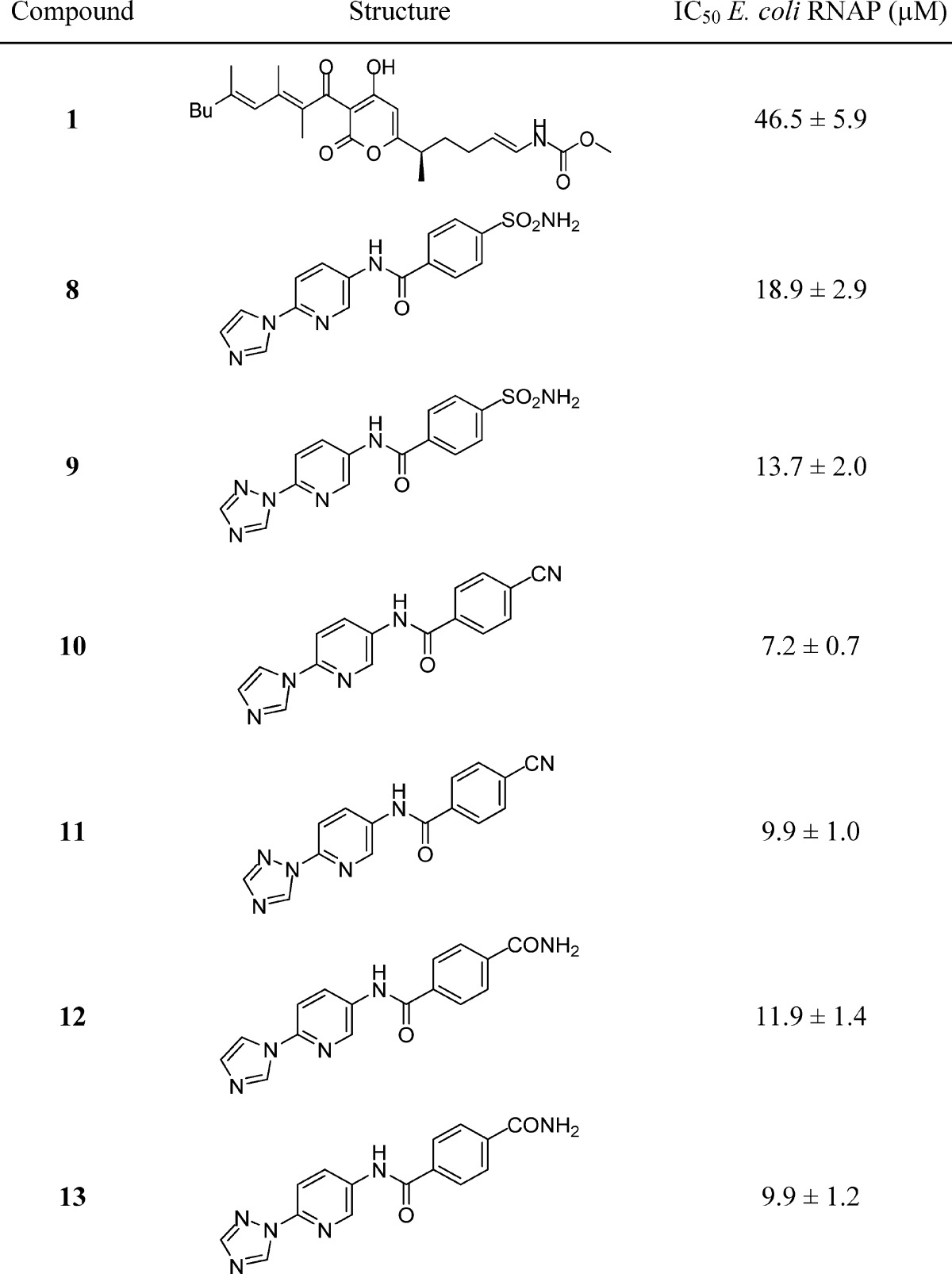
Scheme 1. Synthesis of Pyridyl-Benzamide Compounds 7–13.

Reagents and conditions: (a) POCl3, reflux, 18 h, 97%. (b) Pyridinylmethanol/imidazole/triazole, NaH, DMF, room temperature, 1 h, 70–87%. (c) H2, Pd/C, MeOH, room temperature, 2 h, 60–91%. (d) Substituted benzoic acids, 1-hydroxybenzotriazole, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide, DMF, 35 °C, 16 h, 60–70%. (e) H2O2, 6 M NaOH, EtOH, 55 °C, 4 h, 80–95%.
To further explore the effect of structural variations upon RNAP inhibition, a small number of compounds 10–13 were synthesized where the sulfonamide group present in compound 7 was varied. This indicated that a nitrile group (10 and 11) offered the greatest gain in potency (Table 1). Using AUTODOCK21 to generate potential binding conformations of 8–13, we observed a consistent pose for these inhibitors, different to that proposed for compound 7. This positioned the nitrogen-containing heterocycle in the hydrophobic pocket, which binds the dienone side chain of MyxB (Figure 2c). Compounds 8–13 were now predicted to make hydrogen-bonding interactions to Lys621, and we reasoned that the geometry of this hydrogen bond is optimal when the nitrile group is present, as in compounds 10 and 11. Additionally, the NH2 moiety within sulfonamides 8 and 9 and carboxamides 12 and 13 is needed to make a hydrogen bond contact to the carbonyl of Phe1032.
To further clarify the importance of the various functional groups present within these inhibitors, analogues were synthesized where groups, which were predicted to result in the loss of contacts within the binding cavity, were systematically removed. In keeping with these predictions, compounds without the sulfonamide and terminal N-heterocyclic moieties (14 and 15) were inactive, as was compound 16 containing the “reverse amide” motif.
Extending the structure–activity relationship (SAR) around compounds 8 and 9, we synthesized compounds 17–27 in which the substituents at both ends of the inhibitor were varied (Table 2). The lack of activity observed for compounds 17–20 appears to underline the strict geometrical requirements for hydrogen bonding to Lys621. While 18–20 contain a carbonyl oxygen atom capable of accepting a hydrogen bond from Lys621, they are unable to make the additional hydrogen bond to Phe1032 required for efficient binding of these compounds. Additionally, the nature of the substitution at the opposite end of the inhibitors is also important. Although 21–27 all contain either sulfonamides or nitriles, departure from a five-membered azole ring at the opposite end of the molecule resulted in significantly weaker (21, 22, 25, and 27) or total loss (23, 24, and 26) of activity. We reasoned that both the size of the ring and the position of ring heteroatoms are important for activity. This is clearly seen with compound 27, which contains a furan ring similar in size to imidazole, but the heteroatom at the 2-position is unable to make a hydrogen bond to Thr1443 predicted to interact with the imidazolyl moiety of 10 (Figure 2c).
Table 2. Inhibition of E. coli RNAP of Compounds 14–27.
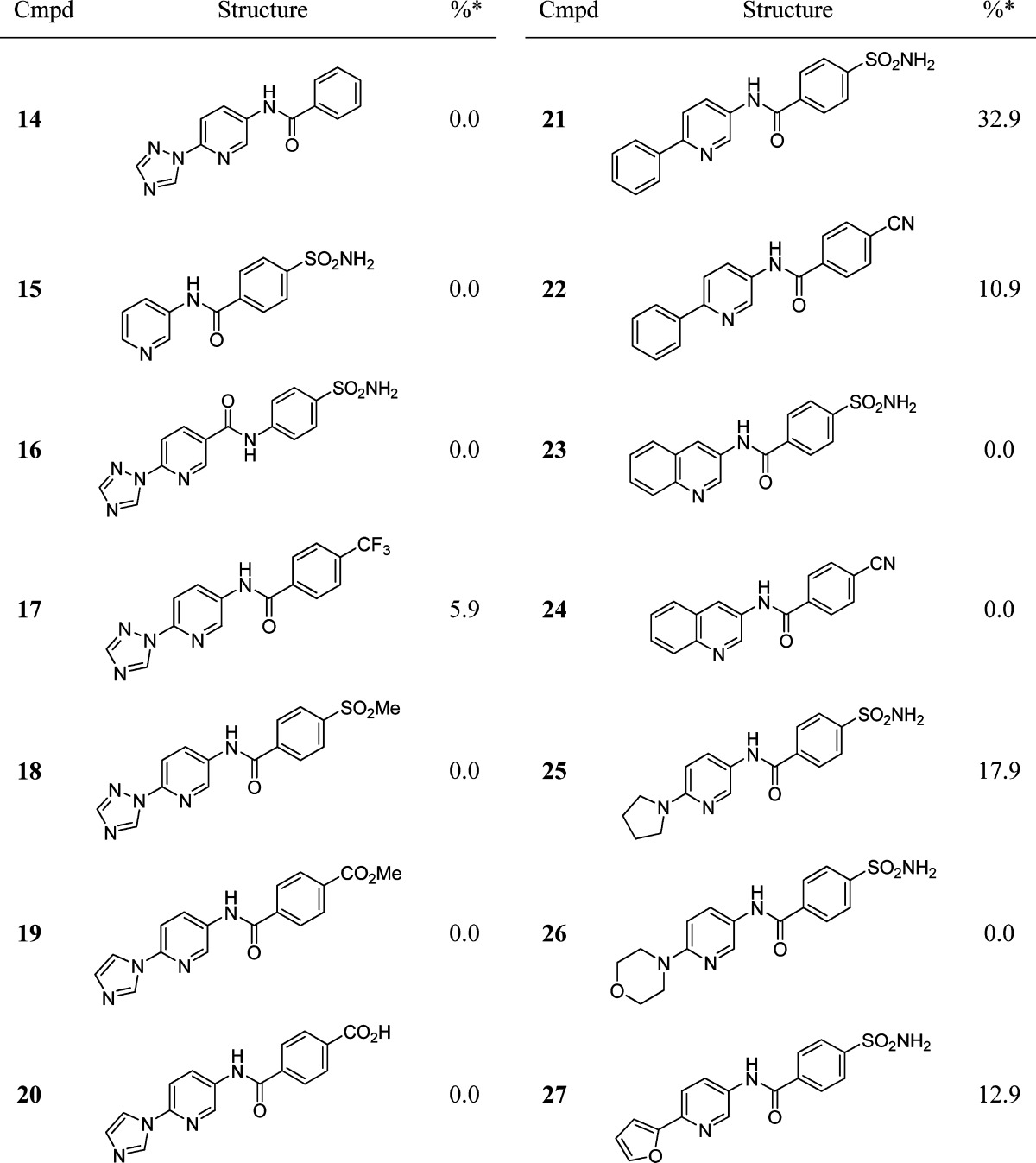
Percentage inhibition at 100 μM E. coli RNAP.
To probe the selectivity of enzyme inhibition, compounds found to be active in the E. coli RNAP assay were subjected to specificity assays using malate dehydrogenase and chymotrypsin to identify promiscuous activity,22 and inhibition against yeast RNA Polymerase II (Pol II) to ascertain whether compounds were selective for bacterial RNAP. Compounds 8–13 exhibited no substantial inhibition of the three enzymes at 100 μM and, therefore, appear to be selective inhibitors of bacterial RNAP (Table 3).
Table 3. Inhibition of Yeast Pol II, Malate Dehydrogenase, and Chymotrypsin by 8–13.
| % inhibition ± SDa |
|||
|---|---|---|---|
| compd | yeast Pol II | malate dehydrogenase | chymotrypsin |
| 8 | 14.7 ± 5.0 | 0.5 ± 0.5 | 0.0 ± 0.0 |
| 9 | 9.3 ± 2.0 | 1.4 ± 1.1 | 0.6 ± 0.4 |
| 10 | 17.6 ± 3.3 | 0.6 ± 0.4 | 1.8 ± 1.4 |
| 11 | 12.0 ± 3.1 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| 12 | 10.5 ± 4.2 | 0.0 ± 0.0 | 0.1 ± 0.1 |
| 13 | 5.2 ± 3.6 | 0.0 ± 0.0 | 1.5 ± 1.5 |
Percentage inhibition at 100 μM.
MICs were determined for all of the compounds described using standard susceptibility tests, in triplicate, against S. aureus, Bacillus subtilis, and E. coli, strains according to the British Society for Antimicrobial Chemotherapy guidelines (see the Supporting Information for further details).23 None of the compounds were found to possess antibacterial activity (>256 μg/mL in all cases), even in the presence of the outer membrane permeabilizer polymyxin B nonapeptide (PMBN) and E. coli deficient in the AcrAB multidrug efflux pump component.
In summary, we have used a de novo molecular design approach to identify a new series of inhibitors targeting bacterial RNAP. To our knowledge, they are the first examples of designed small molecule inhibitors predicted to bind to the MyxB binding region of RNAP. Compounds 8–13 are of low molecular mass, have improved potency and ligand efficiency over MyxB, are selective for bacterial RNAP, and do not appear to be promiscuous inhibitors. The compounds may not possess the requisite physicochemical properties required for antibacterial activity, since antibacterial agents occupy a unique chemical property space compared to that for other therapeutic drugs.24 Most likely, the lack of antibacterial activity of these compounds is attributable to poor cell penetration, and in this connection, further optimization of this scaffold is in progress. This research advances our understanding of bacterial RNAP inhibitors with binding sites in nonessential prokaryotic domains that are distant from the catalytic site.
Acknowledgments
We thank the BBSRC and MRC for support. We also thank T. Moy for the gift of MyxB, D. Bushnell for yeast Pol II, and K. Simmons and M. Migliore for discussion.
Supporting Information Available
Details of compound syntheses and characterization and of the biological assay protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Medicinal Chemistry and Chemical Biology Technology Group, School of Chemistry.
Supplementary Material
References
- Villain-Guillot P.; Bastide L.; Gualtieri M.; Leonetti J. P. Progress in targeting bacterial transcription. Drug Discovery Today 2007, 12, 200–208. [DOI] [PubMed] [Google Scholar]
- Darst S. A. New inhibitors targeting bacterial RNA polymerase. Trends Biochem. Sci. 2004, 29, 159–162. [DOI] [PubMed] [Google Scholar]
- Ho M. X.; Hudson B. P.; Das K.; Arnold E.; Ebright R. H. Structures of RNA polymerase-antibiotic complexes. Curr. Opin. Struct. Biol. 2009, 19, 715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra I. Bacterial RNA polymerase: A promising target for the discovery of new antimicrobial agents. Curr. Opin. Invest. Drugs 2007, 8, 600–607. [PubMed] [Google Scholar]
- Barker J. J. Antibacterial drug discovery and structure-based design. Drug Discovery Today 2006, 11, 391–404. [DOI] [PubMed] [Google Scholar]
- Chopra I. Research and development of antibacterial agents. Curr. Opin. Microbiol. 1998, 1, 495–501. [DOI] [PubMed] [Google Scholar]
- Belogurov G. A.; Vassylyeva M. N.; Sevostyanova A.; Appleman J. R.; Xiang A. X.; Lira R.; Webber S. E.; Klyuyev S.; Nudler E.; Artsimovitch I.; Vassylyev D. G. Transcription inactivation through local refolding of the RNA polymerase structure. Nature 2009, 457, 332–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay J.; Das K.; Ismail S.; Koppstein D.; Jang M. Y.; Hudson B.; Sarafianos S.; Tuske S.; Patel J.; Jansen R.; Irschik H.; Arnold E.; Ebright R. H. The RNA Polymerase “Switch Region” Is a Target for Inhibitors. Cell 2008, 135, 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irschik H.; Gerth K.; Hofle G.; Kohl W.; Reichenbach H. The Myxopyronins, New Inhibitors of Bacterial RNA-Synthesis from Myxococcus-Fulvus (Myxobacterales). J. Antibiot. 1983, 36, 1651–1658. [DOI] [PubMed] [Google Scholar]
- Doundoulakis T.; Xiang A. X.; Lira R.; Agrios K. A.; Webber S. E.; Sisson W.; Aust R. M.; Shah A. M.; Showalter R. E.; Appleman J. R.; Simonsen K. B. Myxopyronin B analogs as inhibitors of RNA polymerase, synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2004, 14, 5667–5672. [DOI] [PubMed] [Google Scholar]
- Lira R.; Xiang A. X.; Doundoulakis T.; Biller W. T.; Agrios K. A.; Simonsen K. B.; Webber S. E.; Sisson W.; Aust R. M.; Shah A. M.; Showalter R. E.; Banh V. N.; Steffy K. R.; Appleman J. R. Syntheses of novel myxopyronin B analogs as potential inhibitors of bacterial RNA polymerase. Bioorg. Med. Chem. Lett. 2007, 17, 6797–6800. [DOI] [PubMed] [Google Scholar]
- Mauser H.; Guba W. Recent developments in de novo design and scaffold hopping. Curr. Opin. Drug Discovery Dev. 2008, 11, 365–374. [PubMed] [Google Scholar]
- Simmons K. J.; Chopra I.; Fishwick C. W. G. Structure-based discovery of antibacterial drugs. Nat. Rev. Microbiol. 2010, 8, 501–510. [DOI] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 2007, 6, 29–40. [DOI] [PubMed] [Google Scholar]
- Gillet V.; Johnson A. P.; Mata P.; Sike S.; Williams P. Sprout—A Program for Structure Generation. J. Comput.-Aided Mol. Des. 1993, 7, 127–153. [DOI] [PubMed] [Google Scholar]
- Woon E. C. Y.; Zervosen A.; Sauvage E.; Simmons K. J.; Zivec M.; Inglist S. R.; Fishwick C. W. G.; Gobec S.; Charlier P.; Lexen A.; Schofield C. J. Structure Guided Development of Potent Reversibly Binding Penicillin Binding Protein Inhibitors. ACS Med. Chem. Lett. 2010, 2, 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen D.; Bedingfield P.; McConkey G. A.; Fishwick C. W. G.; Johnson A. P. A study of the effects of substituents on the selectivity of the binding of N-arylaminomethylene malonate inhibitors to DHODH. Bioorg. Med. Chem. Lett. 2010, 20, 1284–1287. [DOI] [PubMed] [Google Scholar]
- Agarwal A. K.; Johnson A. P.; Fishwick C. W. G. Synthesis of de novo designed small-molecule inhibitors of bacterial RNA polymerase. Tetrahedron 2008, 64, 10049–10054. [Google Scholar]
- Mariner K. R.; Trowbridge R.; Agarwal A. K.; Miller K.; O'Neill A. J.; Fishwick C. W. G.; Chopra I. Furanyl-Rhodanines Are Unattractive Drug Candidates for Development as Inhibitors of Bacterial RNA Polymerase. Antimicrob. Agents Chemother. 2010, 54, 4506–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy T. I.; Daniel A.; Hardy C.; Jackson A.; Rehrauer O.; Hwang Y. S.; Zou D.; Nguyen K.; Silverman J. A.; Li Q. Y.; Murphy C. Evaluating the activity of the RNA polymerase inhibitor myxopyronin B against Staphylococcus aureus. FEMS Microbiol. Lett. 2011, 319, 176–179. [DOI] [PubMed] [Google Scholar]
- http://www.schrodinger.com.
- Seidler J.; McGovern S. L.; Doman T. N.; Shoichet B. K. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J. Med. Chem. 2003, 46, 4477–4486. [DOI] [PubMed] [Google Scholar]
- A Guide to Sensitivity Testing: Report of the Working Party on Antibiotic Sensitivity Testing of the British Society for Antimicrobial Chemotherapy. J. Antimicrob. Chemother. 1991, 27 (Suppl. D), pp 1–50. [PubMed] [Google Scholar]
- O'Shea R.; Moser H. E. Physicochemical properties of antibacterial compounds: Implications for drug discovery. J. Med. Chem. 2008, 51, 2871–2878. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.