

Abstract
A series of cyclitol derivatives with myo-configuration are β-glucocerebrosidase (GCase) inhibitors and show excellent characteristics for the development of pharmacological chaperones for enzyme deficiency in Gaucher disease (GD). The most potent inhibitor, (1S,2R,3R,4S,5R,6S)-5,6-bis(nonylamino)cyclohexane-1,2,3,4-tetraol, displayed a Ki value of 26 nM in isolated enzyme and also inhibited GCase in wild-type (wt) human fibroblasts at nanomolar concentrations. This diaminocyclitol produced maximum increases of GCase activities of 60% in N370S lymphoblasts at 100 nM and 30% in L444P at 1 nM following a 3-day incubation, showing the permeability, subcellular distribution, and cell metabolism characteristics for use as pharmacological chaperone.
Keywords: Gaucher disease, β-glucocerebrosidase, pharmacological chaperone, L444P mutation, N370S mutation, cyclitol
Gaucher disease (GD) is the most prevalent lysosomal storage disorder caused by inherited mutations in the gene encoding acid β-glucosidase (GCase, β-glucocerebrosidase, EC 3.2.1.45),1 the lysosomal enzyme responsible for glucosylceramide metabolism into glucose and ceramide. These mutations lead to significant protein misfolding during translation in the endoplasmic reticulum (ER) and reduced enzyme trafficking to the lysosome.2 GCase deficiency results in the accumulation of undegraded substrate in the lysosomes of macrophages, which often leads to hepatosplenomegaly, anemia, bone lesions, respiratory failure, and, in more severe cases, central nervous system involvement.1 The two most prevalent GCase missense mutant forms reported in GD patients are N370S, which typically results in non-neuronopathic disease, and L444P, which causes a more severe neuronopathic form.
Currently, enzyme replacement therapy and substrate reduction therapy are the only approved treatments for patients with the non-neuronopathic GD.3 Recently, a third promising therapeutic option, the pharmacological chaperone therapy (PCT), has emerged.4,5 PCT is based on the use of reversible competitive GCase inhibitors that are capable of enhancing its residual hydrolytic activity at subinhibitory concentrations by stabilizing the functional form of the misfolded protein and preventing its premature degradation in the ER. This improves enzyme trafficking to the lysosome and enhances its hydrolytic activity. Thus, PCT is highly promising for GD, because it combines the benefits of the small-molecule approach, including oral bioavailability and the potential to cross the blood–brain barrier, with the specificity of an enzyme-directed approach. In addition, a combined therapy based on the coadministration of pharmacological chaperones and recombinant enzyme is undergoing clinical trials.4
In recent years, several distinct structural classes of pharmacological chaperones have been reported, such as N-nonyldeoxynojirimycin (NN-DNJ),6 isofagomine,7 and α-1-C-octyl-1-deoxynojirimycin (CO-DNJ)8 (Figure 1). In this context, we have been actively working on the development of new aminocyclitol derivatives with potential applicability as pharmacological chaperones of this enzyme.9−11 Recently, we have described a family of bicyclic compounds that inhibit GCase in human fibroblasts at nanomolar concentrations.12 In addition, these compounds increased GCase activity in GD lymphoblasts derived from N370S and L444P variants at low concentrations, showing the potency and cellular properties required for GCase pharmacological chaperones. These included some bicyclic guanidines that define a new diaminocyclitol scaffold as possible GCase pharmacological chaperone (Figure 1). This scaffold can also be regarded as an inositol derivative arising from the replacement of two contiguous hydroxyls by amino functionalities, namely 1d-1,2-diamino-1,2-dideoxy-myo-inositol. In order to verify the myo-diaminocyclitol GCase activity hypothesis, we obtained compounds 1–5 and the related ether 6, to be tested on GCase enzymes including N370S and L444P lymphoblasts of GD patients.
Figure 1.

Chemical structures of cyclitol derivatives (1–6) and other reported potential GCase pharmacological chaperones.
Our group reported9,13,14 synthetic methodologies for aminocyclitols, based on the regioselective opening of conduritol B epoxide, which have been applied to the stereocontrolled synthesis of aminocyclitols and 1,2-diaminocyclitols. Diaminocyclitol 1 was obtained by reductive amination of nonanal with the enantiomerically pure 2-azidocyclohexylamine 12, which was obtained from azido alcohol 7(14) by a procedure described previously for racemic 12,13 followed by catalytic hydrogenation (Scheme 1).
Scheme 1. Synthesis of Diaminocyclitol 1.

Reagents and conditions: (a) LiAlH4, THF, rt, 91%. (b) Boc2O, Et3N, CH2Cl2, rt, 82%. (c) MsCl, Et3N, THF, rt, 63%. (d) NaN3, DMF, 90 °C, 65%. (e) CF3CO2H, CH2Cl2, rt, 86%. (f) C8H17CHO, NaBH3CN, AcOH (cat.), MeOH, rt, 75%. (g) Pd/C, MeOH, HCl, H2 (2 atm), rt, 77%.
The synthesis of compounds 2–5 started from (+)-conduritol B 14(14) (Scheme 2), which was benzylated and dihydroxylated to give myo-inositol derivative 16.15 Dimesylation of 16 followed by substitution with NaN3 afforded diazide 17.16 In our hands, the reported17 conversion of 17 to 19 using catalytic hydrogenation in the presence of Boc2O was unsuccessful. However, LiAlH4 reduction of diazide 17 to 18 followed by conventional N-Boc protection gave dicarbamate 19.
Scheme 2. Synthesis of Compounds 2–6 from (+)-Conduritol B 14.

Reagents and conditions: (a) NaH, BnBr, DMF, 30 °C, 75%. (b) OsO4, acetone–H2O, N-methylmorpholine N-oxide, rt, 83%. (c) (1) MsCl, pyridine, rt; (2) NaN3, DMF, 85 °C, 77% for two steps. (d) LiAlH4, THF, rt, 93%. (e) Boc2O, Et3N, CH2Cl2, rt, 53%. (f) NaH, C9H19I, DMF, 80 °C, 21% (20), 24% (21). (g) CF3CO2H, CH2Cl2, rt, 86%. (h) N,N′-carbonyldiimidazole, CH2Cl2, reflux, 83%. (i) NaH, C9H19I, DMF, 0 °C, 78%. (j) Pd/C, MeOH, HCl, H2 (2 atm), rt, 85%. (k) Pd/C, MeOH, H2 (2 atm), rt, 88–93%. (l) BCl3, CH2Cl2, −78 °C, 87%.
Unfortunately, we were unable to effectively dialkylate 19 with nonyl iodide to produce 20. This reaction afforded a 1:1 mixture of the desired Boc-protected diamine 20 and imidazolidinone 21. On one hand, compound 20 was transformed into diaminocyclitol 2 after removing protecting groups by conventional methods. On the other hand, the hydrogenolysis of 21 with Pd/C provided the 1,3-di-N-nonylimidazolidinone 3.
The intermediates 16 and 18 were used to synthesize compounds 4–6 (Scheme 2). The free diamine 4 was obtained after BCl3O-debenzylation of cis-diamine 18. The imidazolidinone 5 was prepared by reaction of cis-diamine 18 with N,N′-carbonyldiimidazole to give the corresponding imidazolidinone 23, which was transformed into the final compound 5 after O-debenzylation. Finally, 1,2-di-O-nonyl-myo-inositol 6 was obtained by alkylation of diol 16 with nonyl iodide, followed by O-benzyl removal.
Compounds 1–6 were evaluated as inhibitors of recombinant GCase (imiglucerase, Cerezyme) at the pH values 5.2 and 7.0. For comparative purposes, the iminosugar NN-DNJ (Figure 1) was also tested at pH 7.0. As shown in Table 1, compounds 1–3 were found to be potent imiglucerase inhibitors. On the other hand, the free diamine 4, imidazolidinone 5, and 1,2-di-O-nonyl-myo-inositol 6 were weak imiglucerase inhibitors.
Table 1. GCase Inhibitory Activity and Maximum Observed Increase in GCase Activity Using Pharmacological Chaperones and NN-DNJ.
| imiglucerase |
||||||
|---|---|---|---|---|---|---|
| IC50 (μM) |
||||||
| compd | pH 7.0 | pH 5.2 | Kia (μM) | % GCase inhibitionb | N370S GCase activity increasec | L444P GCase activity increasec |
| 1 | 0.081 | 0.64 | 0.17 | 50 | 1.6 ± 0.1 (1 μM) | 1.3 ± 0.2 (1 μM) |
| 2 | 0.056 | 0.09 | 0.026 | 96 | 1.6 ± 0.2 (0.1 μM) | 1.3 ± 0.1 (1 nM) |
| 3 | 1.21 | 1.61 | 0.70 | 36 | 2.0 ± 0.1 (10 μM) | no activity |
| 4 | 44.4 | 53.4 | n.d.d | 17 | 1.2 ± 0.1 (10 μM) | n.d. |
| 5 | n.d. | 46%e | n.d. | n.d | n.d. | n.d. |
| 6 | 49.3 | 144.0 | n.d. | n.d. | n.d. | n.d. |
| NN-DNJ | 0.30 | 0.66f | 0.30f | 56g | 1.4 ± 0.1 (5 μM) | no activity |
Competitive inhibitor (pH 5.2).
Incubation for 24 h at 5 μM inhibitor and 5 mM substrate in wild-type (wt) human fibroblast.
N370S and L444P lymphoblasts from Gaucher patients were incubated with test compounds for 3 days before being used for enzyme assay. Data in parentheses correspond to the concentration of the tested compound. Experiments were performed in triplicate, and the mean ± SD is shown. The relative activity was obtained by normalizing the activity corresponding to each compound concentration tested to the activity of untreated cells.
n.d.: not determined.
% inhibition at 1 mM inhibitor.
See ref (22).
Incubation for 24 h at 50 μM inhibitor and 5 mM substrate.
The N-alkylated diamines 1 and 2 were found to be better inhibitors than the free diamine 4, indicating that the presence of an N-alkyl chain is favorable for imiglucerase inhibition, in agreement with the correlation between lipophilicity and inhibitory activity that was observed in other glycomimetic inhibitors of this enzyme.9−12,18−21 Similarly, the 1,3-di-N-nonylimidazolidinone 3 was significantly more active than the imidazolidinone 5, which shows only marginal inhibition of the enzyme. It is remarkable that the inhibitory potency of compound 3 is in the nanomolar range in spite of the differences in structure and protonation state compared with related 1,2-diaminocyclohexanes or guanidines (see Figure 1). Inhibition constants (Ki) for the most active compounds were determined, and in all cases, a competitive inhibition mode was found (see Figures S1–S3 of the Supporting Information). The N,N′-dinonyl diaminocyclitol 2 is the most potent inhibitor found in this work, having a remarkable Ki value of 26 nM, which is 7-fold more potent than N-nonyl diaminocyclitol 1 and 27-fold more potent than 1,3-di-N-nonylimidazolidinone 3. This is the most potent aminocyclitol derivative found in our series and compared favorably with other cyclitol compounds, including the bicyclic derivatives.12
A pharmacological chaperone is expected to bind to the enzyme active site at the neutral pH of the ER to assist folding, but it should dissociate at the lysosome low-pH to facilitate glucosylceramide binding and the subsequent enzymatic reaction. Therefore, compounds that exhibit higher inhibitory activity at lumenal ER pH (7.0) than at lysosomal pH (5.2) would be better pharmacological chaperones. Interestingly, compounds 1–4 and 6 showed lower IC50 values at neutral pH, which denote an advantageous property for pharmacological chaperone candidate molecules. A similar pH dependence of the inhibitory activity has been previously reported for other compounds which showed enzyme enhancement in cells with various GCase mutations.23,24
Candidate compounds for PCT should display high selectivity for GCase enzyme without inhibiting other glycosidases. To address this subject, the cyclitol derivatives were assayed as inhibitors of commercial α- and β-glycosidases, showing negligible effects at 100 μM (see Table S1 of the Supporting Information).25 In addition, none of the compounds displayed significant inhibition on glucosylceramide synthase at 250 μM, thus indicating a good selectivity toward GCase.26
The resistance to thermal denaturation in the presence of a potential pharmacological chaperone is a method used to assess the enzyme stabilization effects due to compound binding.6,9−12,24 In this context, the effect of compounds 1–4 and NN-DNJ on the stabilization of recombinant GCase activity during thermal denaturation at 48 °C was determined. The stabilization ratio27 values found for each compound at different concentrations and incubation times are plotted in Figure 2A (see also Table S2 of the Supporting Information). Diaminocyclitols 1 and 2 exhibited stabilization ratios of 5.5 and 34.7, respectively, at 10 μM after 1 h of incubation time at 48 °C, comparing favorably with NN-DNJ, which showed a 9.2 stabilization ratio at 100 μM. The enzyme stabilization effects of compounds 3 and 4 were much weaker.
Figure 2.
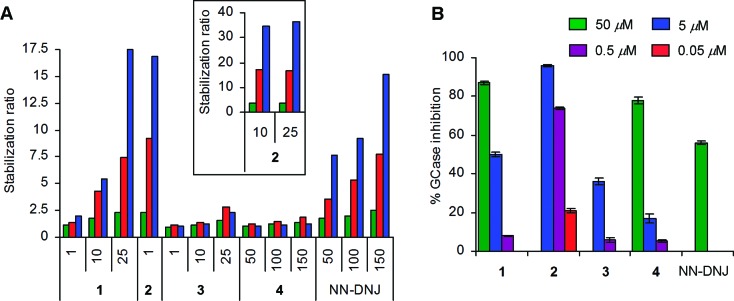
(A) Stabilization ratios of compounds 1–4 and NN-DNJ after thermal denaturation (48 °C) for 20 min (green bar), 40 min (red bar), and 60 min (blue bar) at the indicated inhibitor concentrations (μM). (B) Intact cell GCase inhibition in wt human fibroblasts. Compounds 1–4 and NN-DNJ were incubated for a 24 h period at the indicated inhibitor concentrations. Experiments were performed in triplicate, and bars represent the mean ± SD.
The cellular GCase inhibition by compounds 1–4 and NN-DNJ was initially studied in wt human fibroblasts after 24 h of incubation at nontoxic concentrations: 5 μM for 2–3 and 50 μM for 1 and 4, well below the CC50 determined for these compounds (see Table S3 of the Supporting Information). The high potency of compounds 1–4 required analysis at lower concentrations (see Figure 2B and Table 1). A good correlation between the Ki values against imiglucerase and GCase inhibition in cell culture was observed, except for compound 4, which showed a noticeable inhibition in cellular assays, higher than could be expected from data obtained in isolated enzyme inhibition experiments. The reasons for this behavior are at present unclear and could be related to a favorable permeability and stability or a synergistic effect in cells.
Interestingly, compounds 1–3 also behaved as potent GCase inhibitors in these cells at low-micromolar concentrations. As shown in Figure 2B, diaminocyclitol 2 was the most potent inhibitor with 74% and 21% of GCase inhibition at 500 and 50 nM, respectively. These results show that these compounds are powerful GCase inhibitors in live cells, reflecting good membrane permeability and cellular stability to inhibit the cellular enzyme.
The potential for pharmacological chaperone activity of compounds 1–4 was further evaluated in human lymphoblasts derived from Gaucher patients homozygous for N370S or L444P mutations. The iminosugar NN-DNJ, an activator for N370S GCase but not for the L444P variant, was used as a control.6 The compounds were incubated at different concentrations for 3 days with the Gaucher cells, and the GCase activity was measured to determine the increase or reduction of the enzyme activity in compound treated cells compared with untreated cells. The results obtained are summarized in Table 1 (see also Figure 3).
Figure 3.
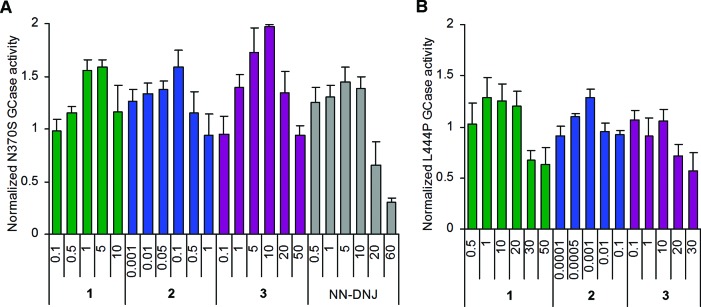
Effects of compounds 1–3 on GCase activity in N370S (A) and L444P (B) lymphoblasts from Gaucher patients. Cells were cultured for 3 days in the absence or presence of increasing concentrations (μM) of compounds before GCase activity was measured. Experiments were performed in triplicate, and each bar represents the mean ± SD. Enzyme activity is normalized to untreated cells, assigned a relative activity of 1.
In the analysis of N370S GCase activation, diaminocyclitol 1 showed a 60% activity increase at 1 μM and 1,3-di-N-nonylimidazolidinone 3 caused a 2.0-fold increase at 10 μM. Similarly, NN-DNJ showed a 40% activity increase at 5 μM. Remarkably, the treatment of N370S lymphoblasts with diaminocyclitol 2 led to a 1.6-fold maximal increase in N370S GCase activity at the very low concentration of 100 nM (see Figure 3A). Compound 4 showed a nonsignificant enzyme activity enhancement on N370S lymphoblasts, and it was not tested in the L444P variant cells.
The effects of compounds 1–3 and NN-DNJ on L444P GCase activity, which characterize a more severe disease phenotype than N370S, were measured using the type 2 GD lymphoblast cell line. After a 3-day incubation, NN-DNJ and imidazolidinone 3 showed no GCase activation at low concentrations and inhibition at high concentrations. In contrast, diaminocyclitols 1 and 2 maximally increased the L444P GCase activity by about 30% at 1 μM and 1 nM, respectively (see Table 1 and Figure 3B).
In summary, different diaminocyclitol derivatives with myo-configuration have been synthesized as potential GCase pharmacological chaperones. These compounds have been tested as GCase inhibitors in imiglucerase and wt human fibroblasts. Among them, diaminocyclitols 1–2 and imidazolidinone 3 were found to be potent inhibitors of recombinant GCase with Ki values in the nanomolar range and also behaved as strong inhibitors of GCase in wt fibroblasts. Furthermore, cyclitol derivatives 1–3 are able to increase the activity of N370S GCase between 1.6- and 2-fold at low micromolar or even nanomolar concentrations, and diaminocyclitols 1–2 maximally increase the GCase activity of the L444P cell line by about 30%. It is worth noting the unusual potency of N,N′-dinonyl diaminocyclitol 2, which increased GCase activity in L444P and N370S lymphoblasts at 1 nM. In spite of the reduced number of compounds tested, the results described establish that the myo-1,2-diaminocyclitol structure characterizes a promising family in the search for optimal compounds for the potential treatment of GD.
Acknowledgments
The authors thank Drs. J. Casas and A. Delgado for helpful discussions, Dr. M. Egido-Gabás, E. Dalmau, and N. Guillem for experimental contributions, and Genzyme Corp. for a generous imiglucerase supply.
Glossary
Abbreviations
- GD
Gaucher disease
- GCase
β-glucocerebrosidase
- ER
endoplasmic reticulum
- PCT
pharmacological chaperone therapy
- NN-DNJ
N-nonyldeoxynojirimycin
- CO-DNJ
α-1-C-octyl-1-deoxynojirimycin
- wt
wild-type
- CC50
compound concentration required to induce 50% cytotoxicity
Supporting Information Available
Experimental details, characterization data for all new compounds, and biological assay protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by MICINN (CTQ2008-01426/BQU), CSIC, and Generalitat de Catalunya (2009-SGR-1072). A.T. thanks MICINN for a predoctoral fellowship.
Author Contributions
Ana Trapero performed chemistry and biology experiments, analyzed data and wrote the paper. Amadeu Llebaria designed research, analyzed data and wrote the paper.
Supplementary Material
References
- Zhao H.; Grabowski G. A. Gaucher disease: Perspectives on a prototype lysosomal disease. Cell. Mol. Life Sci. 2002, 59, 694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman A. H.; van Meer G. The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 2004, 5, 554–565. [DOI] [PubMed] [Google Scholar]
- Platt F. M.; Lachmann R. H. Treating lysosomal storage disorders: Current practice and future prospects. Biochim. Biophys. Acta, Mol. Cell Res. 2009, 1793, 737–745. [DOI] [PubMed] [Google Scholar]
- Parenti G. Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. EMBO Mol. Med. 2009, 1, 268–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Sawkar A. R.; Kelly J. W. Pharmacologic chaperoning as a strategy to treat Gaucher disease. FEBS J. 2007, 274, 4944–4950. [DOI] [PubMed] [Google Scholar]
- Sawkar A. R.; Cheng W.-C.; Beutler E.; Wong C.-H.; Balch W. E.; Kelly J. W. Chemical chaperones increase the cellular activity of N370S β-glucosidase: A therapeutic strategy for Gaucher disease. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15428–15433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R.; Benjamin E. R.; Pellegrino L.; Schilling A.; Rigat B. A.; Soska R.; Nafar H.; Ranes B. E.; Feng J.; Lun Y.; Powe A. C.; Palling D. J.; Wustman B. A.; Schiffmann R.; Mahuran D. J.; Lockhart D. J.; Valenzano K. J. The Pharmacological Chaperone Isofagomine Increases Activity of the Gaucher Disease L444P Mutant Form of β-Glucosidase. FEBS J. 2010, 277, 1618–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L.; Ikeda K.; Kato A.; Adachi I.; Godin G.; Compain P.; Martin O.; Asano N. α-1-C-octyl-1-deoxynojirimycin as a pharmacological chaperone for Gaucher disease. Bioorg. Med. Chem. 2006, 14, 7736–7744. [DOI] [PubMed] [Google Scholar]
- Egido-Gabás M.; Canals D.; Casas J.; Llebaria A.; Delgado A. Aminocyclitols as Pharmacological Chaperones for Glucocerebrosidase, a Defective Enzyme in Gaucher Disease. ChemMedChem 2007, 2, 992–994. [DOI] [PubMed] [Google Scholar]
- Díaz L.; Bujons J.; Casas J.; Llebaria A.; Delgado A. Click Chemistry Approach to New N-Substituted Aminocyclitols as Potential Pharmacological Chaperones for Gaucher Disease. J. Med. Chem. 2010, 53, 5248–5255. [DOI] [PubMed] [Google Scholar]
- Díaz L.; Casas J.; Bujons J.; Llebaria A.; Delgado A. New Glucocerebrosidase Inhibitors by Exploration of Chemical Diversity of N-Substituted Aminocyclitols Using Click Chemistry and in Situ Screening. J. Med. Chem. 2011, 54, 2069–2079. [DOI] [PubMed] [Google Scholar]
- Trapero A.; Alfonso I.; Butters T. D.; Llebaria A. Polyhydroxylated Bicyclic Isoureas and Guanidines Are Potent Glucocerebrosidase Inhibitors and Nanomolar Enzyme Activity Enhancers in Gaucher Cells. J. Am. Chem. Soc. 2011, 133, 5474–5484. [DOI] [PubMed] [Google Scholar]
- Serrano P.; Llebaria A.; Delgado A. Regio- and Stereoselective Synthesis of Aminoinositols and 1,2-Diaminoinositols from Conduritol B Epoxide. J. Org. Chem. 2005, 70, 7829–7840. [DOI] [PubMed] [Google Scholar]
- González-Bulnes P.; Casas J.; Delgado A.; Llebaria A. Practical synthesis of (−)-1-amino-1-deoxy-myo-inositol from achiral precursors. Carbohydr. Res. 2007, 342, 1947–1952. [DOI] [PubMed] [Google Scholar]
- Luchetti G.; Ding K.; d’Alarcao M.; Kornienko A. Enantio- and Diastereodivergent Synthetic Route to Multifarious Cyclitols from d-Xylose via Ring-Closing Metathesis. Synthesis 2008, 19, 3142–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guédat P.; Spiess B.; Schlewer G. Synthesis of (±)-1,2-Dideoxy-1,2-Diamino-myo-Inositol. Tetrahedron Lett. 1994, 35, 7375–7378. [Google Scholar]
- Azev V. N.; d’Alarcao M. Synthesis of 1,2-Diamino-1,2-dideoxy-myo-inositol-Derived Ligands for the Investigation of Metal Complex Reactivity. J. Org. Chem. 2004, 69, 4839–4842. [DOI] [PubMed] [Google Scholar]
- Overkleeft H. S.; Renkema G. H.; Neele J.; Vianello P.; Hung I. O.; Strijland A.; van der Burg A. M.; Koomen G.-J.; Pandit U. K.; Aerts J. M. Generation of Specific Deoxynojirimycin-type Inhibitors of the Non-lysosomal Glucosylceramidase. J. Biol. Chem. 1998, 273, 26522–26527. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Sheth K. A.; Li S.; Chang H.-H.; Fan J.-Q. Rational Design and Synthesis of Highly Potent β-Glucocerebrosidase Inhibitors. Angew. Chem., Int. Ed. 2005, 117, 7616–7619. [DOI] [PubMed] [Google Scholar]
- Aguilar M.; Gloster T. M.; García-Moreno M. I.; Ortiz Mellet C.; Davies G. J.; Llebaria A.; Casas J.; Egido-Gabás M.; García Fernández J. M. Molecular Basis for β-Glucosidase Inhibition by Ring-Modified Calystegine Analogues. ChemBioChem 2008, 9, 2612–2618. [DOI] [PubMed] [Google Scholar]
- Ogawa S.; Kobayashi Y.; Kabayama K.; Jimbo M.; Inokuchi J. Chemical Modification of β-Glucocerebrosidase Inhibitor N-Octyl-β-valienamine: Synthesis and Biological Evaluation of N-Alkanoyl and N-Alkyl Derivatives. Bioorg. Med. Chem. 1998, 6, 1955–1962. [DOI] [PubMed] [Google Scholar]
- Compain P.; Martin O. R.; Boucheron C.; Godin G.; Yu L.; Ikeda K.; Asano N. Design and Synthesis of Highly Potent and Selective Pharmacological Chaperones for the Treatment of Gaucher’s Disease. ChemBioChem 2006, 7, 1356–1359. [DOI] [PubMed] [Google Scholar]
- Bendikov-Bar I.; Ron I.; Filocamo M.; Horowitz M. Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol. Dis. 2011, 46, 4–10. [DOI] [PubMed] [Google Scholar]
- Maegawa G. H.; Tropak M. B.; Buttner J. D.; Rigat B. A.; Fuller M.; Pandit D.; Tang L.; Kornhaber G. J.; Hamuro Y.; Clarke J. T.; Mahuran D. J. Identification and Characterization of Ambroxol as an Enzyme Enhancement Agent for Gaucher Disease. J. Biol. Chem. 2009, 284, 23502–23516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compounds were screened against the following glycosidases: sweet almond β-glucosidase, yeast α-glucosidase, rice α-glucosidase, green coffee beans α-galactosidase, and bovine liver β-galactosidase. Cyclitols 1–3 showed between 44 and 92% inhibition of β-galactosidase at 100 μM. For further details, see the Supporting Information.
- Only diaminocyclitol 2 showed a significant inhibition (73%) on glucosylceramide synthase at 250 μM, but was inactive when tested at 50 μM. For further details, see the Supporting Information.
- This parameter is defined as the ratio of the relative enzymatic activities (inhibitor vs control) at a given inhibitor concentration and incubation time.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.