

Abstract

We have discovered novel benzofuran-based S1P1 agonists with excellent in vitro potency and selectivity. 1-((4-(5-Benzylbenzofuran-2-yl)-3-fluorophenyl)methyl) azetidine-3-carboxylic acid (18) is a potent S1P1 agonist with >1000× selectivity over S1P3. It demonstrated a good in vitro ADME profile and excellent oral bioavailability across species. Dosed orally at 0.3 mg/kg, 18 significantly reduced blood lymphocyte counts 24 h postdose and demonstrated efficacy in a mouse EAE model of relapsing MS.
Keywords: Sphingosine-1 phosphate receptor, S1P1, S1P3, benzofuran, relapsing MS, immunomodulators, lymphopenia
Current therapeutic options for multiple sclerosis (MS) are all injectable, and a need for an effective oral agent exists. Myelin reactive T cells (lymphocytes) in the peripheral immune system play a key role in MS.1−6 Regulation of sphingosine-1 phosphate receptor subtype-1 (S1P1), a G-protein-coupled receptor (GPCR) expressed on T lymphocytes, could produce a new class of immunomodulators with a novel mechanism of action. Lymphocyte egress from lymphoid tissues requires the lipid mediator S1P1 and is required for the emigration of lymphocytes from the thymus and the trafficking of lymphocytes in secondary lymphoid organs.7−9
The pro-drug FTY-720 upon phosphorylation activates the S1P1 receptor.10 It was successful in advanced clinical trials for relapsing−remitting MS and was recently recommended for approval by an FDA advisory committee for relapsing MS.11,12 It is, however, a nonselective S1P receptor agonist with potent affinity for four of five S1P receptor subtypes. Agonism of S1P3 is believed to cause chronotropic side effects.13−16 We aimed to develop a novel, selective S1P1 agonist from knowledge-based lead design and computational chemistry approaches.
Findings from Merck17,18 revealed that unlike FTY-720-P and related compounds requiring intracellular phosphorylation, hydrophobic S1P screening hits such as oxadiazole SEW-2871 when decorated with a polar headgroup not only demonstrate strong binding to S1P1 receptors but are far more selective against S1P3. The structure−activity relationship (SAR) that emerged indicated that minor variation of the core scaffold could influence receptor binding potency as well as selectivity (Figure 1). For example, compounds A, B, and C differ very little (see oxadiazole ring) yet demonstrate >10-fold difference in S1P1 potency between each of them. Selectivity against S1P3 receptor of course improves significantly as a consequence.
Figure 1.

FTY-720-P, early S1P1 agonists, and Predix scaffold. aDisplacement of [33P]-labeled sphingosine-1-phosphate (S1P) from human S1P receptors expressed on CHO cell membranes.
Our in silico group developed models of the S1P1 receptor using PREDICT methodology.19 Docking of the Merck inhibitors provided insight and supported the medicinal chemistry observation that small changes to the core five-member ring structures could lead to potent, selective, and novel new S1P1 agonists. Utilizing this SAR insight, we designed several novel scaffolds each consisting of the azetidine carboxylate, a moderately polar core that lacks an H-bond donor and hydrophobic tail group. The scaffolds were designed with the additional consideration that the initial hit exploration library must be synthesizable using cluster synthesis approach. Mol wt, CLog P, and calculated ADME properties were evaluated prior to synthesis decisions. Among conceived designs, scaffold D (Figure 1), with its fused 5,6-heterocycle core with several combinations of the X and Y atoms, was favored due to overall superior match with our pharmacophore hypothesis. In this paper, we describe the successful discovery of benzofuran (X = O; Y = C)-based potent, selective S1P1 agonists.
Benzofuran 1 with the tail 5-n-butyl group, one of the first members of initial benzofuran lead-seeking compounds, demonstrated submicromolar potency in the GTPγS assay for the S1P1 receptor (Table 1). It had no activity at hS1P3 as determined by Ca2+ mobilization in hS1P3- in Gq/i5- transfected CHO-K1 cells. In an assay measuring receptor internalization (RI) of an hS1P1-GFP fusion protein in U2Os cells, compound 1 demonstrated micromolar potency. The RI data were deemed to be functionally more relevant and were run in parallel whenever feasible. The two assays generally correlated with each other. For SAR purposes, however, the GTPγS assay for the S1P1 receptor remained a primary screen. An effort was undertaken to investigate the benzofuran motif over other exploratory scaffolds in view of its novelty, initial potency, and generally good early ADME properties. Initial changes focused on the tail region. Analogue 2 with a 5-n-butyloxy group retained submicromolar S1P1 potency. Compounds 3 with 5-benzyl group, 4 with 5-cyclohexyl, and 5 with 5-isobutyl all demonstrated submicromolar activities in the GTPγS assay. Compounds 3 and 4 were full agonists in the RI screen (data not shown). Substitution with a 5-phenethyl group (6) or a phenoxy (8) was tolerated. Introduction of more polar groups such as a pyridyl (7) led to substantial loss of S1P1 potency. Interestingly, the piperidine derivative 9 retained micromolar S1P1 potency; it, however, was poorly selective (∼2×) over S1P3 receptor. This initial data demonstrated that hydrophobic groups were preferred in the tail region of the benzofuran core.
Table 1. SAR of 5-Substituted Benzofuran Corea.
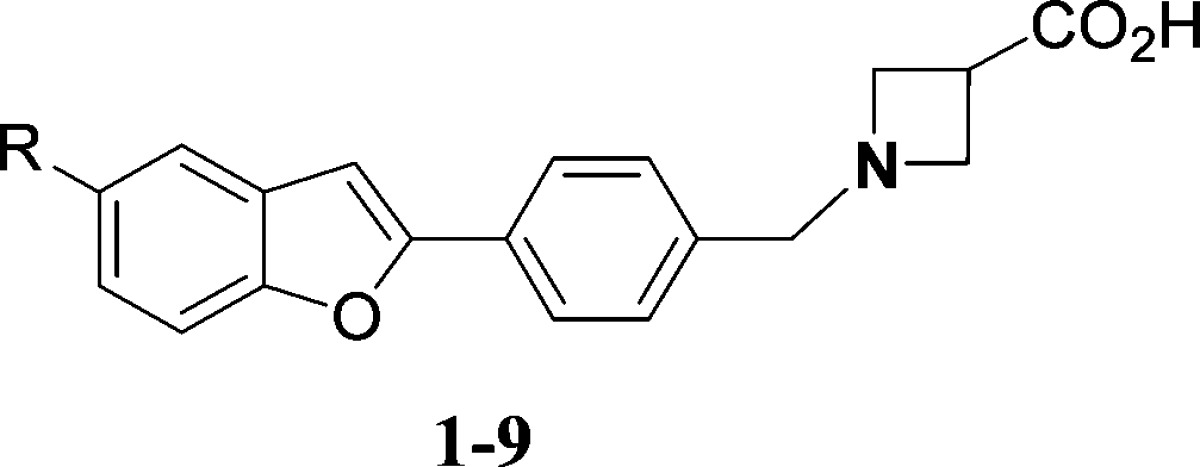
| no. | R | GTPγSb hS1P1 EC50 (μM) | hS1P3 Ca2+ EC50 (μM) | hS1P1 RI EC50 (μM) |
|---|---|---|---|---|
| 1 | n-Bu | 0.35 | >25 | 2.48 |
| 2 | O−n-Bu | 0.52 | >7.2 | NA |
| 3 | CH2Ph | 0.44 | >4.3 | 2.07 |
| 4 | cHex | 0.6 | >25 | 0.46 |
| 5 | CH2CH(CH3)2 | 0.38 | 1.9 | 0.15 |
| 6 | CH2CH2Ph | 0.58 | >25 | 0.354 |
| 7 | 3-Pyr | >1 | >25 | 5.03 |
| 8 | O−Ph | 0.51 | >25 | NA |
| 9 | N(CH2)5 | 1.5 | 1.44 | 0.62 |
Data represents an average of at least two determinations.
Compound induced binding of [35S]-GTPγS to human S1P-1 receptors expressed on CHO cell membranes (EC50). NA, not available.
Data from the lead optimization phase of benzofuran S1P modulators are shown in Table 2. Moving the tail substituent from the 5-position to the benzofuran-6-position led to loss in S1P1 receptor potency (10). Replacement of the core phenyl ring with 5-atom heteroaromatic moieties such as thiazole (11) also led to loss of potency. The thiophene (12) retained potency. We next studied substituents in the core phenyl ring. Small groups such as fluoro (13), chloro (15), and methoxy (14) at the 3-position were tolerated. Interestingly, a 3-pyridyl replacement (16) of phenyl demonstrated retention of in vitro potency and selectivity. Combining the 5-benzyl tail substituent with the core 3-fluoro-phenyl ring produced compound 18 with less than 200 nM potency in the GTPγS screen. Furthermore, it demonstrated less than 100 nM potency in the hS1P1 RI assay. The 2-fluorophenyl analogue 17 showed a 4−5-fold reduced hS1P1 potency relative to 3-fluorophenyl analogue 18. The 3-fluorophenyl group when combined with 5-cyclo-hexyl tail moiety demonstrated potency similar to analogue 18. Retaining the optimal 3-fluorophenyl ring but combining with somewhat more polar tail groups (20 and 21) led to loss in potency.
Table 2. Lead Optimization of Benzofuran Corea.
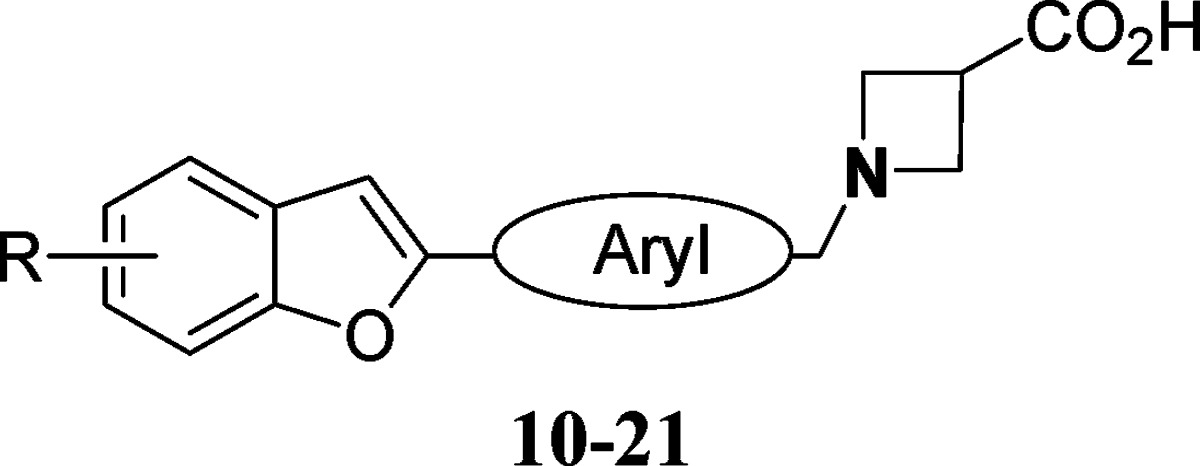
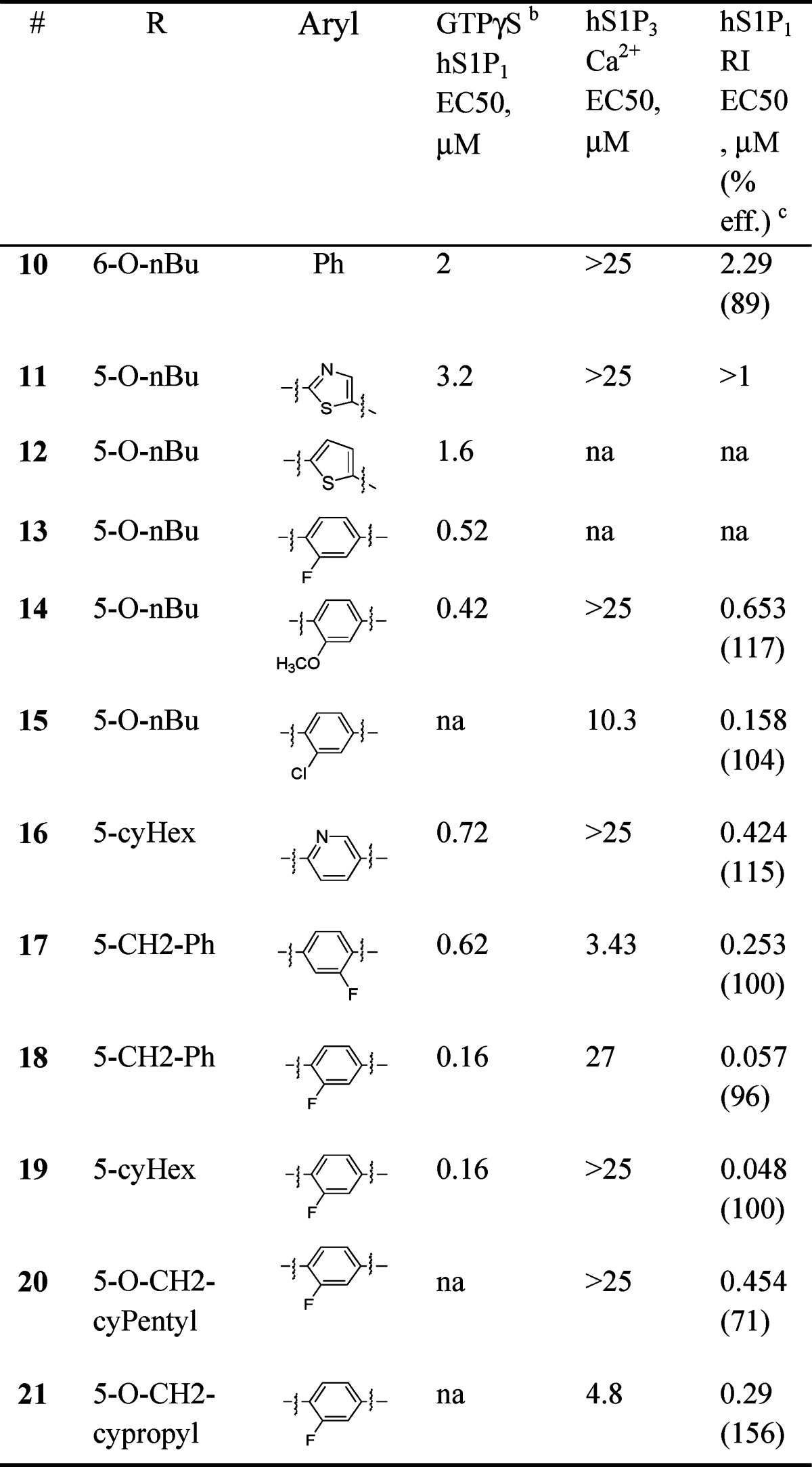 |
Data represents an average of at least two determinations.
Compound induced binding of [35S]-GTPγS to human S1P-1 receptors expressed on CHO cell membranes.
Percent efficacy is reported relative to 1.0 μM 1-(4-(6-benzylbenzofuran-2-yl)-3-fluorobenzyl)azetidine-3-carboxylic acid 18 (for hS1P1 RI); NA, not available.
One of our key preclinical lead criteria was >1000-fold S1P1 selectivity over S1P3. Benzofuran analogue 18 achieved highest selectivity with demonstrated EC50 values of 16 nM for hS1P1 in the GTPγS binding assay and 27 μM for hS1P3 in the Ca2+-flux assay, respectively. The ADME profile was favorable with a half-life in human liver microsomes of over 90 min. Clearance in the rat appeared to be primarily metabolic. Permeability across Caco-2 monolayers was moderate to good with low efflux characteristics.
Compound 18 produced comparable lymphopenia activity to FTY720 in Lewis rats with dose-dependent reduction in circulating blood lymphocytes 24 h postdose. Near maximal lymphopenia (75% reduction in lymphocytes vs vehicle) was achieved at doses of 1 and 3 mg/kg (Figure 2). Onset of lymphopenia was rapid across species in mice, rat, and nonhuman primates, with short recovery times in lymphocytes counts (less than 48 h).
Figure 2.
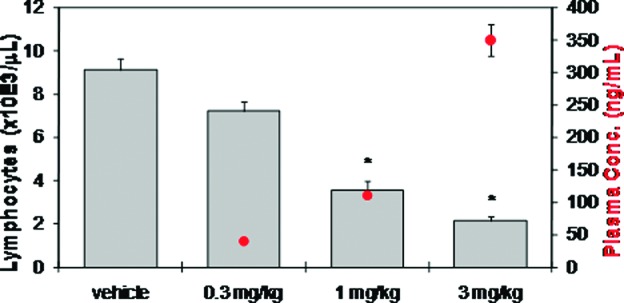
Compound 18 dosed orally reduces blood lymphocyte counts in female Lewis rats 24 h postdose (N = 5/group; bars represent average blood lymphocyte counts + SE; circles represent average plasma concentration ± SE; *P < 0.05 vs vehicle by ANOVA/Dunnett's multiple comparison test; vehicle = 20% captisol, pH 2).
Compound 18 demonstrated efficacy comparable to FTY-720 at (10 mg/kg po) nearly completely reversing the course of disease in Experimental Autoimmune Encephalitis (EAE) SJL mice model of multiple sclerosis (Figure 3). Preclinical pharmacokinetic (PK) in rat, nonhuman primate, and canine indicated that compound 18 had low clearance (corresponding to 4−15% of liver blood flow), a moderate steady state volume of distribution (1.0−2.2 L/kg), a moderate to long half-life (5−21 h), and good oral bioavailability (51−88%; Table 3). In vitro studies established that compound 18 neither inhibited nor induced human cytochrome P450 enzymes. Compound 18 was nonmutagenic (Ames and micronucleus negative) and did not significantly inhibit the hERG channel (patch clamp: IC50, 17 μM). In humans, it was predicted to have low clearance and a 5−10 h half-life, which would make it suitable for once a day dosing.
Figure 3.

Compound 18 data in experimental autoimmune encephalitis (EAE) SJL mice model of MS.
Table 3. Mean Compound 18 PK Parameters in Pie Clinical Species.
| species | CL (L/h/kg) | Vss (L/kg) | T1/2,Z (h) | MRT (h) | F % |
|---|---|---|---|---|---|
| rata | 0.138 | 1.7 | 12.1 | 12.7 | 80 |
| NHPb | 0.309 | 1 | 4.7 | 3.4 | 88 |
| caninec | 0.266 | 2.2 | 21.1 | 9 | 51 |
Male Sprague−Dawley (iv: 1 mg/kg, DMSO, N = 3; po: 3 mg/kg, 20% captisol, pH 4.0, N = 3).
NHP = nonhuman primate, male cynomolgus (iv: 1 mg/kg, 5% hydroxypropyl-β-cyclodextrin, N = 3; po: 5 mg/kg, 5% hydroxypropyl-β-cyclodextrin, N = 3.).
Male beagle (iv: 2 mg/kg, 20% captisol, pH 4.0, N = 3; po: 3 mg/kg, 20% captisol, pH 4.0, N = 3).
In a 4 day repeated dose toxicity study in rats, exposure was dose proportional up to 60 mg/kg. The brain/serum ratio was consistent across doses (10−16-fold), while the volume of distribution was 10-fold lower than FTY720. The NOAEL was 20 mg/kg. To examine if lack of S1P3 potency corresponded to an absence of chronotropic effect, compound 18 was dosed in telemetered female SD rats as an oral bolus in a volume of 5 mg/mL in 20% captisol. The mean arterial pressure (MAP) was moderately elevated at 20 and 40 mg/kg from 4 to 6 h after dosing, with peak effects corresponding to Cmax. There was no effect on heart rate at all doses, confirming a no-effect level for HR changes at 40 mg/kg po, indicating a wide margin for S1P3-mediated cardiovascular toxicity. The mechanism underlying the elevated MAP is uncertain.
The synthesis of compound 18 is described in Scheme 1. The 5-benzyl benzofuran 25 was prepared via a two-step reaction, starting from the alkylation of phenol 22 with 2-bromo-1,1- diethoxyethane 23, followed by cyclization with polyphosphoric acid (PPA) in benzene at reflux temperature. The boronic acid functionality was selectively incorporated at the 2-position of the benzofuran ring of 26 under our modified conditions using n-BuLi and B(iPrO)3 at low temperature. Suzuki C−C coupling of boronic acid 26 and aryl halide 27 was carried out in a microwave apparatus using 5 mol % palladium(II) catalyst, triethyl amine, and ethanol as the solvent at 100 °C, cleanly yielding 4-(5-benzylbenzofuran-2-yl)-3-fluorobenzaldehyde 28. Reductive amination of the aldehyde with azetidine-3-carboxylic acid 29 provided the desired compound 18 as its zwitterion.
Scheme 1. Synthesis of 18.

(a) KOH, DMSO, 20 h, 84%. (b) PPA, benzene, 89%. (c) n-BuLi, THF, −78 °C. (d) B(iPrO)3. (e) 2 N HCl or NH4Cl (66% yield after steps c−e). (f) PdCl2(Ph3P)2 (5 mol %) Et3N, EtOH, 100 °C, microwave, 65%. (g) AcOH, NaCNBH3, DCM/MeOH, 56%.
In summary, knowledge-based scaffold hopping combined with computation chemistry tools led to identification of the benzofuranyl core as a novel new S1P1 lead series. Rapid optimization efforts designed to enhance potency, while balancing lipophilicity led to 3-fluorophenyl benzofuran 18, which was found to have potent S1P1 activity and excellent S1P3 selectivity. Oral dosing of 18 in rats (1 mg/kg) resulted in a statistically significant reduction in circulating lymphocytes 24 h postdose, as well as efficacy in the mice EAE model of MS. The overall profile of compound 18 led to nomination for preclinical development including follow-on GLP toxicology studies. Such studies revealed pro-convulsive activity at doses of 40 mg/kg and higher. Optimization efforts toward scaffolds that avoid such toxic properties will be the subject of upcoming manuscripts.
Acknowledgments
We thank Sheila Dewitt, Epix, and Sharon Pick and Victor Cee, Amgen, for assistance in coordinating completion of this manuscript. We also thank Pini Orbach, Sharon Shacham, and Oren Becker for their contributions to the S1P program at Epix.
Supporting Information Available
Experimental procedures and characterization data for 1−21 and details of in vitro and in vivo assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Chembiotek, A TCG Life Sciences Company, Block BN, Plot 7, Saltlake Electronics Complex, Sector V, Kolkata 700091, India.
Supplementary Material
References
- Chun J; Hartung H. Mechanism of Action of Oral Fingolimod (FTY720) in Multiple Sclerosis. Clin. Neuropharmacol. 2010, 33291–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappos L.; Radue E.-W.; O'Connor P.; Polman C.; Hohlfeld R.; Calabresi P; Selmaj K.; Agoropoulou C.; Leyk M.; Zhang-Auberson L.; Burtin P. A. Placebo-Controlled Trial of Oral Fingolimod in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [DOI] [PubMed] [Google Scholar]
- Cohen J. A.; Barkhof F.; Comi G.; Hartung H.-P.; Khatri B. O.; Montalban X.; Pelletier J.; Capra R.; Gallo P.; Izquierdo G.; Tiel-Wilck K.; de Vera A.; Jin J.; Stites T.; Wu S.; Aradhye S.; Kappos L. Oral Fingolimod or Intramuscular Interferon for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 402–415. [DOI] [PubMed] [Google Scholar]
- Brinkmann V. FTY720 (fingolimod) in multiple sclerosis: therapeutic effects in the immune and the central nervous system. Br. J. Pharmacol. 2009, 15851173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster C. A.; Howard L. M.; Schweitzer A.; Persohn E.; Hiestand P. C.; Balatoni B.; Reuschel R.; Beerli C.; Schwartz M.; Billich A. Brain Penetration of the Oral Immunomodulatory Drug FTY720 and its Phosphorylation in the Central Nervous System During Experimental Autoimmune Encephalomyelitis: Consequences for Mode of Action in Multiple Sclerosis. J. Pharmacol. Exp. Ther. 2007, 3232469–476. [DOI] [PubMed] [Google Scholar]
- Kataoka H.; Sugahara K.; Shimano K.; Teshima K.; Koyama M.; Fukunari A.; Chiba K. FTY720, Sphingosine 1-Phosphate Receptor Modulator, Ameliorates Experimental Autoimmune Encephalomyelitis by Inhibition of T Cell Infiltration. Cell. Mol. Immunol. 2005, 2, 439–448. [PubMed] [Google Scholar]
- Baumruker T; Billich A; Brinkmann V. FTY720, an Immunomodulatory Sphingolipid Mimetic: Translation of a Novel Mechanism into Clinical Benefit in Multiple Sclerosis. Expert Opin. Invest. Drugs 2007, 163283–289. [DOI] [PubMed] [Google Scholar]
- Chiba K.; Matsuyuki H.; Maeda Y.; Sugahara K. Role of Sphingosine 1-Phosphate Receptor type 1 in Lymphocyte Egress from Secondary Lymphoid Tissues and Thymus. Cell. Mol. Immunol. 2006, 3111–19. [PubMed] [Google Scholar]
- Brinkmann V. FTY720: Mechanism of Action and Potential Benefit in Organ Transplantation. Yonsei Med. J. 2004, 456991–997. [DOI] [PubMed] [Google Scholar]
- Verzijl D.; Peters S. L. M.; Alewijnse A. E. Sphingosine-1-Phosphate Receptors: Zooming in on Ligand-Induced Intracellular Trafficking and Its Functional Implications. Mol. Cells 2010, 29299–104. [DOI] [PubMed] [Google Scholar]
- Swedin H. FDA Advisory Committee Unanimously Recommends Approval of Novartis Investigational Treatment FTY720 to Treat Relapsing Remitting MS. Novartis Press Release, PR Newswire, June 10, 2010. [Google Scholar]
- Cohen J. A.; et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 2010, 3625402–415. [DOI] [PubMed] [Google Scholar]
- Kovarik J. M.; Lu M.; Riviere G.; Barbet I.; Maton S.; Goldwater D. R.; Schmouder R. L. The Effect on Heart Rate of Combining Single-dose Fingolimod with Steady-state Atenolol or Diltiazem in Healthy Subjects. Eur. J. Clin. Pharmacol. 2008, 645457–463. [DOI] [PubMed] [Google Scholar]
- Schmouder R.; Serra D.; Wang Y.; Kovarik J. M.; DiMarco J.; Hunt T. L.; Bastien M.-C. FTY720: Placebo-Controlled Study of the Effect on Cardiac Rate and Rhythm in Healthy Subjects. J. Clin. Pharmacol. 2006, 46, 895–904. [DOI] [PubMed] [Google Scholar]
- Forrest M.; Sun S.-Y.; Hajdu R.; Bergstrom J.; Card D.; Doherty G.; Hale J.; Keohane C.; Meyers C.; Milligan J.; Mills S.; Nomura N.; Rosen H.; Rosenbach M.; Shei G.-J.; Singer I. I.; Tian M.; West S.; White V.; Xie J.; Proia R. L.; Mandala S. Immune Cell Regulation and Cardiovascular Effects of Sphingosine-1-phosphate Receptor Agonists in Rodents are Mediated via Distinct Receptor Subtypes. J. Pharmacol. Exp. Ther. 2004, 309, 758–768. [DOI] [PubMed] [Google Scholar]
- Sanna M. G.; Liao J.; Jo E.; Alfonso C.; Ahn M.-Y.; Peterson M. S.; Webb B.; Lefebvre S.; Chun J.; Gray N.; Rosen H. Sphingosine 1-Phosphate (S1P) Receptor Subtypes S1P1 and S1P3, Respectively, Regulate Lymphocyte Recirculation and Heart Rate. J. Biol. Chem. 2004, 279, 13839–13848. [DOI] [PubMed] [Google Scholar]
- Hale J. J.; Lynch C. L.; Neway W.; Mills S. G.; Hajdu R.; Keohane C.; Ann; Rosenbach M. J.; Milligan J. A.; Shei G.-J.; Parent S. A.; Chrebet G.; Bergstrom J.; Card D.; Ferrer M.; Hodder P.; Strulovici B.; Rosen H.; Mandala S. A Rational Utilization of High-Throughput Screening Affords Selective, Orally Bioavailable 1-Benzyl-3-carboxyazetidine Sphingosine-1-phosphate-1 Receptor Agonists. J. Med. Chem. 2004, 47276662–6665. [DOI] [PubMed] [Google Scholar]
- Li Z.; Chen W.; Hale J. J.; Lynch C. L.; Mills S. G.; Hajdu R.; Keohane C. A.; Rosenbach M. J.; Milligan J. A.; Shei G.-J.; Chrebet G.; Parent S. A.; Bergstrom J.; Card D.; Forrest M.; Quackenbush E. J.; Wickham L.; Alexandra V.; Hugo E.; Rose M.; Rosen H.; Mandala S. Discovery of Potent 3,5-Diphenyl-1,2,4-oxadiazole Sphingosine-1-phosphate-1 (S1P1) Receptor Agonists with Exceptional Selectivity against S1P2 and S1P3. J. Med. Chem. 2005, 48206169–6173. [DOI] [PubMed] [Google Scholar]
- Shacham S.; Marantz Y.; Bar-Haim S.; Kalid O.; Warshaviak D.; Avisar N.; Inbal B.; Heifetz A.; Fichman M.; Topf M.; Naor Z.; Noiman S.; Becker O. M. PREDICT Modeling and In-Silico Screening for G-Protein Coupled Receptors. Proteins 2004, 57, 51–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.