

Abstract

The discovery of MK-1220 is reported along with the development of a series of HCV NS3/4A protease inhibitors containing a P2 to P4 macrocyclic constraint with improved preclinical pharmacokinetics. Optimization of the P2 heterocycle substitution pattern as well as the P3 amino acid led to compounds with greatly improved plasma exposure following oral dosing in both rats and dogs while maintaining excellent enzyme potency and cellular activity. These studies led to the identification of MK-1220.
Keywords: Hepatitis C, NS3/4A, HCV, macrocycle, ring-closing metathesis
Hepatitis C virus (HCV) is a chronic infection that affects an estimated 170−200 million people worldwide.1 The positive RNA strand flaviviridae family virus replicates primarily in the liver, and while the disease progresses over of a number of years, a significant fraction of those infected develop serious liver disease, including cirrhosis and hepatocellular carcinoma.2 HCV is also the most common indication for liver transplantation.3 HCV displays a high degree of genetic heterogeneity, with genotypes 1, 2, and 3 accounting for more than 90% of the infections in the developed world and with genotype 1 being predominant (∼70%) in the U.S., Europe, and Japan. The current standard of care treatment for chronic HCV infection is combination therapy with pegylated Interferon-α and Ribavirin4−6 for up to 48 weeks. Sustained viral response (SVR) rates vary by genotype, with ∼45% of HCV genotype 1-infected patients and ∼80% of genotype 2- and 3-infected patients achieving SVR. The limited efficacy, coupled with a number of serious side-effects, limits the number of patients who complete treatment.7,8
Efforts toward improved HCV treatment include the development of direct antivirals which inhibit key steps in the viral replication process.9 One of the more promising targets is the serine protease NS3/4A,10−14 with clinical proof of concept established by Boehringer-Ingelheim with BILN-2061 (Figure 1).15 Subsequently, both Vertex (VX-950, telaprevir)16 and Schering-Plough (SCH-503034, boceprevir)17 have moved α-keto-amide HCV NS3/4A inhibitors into phase 3 clinical trials, while a number of noncovalent inhibitors (danoprevir (ITMN-191),18,19 TMC435350,20−22 BI201335,23 and vaniprevir24 (Figure 1)) have also advanced into late stage trials.
Figure 1.

Macrocycle HCV NS3/4A protease inhibitors.
Beginning in 2005,24−27 our program focused on a series of macrocyclic inhibitors of HCV NS3/4A protease which contain a linker from P2 to P4, in contrast to the P1 to P3 linker present in BILN-2061 and several other known inhibitors (Figure 1). In 2008, we disclosed the general strategy for the discovery of this series and the subsequent optimization of genotype 1b potency and liver exposure, which led to compound 1(26) and vaniprevir (MK-7009) (Figure 1).24 Compound 1 has picomolar binding affinity for HCV NS3/4A protease (1b Ki = 0.04 nM), good cellular potency (1b replicon IC50 = 4.5 nM), and excellent liver exposure in rat following a 5 mg/kg oral dose ([liver]4h = 18,600 nM).26 Obtaining high liver exposure may be critical to the success of any anti-HCV compound because the primary HCV replication site is in hepatocytes.28 However, high systemic exposure would also be desirable given the evidence of extra-hepatic replication sites,28 and unfortunately, compound 1 does not achieve significant plasma levels following oral dosing in preclinical species (rat plasma AUC = 0.36 μM·h; 5 mg/kg, PO).26 With a goal of finding compounds in this series, with high liver exposure and improved plasma exposure following oral dosing in preclinical species while maintaining excellent potency, we decided to further explore the effect of substituents on the P2 isoquinoline group as well as various P3 and P4 groups. These efforts, which ultimately led to the discovery of MK-1220, are described herein.
The synthesis of the various P2-isoquinoline analogues was carried out using the modular approach detailed in Scheme 1. N-Boc hydroxyproline 4 was reacted with either 6-substituted or unsubstituted bromo-chloroisoquinolines 2 and 3, which, following esterification, gave 5 and 6. Boc-deprotection, followed by coupling with acids 6a−k, gave 7a−i, 7k and 8a, 8c, 8j, which could be vinylated under Stille conditions and then cyclized using the Zhan 1b metathesis catalyst29 to give 9a−i, 9k and 10a, 10c, and 10j. The resulting double bond could alternatively be left intact or saturated under standard conditions to give 11a and b, 11d, 11f, 11i, 11k, and 12a, 12c, and 12j. The targeted compounds 1, 13b and c, 13e, 13g−i, 14a, 14b, 14d, 14f, 14i, 14k, and 15a, 15c, and 15j were then prepared via hydrolysis of the ester and coupling with amine 16.30 5-Substituted analogues (see Supporting Information Scheme 2 for general scheme and experimental details) were prepared by a regioselective halogenation/cross-coupling sequence from 11a, which provided the desired acyl-sulfonamides 18a−h after hydrolysis and coupling with 16. 5-Trifluoromethoxy analogue 18i was prepared analogously to the compounds in Scheme 1 using the corresponding trifluoromethoxy isoquinoline, and P3-cyclohexyl analogue 19 was prepared analogously to 18c using 6b.
Scheme 1. Preparation of 6-Substituted and Unsubstituted Isoquinoline Derivatives.

Reagents and conditions: (a) t-BuOK, DMSO, then HCl, EtOH. (b) HCl, dioxane. (c) HATU, DIEA, DMF. (d) vinylSnBu3, Pd(PPh3)4. (e) Zhan 1b, DCM. (f) Pd/C, H2. (g) LiOH, THF, EtOH. (h) 16, HATU, DIEA, DMF.
Compounds were evaluated in a genotype 1b enzyme binding assay,31 and cellular activity was determined using the genotype 1b replicon system in the presence of 10% fetal bovine serum (FBS) and 50% normal human serum (NHS).32,33 The plasma and liver pharmacokinetics of lead HCV protease compounds have been characterized and shown to result from the complex interplay of absorption, uptake transport, efflux transport, protein binding, and metabolism. Since fully characterizing each of these individual components in vitro may or may not translate into accurate in vivo predictions, the team took an empirical approach and differentiated potential lead compounds using initial pharmacokinetic analysis focused on plasma AUC and liver concentration at 4 h after a 5 mg/kg oral dose in rats.
As Table 1 illustrates, the effect on rat exposure in both liver and plasma of various P3 groups can be dramatic. Simple substitution the t-Bu of compound 1(26) for a cyclohexyl group (13b) led to a 2-fold improvement in rat plasma AUC and liver concentration, while reduction of the styryl-olefin (14b) led to a nearly 10-fold improvement in rat plasma AUC (3.1 μM·h). Unfortunately, while 14b had improved rat plasma exposure, the genotype 1b replicon IC50 in the presence of 50% NHS was shifted by greater than 10-fold, which may be due to the increased lipophilicity of the cyclohexyl group. Introduction of a cyclopentyl P3 group (13c) led to even greater plasma exposure (AUC = 4.2 μM·h) with a reduced loss in 50% NHS genotype 1b replicon potency. Given these dramatic effects, a number of other macrocycles with various cyclic P3 groups were prepared. Smaller groups, such as cyclobutyl (14d), led to much reduced potency, while larger groups, such as indane (13e) or benzyl (13g), resulted in compounds with reduced potency and/or moderate rat plasma and liver exposure. Aromatic P3 groups (14f, 13h) resulted in compounds with poor potency. In a striking example of the unpredictable nature of the pharmacokinetics seen in this series, simple substitution of oxygen for carbon in the cyclohexyl ring (compare 13b/14b to 13i/14i) resulted in potent compounds which had almost no rat liver exposure (<0.2 μM @ 4 h) and moderate plasma levels. This type of result emphasizes the potential benefit of screening a large number of compounds for rat oral plasma and liver exposure due to the unpredictable pharmacokinetics of this series of compounds.26
Table 1.
 |
Single or double bond at styryl position.
NS3/4A protease time-resolved fluorescence assay.
Cell-based replicon assay.
5 mg/kg dosed orally in PEG400 (n = 2); AUC calculated based on 4 h experiment.
5 mg/kg dosed orally in PEG400 (n = 2); liver levels after 4 h.
Next, we explored the effect of 5- or 6-substitution on the P2-isoquinoline ring, which in general led to increased plasma exposure with variable effects on potency (Table 2). Interestingly, while the 5-iodo derivative (18b) possessed a moderate 20-fold shift in the replicon assay from 10% FBS to 50% NHS and good plasma exposure (AUC = 1.4 μM·h), the 5-bromo analogue (18a) had a much larger shift in replicon IC50 and only moderate plasma and liver exposure, thus demonstrating that lipophilicity is not necessarily directly coupled to plasma/liver exposure. All the other 5-substituted derivatives tested in the standard rat PK experiment showed good to excellent plasma exposure, with most of the analogues losing significant replicon potency in the presence of 50% NHS (18d and e, 18 g−i). In contrast, thiomethyl analogue 18f displayed excellent in vitro and replicon potency (gt 1b Ki = 0.03 nM; 1b replicon IC50 = 2 nM) and a more modest shift in the presence of 50% NHS (IC50 = 34 nM); however, the rat plasma AUC following a 5 mg/kg PO dose was only moderate (1.8 μM·h). The compound with the most balanced profile was the 5-methoxyisoquinoline 18c, with good gt 1b replicon IC50 (2.7 nM (10% FBS); 35 nM (50% NHS)) and 17.7 μM·h plasma AUC following an oral 5 mg/kg dose in rat, which is a nearly 100-fold improvement in plasma AUC compared to the unsubstituted analogue 14a. A similar plasma exposure enhancing effect was also seen with the 6-methoxyisoquinoline 15a (9.6 μM·h), although this compound did have a larger 50% NHS replicon shift and lower liver exposure.
Table 2.

| 1b Replicon IC50 (nM)b |
||||||
|---|---|---|---|---|---|---|
| compd | R | 1b Ki (nM)a | 10% FBS | 50% NHS | rat PO plasma AUC (μM·h)c | rat [liver] @ 4 h (μM)d |
| 14a | H | 0.18 | 8.7 | 46 | 0.27 | 13.4 |
| 18a | 5-Br | 0.04 | 2 | 170 | 0.28 | 5.8 |
| 18b | 5-I | 0.03 | 2.3 | 56 | 1.4 | 24.5 |
| 18c | 5-OMe | 0.04 | 2.7 | 35 | 17.7 | 33.2 |
| 18d | 5-OEt | 0.37 | 6 | 500 | 6.9 | 15.2 |
| 18e | 5-Et | 0.14 | 2.5 | 90 | 2.1 | 12.5 |
| 18f | 5-SMe | 0.03 | 2 | 34 | 1.8 | 12.6 |
| 18g | 5-CF3 | 0.25 | 21 | 410 | ||
| 18h | 5-SO2Me | 0.11 | 280 | >1000 | ||
| 18i | 5-OCF3 | 0.75 | 50 | 4400 | 1.6 | 8.8 |
| 15a | 6-OMe | 0.04 | 3 | 113 | 9.6 | 3.0 |
NS3/4A protease time-resolved fluorescence assay.
Cell-based replicon assay.
5 mg/kg dosed orally in PEG400 (n = 2); AUC calculated based on 4 h experiment.
5 mg/kg dosed orally in PEG400 (n = 2); liver levels after 4 h.
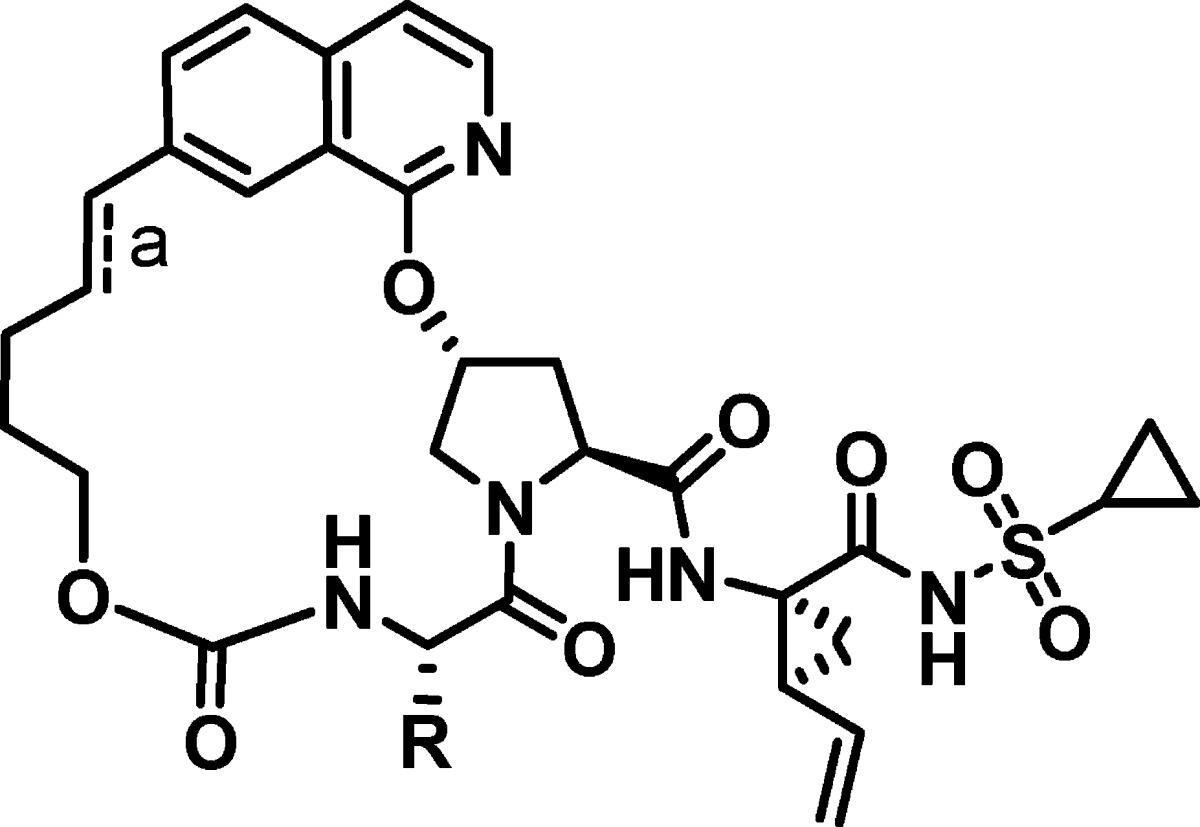
While a number of the most promising analogues (14b, 13c, 18c, 15a) possessed the improved rat plasma exposure we desired, all lost replicon potency in the presence of 50% NHS to a degree that they were less potent than the targeted potency of parent compound 1 (gt 1b replicon IC50 = 14 nM (50% NHS)). We then investigated whether the plasma enhancing effects of P3 cycloalkyl groups and P2-isoquinoline substituents could be combined to produce a compound which maintained high rat plasma while improving replicon potency (Table 3). 5-Methoxyisoquinoline analogue 19 showed reduced rat plasma exposure compared to 18c, while 6-methoxy analogue (15c) showed an additive effect of the P2-methoxy and the P3-cycloalkyl groups with a rat plasma AUC of 18.1 μM·h. This compound still possessed a 40-fold shift in replicon IC50 in the presence of 50% NHS; however, as we demonstrated in the development of MK-7009 (vaniprevir),24 incorporation of longer and/or substituted P2−P4 linkers can both improve potency and decrease the 50% NHS replicon shift. In this case, addition of the gem-dimethyl group into the P2−P4 linker led to a compound (15j, MK-1220)34 with excellent genotype 1 and 2 biochemical and replicon potency and excellent rat plasma (plasma AUC = 11.8 μM·h) and liver exposure (23 μM @ 4 h). The dramatic effect of the 6-methoxy group can be seen with the contrastingly low plasma exposure seen with related P2-unsubstituted analogue 14k (plasma AUC = 0.18 μM·h).
Table 3.

| Replicon IC50 (nM)b |
||||||
|---|---|---|---|---|---|---|
| compd | 1b Ki (nM)a | 1b 10% FBS | 1b 50% NHS | 2a 10% FBS | rat PO plasma AUC (μM·h)c | rat [liver] @ 4 h (μM)d |
| 19 | 0.05 | 3.0 | 60.0 | 6.0 | 2.5 | 13.2 |
| 15c | 0.03 | 2.0 | 80.0 | - | 18.1 | 8.0 |
| 15j (MK-1220) | 0.02 | 4.0 | 11.0 | 5.0 | 11.8 | 23.0 |
| 14k | 0.02 | 2.0 | 10.0 | 11.0 | 0.18 | 16.0 |
NS3/4A protease time-resolved fluorescence assay.
Cell-based replicon assay.
5 mg/kg dosed orally in PEG400 (n = 2); AUC calculated based on 4 h experiment.
5 mg/kg dosed orally in PEG400 (n = 2); liver levels after 4 h.
Given the excellent potency and initial high plasma and liver exposure following oral dosing in rat, 15j was profiled further (Table 4). Compound 15j demonstrates low IV clearance in rat, dog, and rhesus monkey, moderate half-life and bioavailability, and excellent exposure in rat liver even at 24 h (9.9 μM; 5 mg/kg). Plasma and liver exposure in dog are similar to those in rat, with good plasma AUC (12.4 μM·h) and liver levels at both 2 h and 26 h (54 μM, 10.3 μM) following a 5 mg/kg oral dose. In contrast, plasma exposure in rhesus monkey is more limited (Table 4). Of particular note is that the 24 h liver concentration following a 5 mg/kg oral dose in both rats and dogs is ∼1000-fold greater than the 50% NHS-shifted replicon IC50 (11 nM). Following an IV dose to bile-duct cannulated animals, 15j is primarily excreted into the bile as a mixture of parent and oxidative metabolites in rat and dog. Compound 15j also has no significant activity versus other serine proteases (>50,000-fold versus trypsin and chymotrypsin) or in a broad Panlabs screen (>15,000-fold selectivity). Given the excellent potency, favorable preclinical pharmacokinetic properties, and clean off-target profile, 15j was determined to be the optimal compound in this series and was selected for clinical development.
Table 4. Pharmacokinetic Parameters for 15j (MK-1220).

| species | Cla (mL/(min kg)) | T1/2a (h) | %F | PObCmax (μM) | POb AUC (μM·h) | POb [liver] (μM) | POb [liver] (μM) |
|---|---|---|---|---|---|---|---|
| rat | 3.4 | 5.7 | 37% | 4.4 | 12.1 | 23 @ 4 h | 9.9 @ 24 h |
| dog | 3.6 | 2.5 | 41% | 4.0 | 12.4 | 54 @ 2 h | 10.3 @ 26 h |
| monkey | 8.3 | 1.3 | 9% | 0.3 | 1.2 |
IV: Rat (n = 4) and dog (n = 2) at 2 mg/kg and monkey (n = 3) at 1 mg/kg; DMSO.
PO (5 mg/kg, n = 3 in rat and n = 2 in dog and monkey; 10% Tween).
In summary, optimization for potency and rapid screening for rat plasma exposure (following oral dosing) in the P2-isoquinoline series of the P2−P4 macrocyclic HCV NS3/4A protease inhibitors has led to the discovery of several key structural features: notably the plasma exposure enhancing effects of P2-methoxy isoquinolines and P3-cycloalkyl groups. Within this optimized series, MK-1220 (15j) emerged as a candidate suitable for development.
Acknowledgments
We thank Dr. Charles W. Ross, III and Ms. Joan S. Murphy for high-resolution mass spectral data and Sandor L. Varga for interpretation of NMR spectra.
Supporting Information Available
1H NMR spectra and HRMS data for selected compounds, full experimental details for the synthesis of MK-1220, and general scheme and structures for 5-substituted isoquinoline analogues. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Hepatitic C-global prevalence (update). WHO Wkly. Epidemiol. Rec. 1999, 74, 425−427. [PubMed] [Google Scholar]
- Liang T. J.; Heller T. Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology 2004, 127, S62–S71. [DOI] [PubMed] [Google Scholar]
- Brown R. S. Hepatitis C and liver transplantation. Nature (London, U. K.) 2005, 436, 973–978. [DOI] [PubMed] [Google Scholar]
- Hadziyannis S. J.; Sette H. Jr.; Morgan T. R.; Balan V.; Diago M.; et al. Peginterferon-a2a and ribavirin combination therapy in chronic hepatitis C: A randomized study of treatment duration and ribavirin dose. Ann. Intern. Med. 2004, 140, 346–355. [DOI] [PubMed] [Google Scholar]
- Manns M. P.; McHutchison J. G.; Gordon S. C.; Rustgi V. K.; Shiffman M.; et al. Peginterferon alfa-2b plus ribavirin compared with interferon a-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomized trial. Lancet 2001, 358, 958–965. [DOI] [PubMed] [Google Scholar]
- McHutchison J. G.; Gordon S. C.; Schiff E. R.; Shiffman M. L.; Lee W. M.; et al. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. N. Engl. J. Med. 1998, 339, 1485–1492. [DOI] [PubMed] [Google Scholar]
- Aspinall R. J.; Pockros P. J. Review article: The management of side-effects during therapy for hepatitis C. Aliment. Pharmacol. Ther. 2004, 20, 917–929. [DOI] [PubMed] [Google Scholar]
- Fried Michael W. Side effects of therapy of hepatitis C and their management. Hepatology 2002, 36, S237–244. [DOI] [PubMed] [Google Scholar]
- Gordon C. P.; Keller P. A. Control of Hepatitis C: A Medicinal Chemistry Perspective. J. Med. Chem. 2005, 48, 1–20. [DOI] [PubMed] [Google Scholar]
- Chen Kevin X.; Njoroge F. G. A review of HCV protease inhibitors. Curr. Opin. Invest. Drugs 2009, 10, 821–837. [PubMed] [Google Scholar]
- Venkatraman S.; Njoroge F. G. Macrocyclic inhibitors of HCV NS3 protease. Expert Opin. Ther. Pat. 2009, 19, 1277–1303. [DOI] [PubMed] [Google Scholar]
- Thomson J. A.; Perni R. B. Hepatitis C virus NS3.4A protease inhibitors: countering viral subversion in vitro and showing promise in the clinic. Curr. Opin. Drug Discovery Dev. 2006, 9, 606–617. [PubMed] [Google Scholar]
- Chen S.-H.; Tan S.-L. Discovery of small-molecule inhibitors of HCV NS3-4A protease as potential therapeutic agents against HCV infection. Curr. Med. Chem. 2005, 12, 2317–2342. [DOI] [PubMed] [Google Scholar]
- Avolio S.; Summa V. Advances in the development of macrocyclic inhibitors of hepatitis C virus NS3-4A protease. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2010, 10, 1403–1422. [DOI] [PubMed] [Google Scholar]
- Lamarre D.; Anderson P. C.; Bailey M.; Beaulieu P.; Bolger G.; et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature (London, U. K.) 2003, 426, 186–189. [DOI] [PubMed] [Google Scholar]
- Gentile I.; Viola C.; Borgia F.; Castaldo G.; Borgia G. Telaprevir: a promising protease inhibitor for the treatment of hepatitis C virus infection. Curr. Med. Chem. 2009, 16, 1115–1121. [DOI] [PubMed] [Google Scholar]
- Campas C.; Pandian R.; Bolos J.; Castaner R. Boceprevir: NS3 protease inhibitor treatment of chronic hepatitis C. Drugs Future 2009, 34, 697–707. [Google Scholar]
- Seiwert S. D.; Andrews S. W.; Jiang Y.; Serebryany V.; Tan H.; et al. Preclinical characteristics of the hepatitis C virus NS3/4A protease inhibitor ITMN-191 (R7227). Antimicrob. Agents Chemother. 2008, 52, 4432–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan R.; Misialek S.; Stevens S. K.; Myszka D. G.; Brandhuber B. J.; et al. Inhibition and Binding Kinetics of the Hepatitis C Virus NS3 Protease Inhibitor ITMN-191 Reveals Tight Binding and Slow Dissociative Behavior. Biochemistry 2009, 48, 2559–2568. [DOI] [PubMed] [Google Scholar]
- Raboisson P.; de Kock H.; Rosenquist A.; Nilsson M.; Salvador-Oden L.; et al. Structure-activity relationship study on a novel series of cyclopentane-containing macrocyclic inhibitors of the hepatitis C virus NS3/4A protease leading to the discovery of TMC435350. Bioorg. Med. Chem. Lett. 2008, 18, 4853–4858. [DOI] [PubMed] [Google Scholar]
- Lin T.-I.; Lenz O.; Fanning G.; Verbinnen T.; Delouvroy F.; et al. In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor. Antimicrob. Agents Chemother. 2009, 53, 1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz O.; Verbinnen T.; Lin T.-I.; Vijgen L.; Cummings M. D.; et al. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob. Agents Chemother. 2010, 54, 1878–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas-Brunet M.; Bailey M. D.; Goudreau N.; Bhardwaj P. K.; Bordeleau J.; et al. Discovery of a Potent and Selective Noncovalent Linear Inhibitor of the Hepatitis C Virus NS3 Protease (BI 201335). J. Med. Chem. 2010, 53, 6466–6476. [DOI] [PubMed] [Google Scholar]
- McCauley J. A.; McIntyre C. J.; Rudd M. T.; Nguyen K. T.; Romano J. J.; et al. Discovery of Vaniprevir (MK-7009), a Macrocyclic Hepatitis C Virus NS3/4A Protease Inhibitor. J. Med. Chem. 2010, 53, 2443–2463. [DOI] [PubMed] [Google Scholar]
- Holloway M. K.; Liverton N. J.; Ludmerer S. W.; McCauley J. A.; Olsen D. B.. et al. Preparation of macrocyclic compounds as HCV NS3 protease inhibitors. In PCT Int. Appl.; Merck & Co., Inc.: USA, WO 2006/119061, 2006.
- Liverton N. J.; Holloway M. K.; McCauley J. A.; Rudd M. T.; Butcher J. W.; et al. Molecular Modeling Based Approach to Potent P2-P4Macrocyclic Inhibitors of Hepatitis C NS3/4A Protease. J. Am. Chem. Soc. 2008, 130, 4607–4609. [DOI] [PubMed] [Google Scholar]
- Liverton N. J.; Carroll S. S.; Di Muzio J.; Fandozzi C.; Graham D. J.; et al. MK-7009, a potent and selective inhibitor of hepatitis C virus NS3/4A protease. Antimicrob. Agents Chemother. 2009, 54, 305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach B. D.; Rice C. M. Unravelling hepatitis C virus replication from genome to function. Nature (London, U. K.) 2005, 436, 933–938. [DOI] [PubMed] [Google Scholar]
- The Zhan 1b catalyst is available from Strem, catalog # 44-0082; CAS# 918870-76-5.
- Wang X. A.; Sun L.-Q.; Sit S.-Y.; Sin N.; Scola P. M.. et al. Hepatitis C Virus Inhibitors. In U.S. Patent 6,995,174, 2006.
- Mao S.-S.; DiMuzio J.; McHale C.; Burlein C.; Olsen D.; et al. A time-resolved, internally quenched fluorescence assay to characterize inhibition of hepatitis C virus nonstructural protein 3-4A protease at low enzyme concentrations. Anal. Biochem. 2008, 373, 1–8. [DOI] [PubMed] [Google Scholar]
- Migliaccio G.; Tomassini J. E.; Carroll S. S.; Tomei L.; Altamura S.; et al. Characterization of Resistance to Non-obligate Chain-terminating Ribonucleoside Analogs That Inhibit Hepatitis C Virus Replication in Vitro. J. Biol. Chem. 2003, 278, 49164–49170. [DOI] [PubMed] [Google Scholar]
- Lohmann V.; Korner F.; Koch J. O.; Herian U.; Theilmann L.; et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [DOI] [PubMed] [Google Scholar]
- Holloway M. K.; Liverton N. J.; Ludmerer S. W.; McCauley J. A.; Olsen D. B.. et al. Macrocyclic peptides as HCV NS3 Protease Inhibitors. In PCT Int. Appl.; Merck & Co., Inc.: USA, WO 2007/016441, 2007.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.