

Abstract

We report herein the discovery of benzodiazepine sulfonamide-based bombesin receptor subtype 3 (BRS-3) agonists and their unusual chirality. Starting from a high-throughput screening lead, we prepared a series of BRS-3 agonists with improved potency and pharmacokinetic properties, of which compound 8a caused mechanism-based, dose-dependent food intake reduction and body weight loss after oral dosing in diet-induced obese mice. This effort also led to the discovery of a novel family of chiral molecules originated from the conformationally constrained seven-membered diazepine ring.
Keywords: Bombesin receptor subtype 3, BRS-3, agonist, obesity, benzodiazepine sulfonamide, planar chirality, atropisomerism
Obesity is a serious and chronic medical condition that has become a major global health issue. In the United States, obesity rates for adults have doubled, and rates for children have tripled over the last 30 years. Currently, around one-third of U.S. adults and 16% of U.S. children are considered obese.1 In many cases, excessive body weight is the root cause of subsequent comorbidities, including type 2 diabetes, hypertension, cardiovascular disease, cancer, and arthritis.2 The preferred approach to manage obesity calls for lifestyle changes including control of calorie intake and increase in physical activities. However, this approach alone is often insufficient or unsustainable. In the United States, orlistat3 is the only drug approved for the treatment of obesity and has suboptimal tolerability and limited efficacy (the other obesity drug, sibutramine, was recently withdrawn from the market by Abbott Laboratories4). Therefore, new antiobesity agents are highly desirable. Our laboratories are interested in pursuing new mechanisms for the treatment of obesity. The involvement of the bombesin receptor subtype-3 (BRS-3) in regulating energy homeostasis has been demonstrated in animal models. Mice lacking functional BRS-3 develop metabolic defects and obesity.5,5b On the other hand, a BRS-3 agonist caused food intake reduction and an increase in metabolic rate in established diet-induced obese (eDIO) mice, suggesting that the BRS-3 receptor may serve as a potential target for treating obesity.6 Recently, medicinal chemistry efforts to identify small molecule BRS-3 agonists have been reported by others7,7b and our laboratories.8−8e In this letter, we describe our continued efforts in this area that led to the discovery of a novel class of BRS-3 agonists.
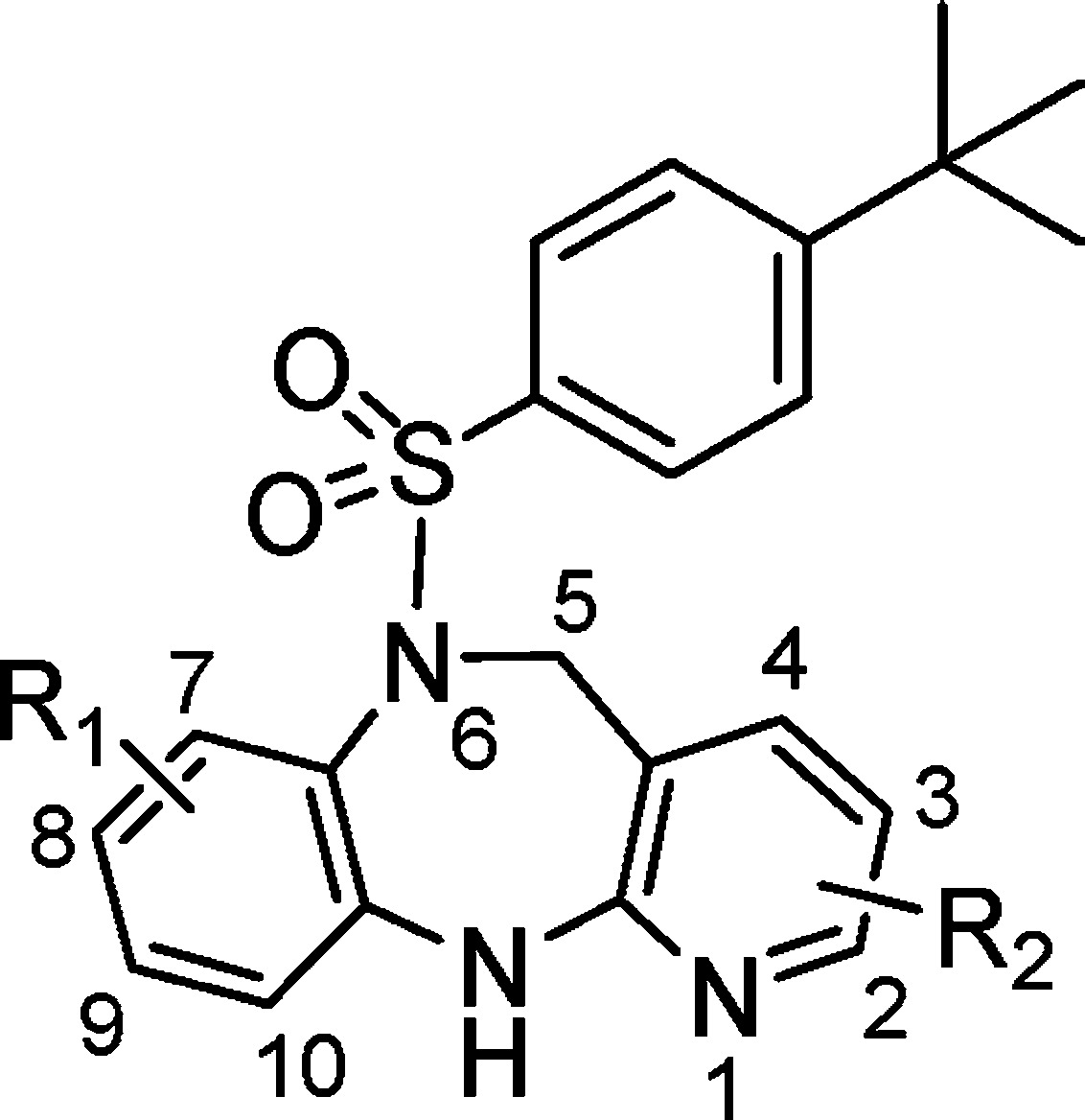
Our efforts started with a high-throughput screening lead (1) as shown in Figure 1. This lead exhibited moderate BRS-3 potency in human and mouse and poor pharmacokinetic properties in the rat (Clp = 164 mL/min/kg, F = 1%, t1/2 = 1.6 h). Our initial aim was to determine whether the diazepine core was optimal. A systematic core structure modification was undertaken, which included moving the pyridine N, breaking the diazepine ring, and saturating the left-hand side phenyl ring. Each case resulted in complete loss of potency. Therefore, the diazepine core was preserved for further structure–activity relationship (SAR) studies that focused on varying the substituents of the aromatic rings.
Figure 1.

Structures of orlistat, sibutramine, and HTS lead 1.
The synthesis of the diazepine sulfonamide analogues was straightforward (Scheme 1). Condensation of substituted phenyldiamine 2 and 2-chloronicotinic acid 3 under microwave or thermal conditions provided a regioisomeric mixture of 4 and 5, favoring the desired isomer 4 in a ratio of 3:1 to 9:1. This mixture was reduced by borane to diazepine 6, which upon reacting with the sulfonyl chloride afforded diazepine sulfonamide 8. Typically, the undesired regioisomer was carried on to the final step where 7 was separated from 8 by silica gel chromatography.
Scheme 1.

However, both silica gel chromatography and reverse-phase high-performance liquid chromatography (HPLC) failed to provide an acceptable separation of 7a from 8. Then, we subjected the mixture to a ChiralCel OD column, expecting two peaks corresponding to the two regioisomers. To our surprise, three peaks were present in the HPLC trace. While one peak corresponded to the undesired regioisomer 7a, the two compounds obtained corresponding to the other two peaks displayed exactly the same 1H NMR but opposite rotation (8a, [α]D = −157°; 8b, [α]D = +155°), suggesting an enantiomeric pair (Figure 2). Because the molecule does not possess a chiral center, possible causes for the observed atropisomerism9 include hindered inversion of the sulfonamide nitrogen, restricted rotation about the N–S single bond, and conformationally constrained seven-membered diazepine ring system. On the basis of X-ray analysis of 8a and 8b, the bond distances from the sulfonamide N to the connected atoms are all typical for single-bond lengths, but the geometry around this N is almost planar (Figure 3). Both molecular modeling and X-ray analysis revealed that no obvious energy barrier existed for either the N–S bond rotation or the inversion of the nearly planar sulfonamide nitrogen. In contrast, the puckered diazepine ring was highly conformationally constrained, and a ring flip would place the sulfonyl group and the 7-Me substituent of the phenyl ring in the same plane, resulting in severe van der Waals interactions between these groups. Modeling suggested a barrier of ∼33 kcal/mol for the ring interconversion. Chiral LC study of 8b afforded a first-order kinetics with a rate constant of 1.35 × 10–8 s–1 at 25 °C and 1.03 × 10–7 s–1 at 40 °C, corresponding to activation energy of 25.2 kcal/mol. Although the racemization is slow at <50 °C (t1/2 = 296 days at 25 °C; 39 days at 40 °C), the rate of interconversion is significantly higher at >100 °C, and a complete racemization was observed after 4 h at 100 °C.10
Figure 2.

Discovery of the atropisomerism.
Figure 3.
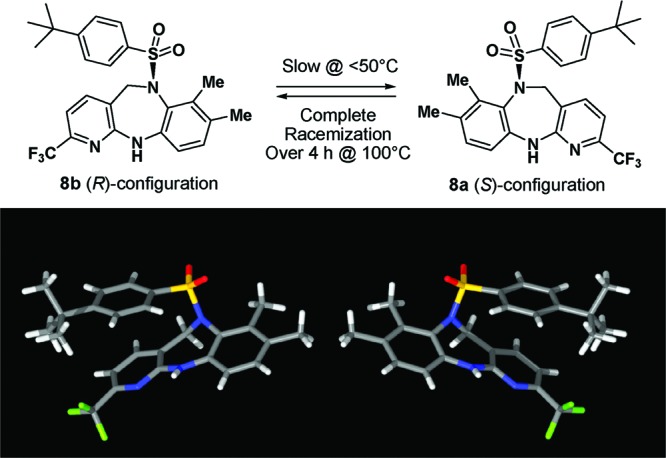
X-ray analysis revealed planar chirality.11
The “SAR” of the atropisomerism is consistent with the planar chirality where the steric hindrance between the sulfonyl group and the neighboring substituent RA on the Ph ring accounted for the interconversion barrier (Figure 4). The atropoisomers of the 7-H and 7-F analogues were not separable by HPLC at room temperature, neither was compound 9 with only an electron lone pair on RA position. Not surprisingly, fused ring analogue 10 exhibited atropisomerism, and the two enantiomers were readily separated.
Figure 4.

“SAR” of the atropisomerism. RA = H (8k) or F (8l), no atropisomerism; 9 did not exhibit atropisomerism. If RA > F, 8 exhibited atropisomerism, e.g., 8i (RA = Cl), 8m (RA = OH), and 10.
As expected, the two atropisomers, 8a and 8b, behaved very differently toward the BRS-3 receptors. The (S)-isomer 8a exhibited hBRS-3 IC50 of 1.4 nM (125I-dY-peptide was used as a radioligand in the binding assay) and EC50 of 97 nM, over 100-fold more potent than the (R)-isomer 8b (Table 1).12 As compared to initial lead 1, CF3 at the 2-position (8a) improved hBRS-3 binding affinity by 30-fold and mouse functional activity by 8-fold. While the 2-position tolerated a variety of functionalities, such as methyl (8c), cyano (8d), and chloro (8e), the 3-position preferred no substituents (8f vs 1; 8g vs 8e). For the left-hand side aromatic ring, deletion of 8-methyl (8h) slightly improved potency; however, removal of 7-methyl (8j) or both methyls (8k) decreased potency. Replacing the 7-methyl group with chloro (8i), fluoro (8l), or hydroxyl (8m) resulted in comparable potency. The fused ring analogue 10 was among the most potent compounds with hBRS-3 IC50 of 0.7 nM.
Table 1. Substitution Effects on the Human and Mouse BRS-3 Activity.
| compda | substitution | hBRS-3b IC50 (nM) | hBRS-3b EC50 (nM) (%Act)c | mBRS-3b EC50 (nM) (% Act)c |
|---|---|---|---|---|
| 1 | 7-Me, 8-Me | 40 ± 21 | 325 ± 117 (91 ± 9%) | 248 ± 42 (101 ± 3%) |
| 7a | 9-Me, 10-Me | 80% inhd at 10 μM | >10000d | >10000d |
| 8a (S) | 7-Me, 8-Me, 2-CF3 | 1.4 ± 0.9 | 97 ± 23 (96 ± 10%) | 33 ± 11 (98 ± 7%) |
| 8b (R) | 7-Me, 8-Me, 2-CF3 | 169 ± 139 | >10000 | >10000 |
| 8c | 7-Me, 8-Me, 2-Me | 10 ± 4.5 | 131 ± 36 (92 ± 13%) | 232 ± 50 (107 ± 10%) |
| 8d (S) | 7-Me, 8-Me, 2-CN | 4.4 ± 1.0 | 50 ± 8 (100 ± 4%) | 50 ± 3 (103 ± 1%) |
| 8e (S) | 7-Me, 8-Me, 2-Cl | 1.2 ± 0.3 | 38 ± 9 (97 ± 2%) | 98 ± 25 (105 ± 7%) |
| 8f | 7-Me, 8-Me, 3-Cl | 430 ± 170 | >10000d | >10000d |
| 8g | 7-Me, 8-Me, 2-Cl, 3-F | 8.9 ± 3.0 | 277 ± 80 (83 ± 1%) | 371 ± 13 (82 ± 6%) |
| 8h (S) | 7-Me, 2-CF3 | 0.9 ± 0.1 | 50 ± 14 (101 ± 10%) | 120 ± 14 (106 ± 1%) |
| 8i (S) | 7-Cl, 2-CF3 | 3.2 ± 0.5 | 63 ± 33 (96 ± 6%) | 155 ± 53 (100 ± 13%) |
| 8j | 8-Me, 2-CF3 | 5.8 ± 0.1 | 199 ± 34 (83 ± 11%) | 377 ± 7 (82 ± 4%) |
| 8k | 2-CF3 | 5.5 ± 0.5 | 110 ± 23 (96 ± 10%) | 161 ± 11 (98 ± 7%) |
| 8l | 7-F, 2-CF3, 8-C(CH3)2OH | 5.0 ± 0.3 | 77 ± 10 (96 ± 2%) | 134 ± 14 (97 ± 0%) |
| 8m (S) | 7-OH, 2-CF3 | 2.2 ± 0.4 | 88 ± 21 (103 ± 1%) | 209 ± 62 (115 ± 14%) |
| 9 | see Figure 4 | 5.3 ± 0.2 | 110 ± 27 (104 ± 0%) | 171 ± 5 (112 ± 12%) |
| 10 (S) | see Figure 4 | 0.7 ± 0.1 | 108 ± 44 (101 ± 8%) | 129 ± 18 (107 ± 4%) |
Racemic mixture or achiral compound (7, 8j, 8k, etc.), except as noted.
Data expressed as mean ± SD (n ≥ 2 independent experiments).
% Act represents the maximum activation of tested compound relative to that of the dY peptide ([d-Tyr,6β-Ala,11 Phe,13 and Nle14]-bombesin(6–14)).
Assayed only once.
Next, the SAR of the aryl sulfonyl group was explored.13 Analogues 11a–i in Table 2 were synthesized as described in Scheme 1 using the appropriate sulfonyl chloride. para-Substituted phenyl was preferred, and a variety of substituents were tolerated. While isopropyl (11a) and trifluoromethoxy (11d) groups could serve as replacements for the tert-butyl (8a), the methylsulfonyl group (11c) caused loss of potency. In addition to phenyl analogues, heterocyclic analogues, such as thiazole (11g) and thiophene analogues (11h and 11i), also exhibited reasonable BRS-3 potency.
Table 2. Human and Mouse BRS-3 Activity of 11.
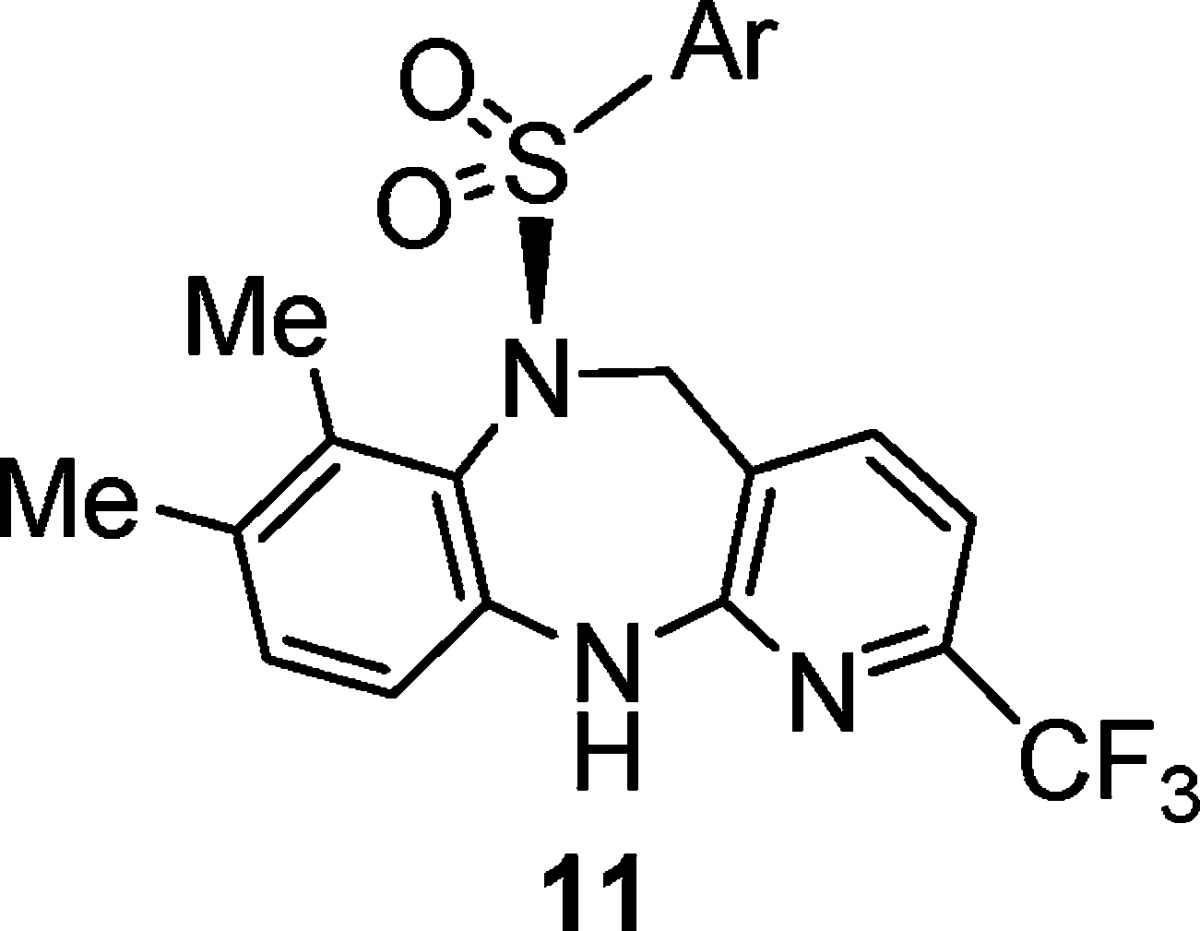
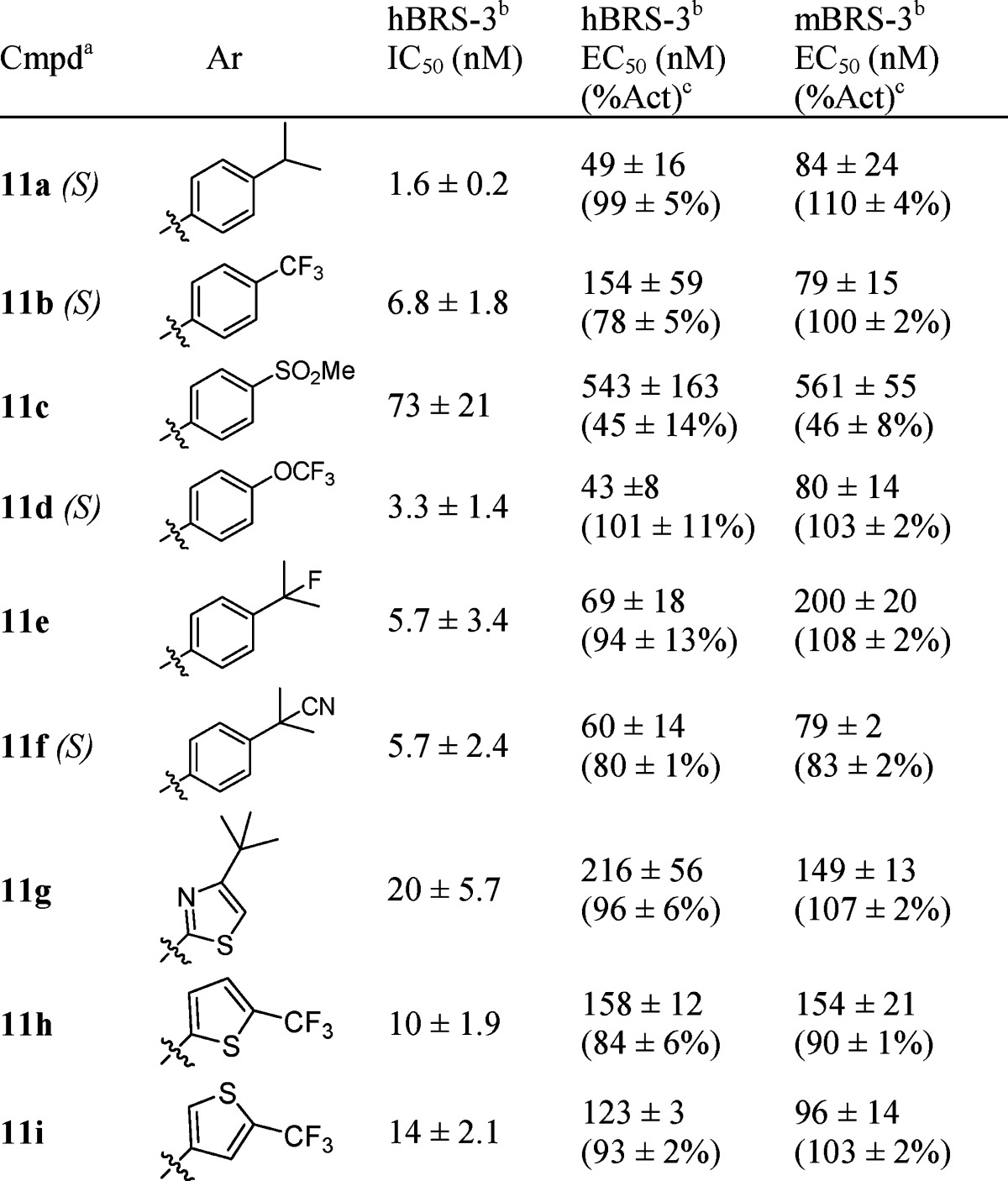
Racemic mixture, except as noted.
Data expressed as mean ± SD (n ≥ 2 independent experiments).
% Act represents the maximum activation of tested compound relative to that of the dY peptide.
A number of more potent BRS-3 agonists were evaluated for their pharmacokinetic properties, and several displayed an improved PK profile (Table 3). As compared to the initial lead 1, these compounds demonstrated lower clearance and higher oral AUC and bioavailability in the rat. The pharmacokinetic properties of 8a were also evaluated in the rhesus monkey where the bioavailability was 17% and the half-life was 1.8 h.
Table 3. Rata Pharmacokinetic Datab of Selected Compounds.
| compd | Clp (mL/min/kg) | T1/2 (h) | po AUCN (μM h kg/mg) | Foral (%) |
|---|---|---|---|---|
| 1 | 164 | 1.6 | 0.007 | 1.2 |
| 8a | 29 | 1.6 | 0.20 | 15 |
| 8h | 38 | 1.6 | 0.13 | 14 |
| 8i | 46 | 1.4 | 0.26 | 34 |
| 8j | 24 | 2.2 | 0.31 | 22 |
| 8l | 24 | 3.0 | 0.19 | 13 |
| 11a | 16 | 3.5 | 0.24 | 10 |
Sprague–Dawley (SD) rat was used.
Plasma clearance (Clp) and half-life (T1/2) calculated following 1 mg/kg iv dose. Normalized oral exposure (po AUCN) and oral bioavailability (Foral) calculated following 4 mg/kg po dose.
Compound 8a exhibited excellent potency against dog and rhesus BRS-3 receptors: dog IC50 = 1.6 nM, EC50 = 36 nM, 97% Act; rhesus IC50 = 3.0 nM, EC50 = 70 nM, 97% Act. This compound was screened against >100 receptors, ion channels, and enzymes; seven off-targets were identified with IC50 values in the range of 1–10 μM, but none was considered significant. Compound 8a was very selective over the two key ion channels, hERG IC50 > 10 μM and DLZ IC50 > 10 μM, but was a potent activator of human PXR (EC50 = 678 nM; 42% Act at 10 μM).14
Compound 8a was assessed for acute efficacy in wild-type (WT) and BRS-3 KO mice. In WT mice, the compound caused significant food intake reduction and body weight loss, in a dose-dependent manner at 3 and 30 mpk 18 h after oral dosing. In KO mice, no effect was observed at 3 and 30 mpk. After subchronic dosing (20 mpk, BID) in e-DIO mice, the compound caused 5.2% body weight loss on day 4 (Figure 5).15 The body weight loss was caused by reduction in food intake as well as increase in fasting metabolic rate.16
Figure 5.
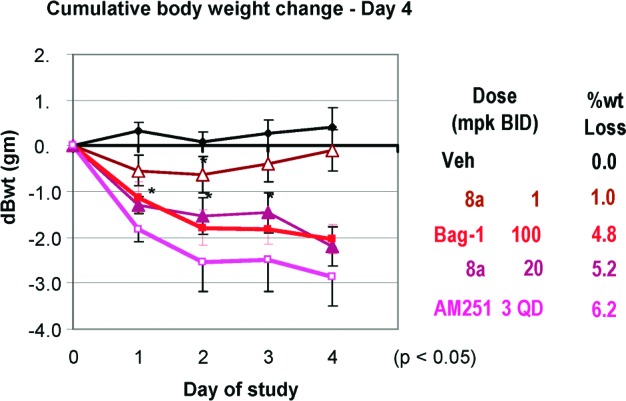
Effect of 8a on subchronic body weight of eDIO mice.
In summary, we have discovered a novel class of benzodiazepine sulfonamide-based BRS-3 agonists along with the unusual planar chirality that originated from a conformationally restrained diazepine ring system. Starting from a high-throughput screening lead, SAR studies led to the synthesis of a series of BRS-3 agonists with improved potency and pharmacokinetic properties, of which compound 8a caused mechanism-based, dose-dependent food intake reduction and body weight loss after oral dosing in eDIO mice.
Acknowledgments
We thank Dr. Timothy A. Blizzard for helpful discussions and the Department of Laboratory Animal Resources for their assistance in animal dosing and sampling.
Supporting Information Available
Synthetic procedures and analytical data of selected BRS-3 agonists, conditions for all of the biological assays, and X-ray, chiral LC, and modeling methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Centers for Disease Control and Prevention: Morbidity and Mortality Weekly Report, Vol. 59, August 3, 2010.
- Olshansky S. J.; Passaro D. J.; Hershow R. C.; Layden J.; Carnes B. A.; Brody J.; Hayflick L.; Butler R. N.; Allison D. B.; Ludwig D. S. A potential decline in life expectancy in the United States in the 21st century. N. Engl. J. Med. 2005, 352, 1138–1145. [DOI] [PubMed] [Google Scholar]
- McNeely W.; Benfield P. Orlistat. Drugs 1998, 56, 241–249. [DOI] [PubMed] [Google Scholar]
- FDA. News Release, October 8, 2010: Abbott Laboratories agrees to withdraw its obesity drug Meridia (sibutramine). See http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm228812.htm.
- Ohki-Hamazaki H.; Watase K.; Yamamoto K.; Ogura H.; Yamano M.; Yamada K.; Maeno H.; Imaki J.; Kikuyama S.; Wada E.; Wada K. Mice lacking bombesin receptor subtype-3 develop metabolic defects and obesity. Nature 1997, 390, 165–169. [DOI] [PubMed] [Google Scholar]
- Ladenheim E. E.; Hamilton N. L.; Behles R. R.; Bi S.; Hampton L. L.; Battey J. F.; Moran T. H. Factors Contributing to Obesity in Bombesin Receptor Subtype-3-Deficient Mice. Endocrinology 2008, 149, 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X.-M.; Chen H.; Dobbelaar P. H.; Dong Y.; Fong T. M.; Gagen K.; Gorski J.; He S.; Howard A. D.; Jian T.; Jiang M.; Kan Y.; Kelly T. M.; Kosinski J.; Lin L. S.; Liu J.; Marsh D.; Metzger J. M.; Miller R.; Nargund R. P.; Palyha O.; Shearman L.; Shen Z.; Stearns R.; Strack A. M.; Stribling S.; Tang Y. S.; Wang S.-P.; White A.; Yu H.; Reitman M. L. Regulation of energy homeostasis by bombesin receptor subtype-3: Selective receptor agonists for the treatment of obesity. Cell Metab. 2010, 11, 101–112. [DOI] [PubMed] [Google Scholar]
- Weber D.; Berger C.; Eickelmann P.; Antel J.; Kessler H. Design of Selective Peptidomimetic Agonists for the Human Orphan Receptor BRS-3. J. Med. Chem. 2003, 46, 1918–1930. [DOI] [PubMed] [Google Scholar]
- Carlton D. L.; Collin-Smith L. J.; Daniels A. J.; Deaton D. N.; Goetz A. S.; Laudeman C. P.; Littleton T. R.; Musso D. L.; Ott Morgan R. J.; Szewczyk J. R.; Zhang C. Discovery of small molecule agonists for the bombesin receptor subtype 3 (BRS-3) based on an omeprazole lead. Bioorg. Med. Chem. Lett. 2008, 18, 5451–5455. [DOI] [PubMed] [Google Scholar]
- He S.; Dobbelaar P. H.; Liu J.; Jian T.; Sebhat I. K.; Lin L. S.; Goodman A.; Guo C.; Guzzo P. R.; Hadden M.; Henderson A. J.; Ruenz M.; Sargent B.; Yet L.; Kelly T. M.; Palyha O.; Kan Y.; Pan J.; Chen H.; Marsh D.; Shearman L. P.; Strack A. M.; Metzger J. M.; Feighner S. D.; Tan C.; Howard A. D.; Tamvakopoulos C.; Peng Q.; Guan X.-M.; Reitman M. L.; Patchett A. A.; Wyvratt M. J.; Nargund R. P. Discovery of substituted biphenyl imidazoles as potent bioavailable bombesin receptor subtype-3 agonists. Bioorg. Med. Chem. Lett. 2010, 20, 1913–1917. [DOI] [PubMed] [Google Scholar]
- Liu J.; He S.; Jian T.; Dobbelaar P. H.; Sebhat I. K.; Lin L. S.; Goodman A.; Guo C.; Guzzo P. R.; Hadden M.; Henderson A. J.; Pattamana K.; Ruenz M.; Sargent B.; Swenson B.; Yet L.; Tamvakopoulos C.; Peng Q.; Pan J.; Kan Y.; Palyha O.; Kelly T. M.; Guan X.-M.; Howard A. D.; Marsh D.; Metzger J.; Reitman M. L.; Wyvratt M. J.; Nargund R. P. Synthesis and SAR of derivatives based on 2-biarylethylimidazole as bombesin receptor subtype-3 (BRS-3) agonists for the treatment of obesity. Bioorg. Med. Chem. Lett. 2010, 20, 2074–2077. [DOI] [PubMed] [Google Scholar]
- Hadden M.; Goodman A.; Guo C.; Guzzo P. R.; Henderson A. J.; Pattamana K.; Ruenz M.; Sargent B.; Swenson B.; Yet L.; Liu J.; He S.; Sebhat I. K.; Lin L. S.; Tamvakopoulos C.; Peng Q.; Kan Y.; Palyha O.; Kelly T. M.; Guan X.-M.; Metzger J.; Reitman M. L.; Nargund R. P. Synthesis and SAR of heterocyclic carboxylic acid isosteres based on 2-biarylethylimidazole as bombesin receptor subtype-3 (BRS-3) agonists for the treatment of obesity. Bioorg. Med. Chem. Lett. 2010, 20, 2912–2915. [DOI] [PubMed] [Google Scholar]
- Sebhat I. K.; Franklin C.; Lo M. M.; Chen D.; Jewell J. P.; Miller R.; Pang J.; Palyha O.; Kan Y.; Kelly T. M.; Guan X.-M.; Marsh D. J.; Kosinski J. A.; Metzger J. M.; Lyons K.; Dragovic J.; Guzzo P. R.; Reitman M. L.; Nargund R. P.; Wyvratt M. J.; Lin L. S. Discovery of (2S)-1,1,1-Trifluoro-2-[4-(1H-pyrazol-1-yl)phenyl]-3-(4-{[1-(trifluoromethyl)cyclopropyl]methyl}-1H-imidazol-2-yl)propan-2-ol (MK-5046), a Potent, Selective Bombesin Receptor Subtype-3 (BRS-3) Agonist for the Treatment of Obesity. ACS Med. Chem. Lett. 2011, 2, 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo M. M.-C.; Chobanian H. R.; Palyha O.; Kan Y.; Kelly T. M.; Guan X.-M.; Reitman M. L.; Dragovic J.; Lyons K. A.; Nargund R. P.; Lin L. S. Pyridinesulfonylureas and Pyridinesulfonylamides as Selective Bombesin Receptor Subtype-3 (BRS-3) Agonists. Bioorg. Med. Chem. Lett. 2011, 21, 2040–2043. [DOI] [PubMed] [Google Scholar]
- Generally speaking, an atropisomer is a stereoisomer where the chiral element is not located on an atom but instead on a molecular plane or axis. The arbitrary definition of atropisomerism is that an atropisomer exists when the half life of interconversion is greater than 1000 s at room temperature. See Oki M. Recent Advances in Atropisomerism. Top. Stereochem. 1983, 14, 1–81. [Google Scholar]
- For a detailed study on the factors (temperature, pH, size of 7-substituents, etc.) influencing the interconversion of this new class of atropisomers, see Welch C. J.; Gong X.; Schafer W.; Chobanian H.; Lin L.; Biba M.; Liu P.; Guo Y; Beard A. Chirality 2009, 21, E105–E109. [DOI] [PubMed] [Google Scholar]
- The assignment of absolute stereochemistry is based on the Cahn–Ingold–Prelog convention. The phenyldiamine plane is viewed as the chiral plane, and the sulfur is designated as the pilot atom; therefore, 8a has an (S)-configuration, and 8b has an (R)-configuration.
- There was a discrepancy between IC50 and EC50 because of the kinetic effects associated with the EC50 assay. For example, when 8a was assayed with a 10 min method, its EC50 value would be 15 nM. The longer the EC50 assay method is , the closer the IC50 and EC50 values are.
- The aliphatic sulfonamides and the corresponding para-tbutylbenzoic acid amide (structures not shown) were not active.
- The human PXR issue will be addressed in a future communication.
- Bag-1 was a BRS-3 agonist from a different lead series; see refs (6) and (8b) (compound 9 in ref (8b)). AM251 was a cannabinoid 1 receptor antagonist [seePhysiol. Behav. 2004, 82 (5), 863–869] that was used here as a positive control. [DOI] [PubMed] [Google Scholar]
- Compound 8a was referred to as Bag-4 in this reference; see Metzger J. M.; Gagen K.; Raustad K.; Yang L.; White A.; Wang S.-P.; Craw S.; Liu P.; Lanza T. J.; Lin L. S.; Nargund R. P.; Guan X.-M.; Strack A. M.; Reitman M. L. Body temperature as a mouse pharmacodynamic response to bombesin receptor subtype-3 (BRS-3) agonists and other potential obesity treatments. Am. J. Physiol. Endocrinol. Metab. 2010, 299, 816–824. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.