

Abstract
Cancer drugs, such as the ovarian cancer drug adriamycin, are effective at slowing disease progression and improving remission rates in patients. However, drug resistance often arises, limiting the activity of these agents in some patients. In particular, efflux pumps, which export drugs out of cells, limit the efficacy of a variety of anticancer agents. While inhibitors to block these pumps currently exist, they are usually not used clinically because they alter other drug properties. Here, we report a novel inhibitor of drug efflux that only reduces pump activity temporarily. This decreases the risk that it will alter drug function and cause nonspecific toxicity. P-glycoprotein efflux pumps are commonly overexpressed by malignant cells and are a major contributing factor to the development of drug resistance. Many therapeutics containing basic nitrogens, hydrophobic character, or aromaticity are efficiently eliminated from cells, and Pgp inhibitors must often be coadministered to limit this process. However, currently available inhibitors often alter the pharmacokinetic profiles of therapeutics or increase off-target toxicity, limiting their clinical utility. Here, we report the development of a novel panel of peptide-chlorambucil conjugates capable of efficiently decreasing efflux of Pgp substrates. These conjugates selectively improve adriamycin toxicity and uptake for short, but not prolonged, periods reducing the risk of altered pharmacokinetics and off-target effects.
Keywords: P-glycoprotein, cell-penetrating peptide, mitochondria-penetrating peptide, chlorambucil, adriamycin
P-glycoprotein (Pgp) efflux pumps are ATP-dependent transporters capable of exporting xenobiotics and toxins out of the cell.1,2 These proteins belong to the ATP-binding cassette (ABC) superfamily3 and are expressed on several cell types.4 In addition to their physiological function,5 upregulation of these transporters by malignant cells has been shown to result in drug resistance by preventing the accumulation of therapeutics to toxic levels.6,7 These pumps efflux compounds that enter the cell passively, as most drugs do, and are active against a wide variety of molecules with seemingly few common physicochemical properties.8 In general, hydrophobic or aromatic compounds, often also containing basic nitrogens, are ideal Pgp substrates; therefore, inhibitors with these properties are also highly active against these pumps.8 For example, adriamycin, a clinically used anticancer drug that is a Pgp substrate,9 has a high degree of aromaticity and hydrophobicity, while cyclosporine A, a known Pgp inhibitor,10 is composed of several hydrophobic amino acids.
Current Pgp inhibitors, such as cyclosporine A, have not been successful in a therapeutic setting either because they alter the pharmacokinetic profile of drugs that they are coadministered with or because they increase off-target toxicity.11,12 For example, ivermectin becomes neurotoxic upon coadministration with Pgp inhibitors but is well tolerated when dispensed alone.13 The ability of an inhibitor to temporarily decrease Pgp substrate efflux could reduce undesirable effects.
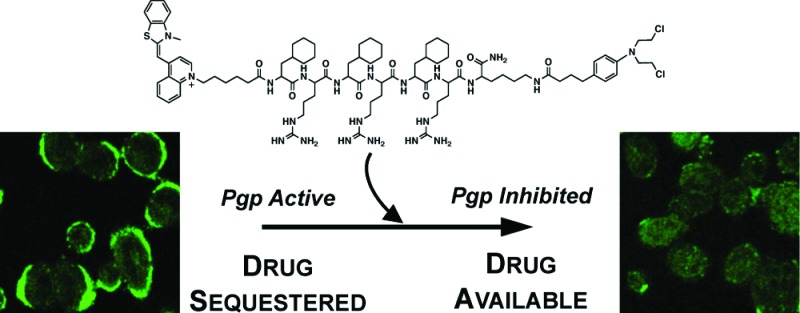
We recently engineered a panel of peptides that possess varying degrees of hydrophobic, aromatic, or cationic character.14,15 These peptides are an alternating array of d-arginine cationic residues (r) and hydrophobic residues, such as cyclohexylalanine (Fx). The peptide shown in Figure 1A was originally designed to deliver small molecules to mitochondria. One of the molecules studied as a mitochondrial cargo was chlorambucil (Cbl), a DNA alkylating agent used clinically as a leukemia drug.16 During the course of studying this peptide-Cbl conjugate that exhibited mitochondrial localization in most cell types (see Figure 1B for localization in a wild-type A2780 ovarian cancer cell line), an interesting observation was made. In the adriamycin-resistant A2780 line (AdrR),17,18 mitochondrial localization was no longer detected, and instead, plasma membrane staining was noted (Figure 1B). In addition, decreased uptake of the mitochondrial stain MitoTracker was noted in this cell line as compared to its wild-type counterpart. Interestingly, the presence of the peptide-Cbl conjugate promoted greater accumulation of MitoTracker (Figure 1B) and restored staining to levels observed in wild-type cells.
Figure 1.

Peptide-Cbl conjugate modulates Pgp activity. (a) Structure of fluorescently labeled peptide-Cbl conjugate used in these studies. Thiazole orange (to) was used for fluorescent labeling of the peptide-Cbl conjugates. The peptide was an alternating array of d-arginine (r) and cyclohexylalanine (Fx). (b) Intracellular localization of 5 μM peptide-Cbl (green), MitoTracker 633 (red), and signal overlay in live wild-type and adriamycin-resistant A2780 cells. (c) Changes in intracellular levels of adriamycin assessed by flow cytometry in cells overexpressing the Pgp efflux pump. All inhibitors were tested at 5 μM. Mean values are plotted, n = 3; error bars are SEM. See Figure S1 in the Supporting Information for basal levels of uptake. (d) Intracellular localization and uptake of adriamycin and MitoTracker 633 as observed by confocal microscopy. All inhibitors were tested at 5 μM. Detector voltages were kept constant for each panel of images to allow for a comparison of signal intensities. Results show that neither peptide nor Cbl alone is sufficient to inhibit Pgp activity, and both components of the peptide-Cbl conjugate are required.
Because the origin of adriamycin resistance in this cell line derives from the overexpression of Pgp efflux pumps, we contemplated whether the enhanced accumulation of MitoTracker reflected a decrease in small molecule efflux. Indeed, when the accumulation of adriamycin, a known Pgp substrate, was monitored by flow cytometry, it was confirmed that in the presence of the peptide-Cbl conjugate, intracellular levels of adriamycin were increased dramatically (Figure 1C). The presence of the alkylating agent Cbl was necessary for activity, as was the conjugation of this chemotherapeutic to the peptide (Figure 1C). Neither of the isolated components of the conjugate could function as an effective Pgp inhibitor. In addition, confocal microscopy showed that upon coincubation with the peptide-Cbl conjugate, both MitoTracker and adriamycin localization shifted from plasma membrane sequestration to intracellular sites (Figure 1D). This increases the concentration of substrate available to interact with its target and achieve bioactivity.
The physiochemical properties for mitochondrial localization14,19,20 appear to be closely related to those for Pgp inhibition.8,21 For both purposes, hydrophobicity, aromaticity, and cationic character are beneficial traits. In addition, basic nitrogens and aromatic rings are thought to act as hydrogen bond acceptors and are essential for Pgp interaction.22 Hydrophobic residues, while not directly involved in Pgp binding, have been shown to be crucial for driving substrates into the lipid membrane. Our peptides are also three repeating units of an amino acid dimer, and recent work has shown that compound duplication increases Pgp inhibitory activity.23,24 Because the peptide-Cbl conjugate possessed all of these features, it was able to efficiently interact with the Pgp pump to slow the efflux of adriamycin. Furthermore, the fact that the conjugate not only interacted with the pump, but also inhibited it, was interesting given that neither the aromatic drug nor the hydrophobic, basic peptide could do so alone.
We sought to examine the specific physiochemical properties of the peptide relevant for Pgp inhibition. To test the role of the peptide, a variety of sequences were conjugated to Cbl and studied. Two cell-penetrating peptide (CPP) sequences, a oligoarginine peptide and one composed of alternating tyrosine and arginine residues, were evaluated as Pgp inhibitors (Figure 2A). These peptides are known to rapidly internalize intracellularly, are capable of transporting a wide array of compounds into cells, and typically localize to the cytoplasm or nucleus.25,26 Because our peptide-Cbl conjugate was originally designed for mitochondrial localization, we reasoned that the oligoarginine or tyrosine-arginine peptides would be better Pgp inhibitors because they would not be sequestered in organelles and be available for Pgp interaction. However, we observed negligible Pgp inhibitory effects with both compounds (Figure 2B), and confocal microscopy showed that these conjugates did not localize to the cell membrane, the site of the Pgp pump (Figure 2D). These results were consistent even when the peptide-Cbl conjugate concentration was increased 4-fold from 5 to 20 μM (Figures S4 and S5 in the Supporting Information). While these peptides possessed cationic and aromatic character, it appears that they did not meet the hydrophobic requirements for Pgp interaction.
Figure 2.

Evaluation of peptide-Cbl physicochemical properties on Pgp activity. (a) Structures of the panel of fluorescently labeled peptide-Cbl conjugates used for optimization of the Pgp inhibitor. d-Enantiomers were used for all arginines (r) and lysines (k) to reduce proteolytic cleavage. Pyridinylalanine and cyclohexylalanine are denoted as FN and FX, respectively. (b) Change in intracellular levels of adriamycin following pretreatment with a 5 μM concentration of various peptide-Cbl conjugates. Analysis was done by flow cytometry in live A2780 cells overexpressing Pgp efflux pumps. Mean values are plotted, n = 3; error bars are SEM. Peptide-Cbl conjugates show Pgp inhibition at comparative levels to cyclosporine A. See Figure S2 in the Supporting Information for basal levels of uptake. (c) Change in intracellular levels of actinomycin D following pretreatment with a 5 μM concentration of various peptide-Cbl conjugates. Mean values are plotted, n = 3; error bars are SEM. See Figure S3 in the Supporting Information for basal levels of uptake. (d) Intracellular localization of fluorescently labeled peptide-Cbl conjugates (5 μM) visualized by confocal microscopy in live cells. Immunofluorescence was used to visualize basal Pgp expression in AdrR cells (left image, red). Intracellular localization of conjugates iii−vi closely matched the staining pattern of the basal Pgp, suggesting that these conjugates were localizing to the efflux pumps. These results illustrate that inhibition efficacy is dependent on plasma membrane localization.
Further investigations of physicochemical properties of the peptide-Cbl Pgp inhibitors were performed by modifying the hydrophobic residues within the sequence. As mentioned above, aromaticity, hydrophobicity, and basic nitrogens have all been identified in the literature as common characteristics of Pgp inhibitors. However, a systematic investigation into which of these properties is most important for activity has not been conducted. Because a wide variety of natural and synthetic amino acids are available commercially, peptide synthesis afforded us the unique opportunity to identify this characteristic, while leaving all other aspects of the conjugate unchanged. Three peptide conjugates were tested: one with a sequence of (FNr)3 that contained basic nitrogens, (Fr)3 with aromatic residues, and (FXr)3 possessing maximal hydrophobicity (Figure 2A). Confocal microscopy showed that all three conjugates localized to the plasma membrane and inhibited Pgp efflux (Figure 2B,D). However, maximal inhibition of adriamycin efflux was observed with the (FXr)3 conjugate, at levels comparable to cyclosporine A, suggesting that hydrophobicity was the most important characteristic for activity. This trend was confirmed with actinomycin, another established Pgp substrate9 (Figure 2C). To determine if these findings were due to a direct interaction between the peptide-Cbl conjugate and the Pgp pump, coimmunoprecipitation was performed. Higher levels of Pgp were detected in cells treated with the conjugate as compared to untreated cells, suggesting a direct interaction (Figure S6 in the Supporting Information). These data clearly illustrate that physicochemical properties of the peptide are essential for Pgp inhibition, and in particular, hydrophobicity and cationicity are key elements for activity.
As a final modification, the properties of the cationic residue were investigated. Arginine was replaced with lysine (Figure 2A), and because both amino acids are in the predominantly protonated state at physiological pH, comparable activity was anticipated. Plasma membrane staining was observed as with (FXr)3 and (FXk)3 showed a slight improvement in activity as compared to the (FXr)3 conjugate (Figure 2B). Therefore, this compound replaced the (FXr)3 conjugate in subsequent experiments.
Following physicochemical optimization of the conjugate, the duration of activity of this panel of inhibitors was assessed. Because many currently available Pgp inhibitors decrease efflux for prolonged periods of time, the coadministered drug is often overly toxic, and off-target effects are observed. Adriamycin efflux and cell toxicity were measured after 1 and 8 h of incubation in the presence and absence of peptide inhibitors and cyclosporine A. At 1 h, increased adriamycin uptake was observed with all of the inhibitors as compared to those cells without inhibitor, and all three peptide conjugates showed comparable activity to cyclosporine A (Figure 3A). In addition, toxicity was elevated from 10% without inhibitor to ∼50% with inhibitors (Figure 3B). This elevated toxicity is due to both the enhanced uptake of drug and also the ability of the drug to reach its biological target rather than succumb to plasma membrane sequestration.
Figure 3.

Time course of adriamycin uptake and toxicity. (a) Uptake of adriamycin modulated by 5 μM peptide-Cbl conjugates or 5 μM cyclosporine A at 1 (left panel) and 8 h (right panel). Mean values are plotted, n = 3; error bars are SEM. Uptake was measured by flow cytometry, and mean values are plotted. (b) Toxicity of adriamycin modulated by 5 μM peptide-Cbl conjugates or 5 μM cyclosporine A at 1 (left panel) and 8 h (right panel). Toxicity was measured by CCK-8 assay, and mean values are plotted. n = 3, error bars are SEM. Results show that peptide-Cbl conjugates increase adriamycin uptake and toxicity for short time periods (1 h) but not long times (8 h). Comparatively, cyclosporine increases uptake and toxicity nonspecifically at both time points.
By 8 h, a clear difference in uptake and toxicity was evident—cells treated with cyclosporine A continued to accumulate adriamycin, while those treated with the peptide inhibitors did not (Figure 3A). In addition, the toxicity of cells treated with cyclosporine A increased from 50% at 1 h to 75% at 8 h, while cells treated with peptide inhibitors exhibited similar toxicity levels at 1 h as at 8 h (Figure 3B). These data show that cyclosporine A nonspecifically increases drug concentrations, while the peptide conjugates developed here specifically inhibit efflux in the short term.
In summary, we report the development of a novel panel of peptide-chlorambucil Pgp inhibitors. These compounds were engineered and optimized for maximal Pgp inhibition and exhibit comparable efficacy to cyclosporine A. In addition, these conjugates selectively inhibit efflux in the short term. This allows the cell to accumulate the drug for controlled periods of time and reduces off-target toxicity. Therefore, these conjugates could be useful at reducing substrate efflux without altering the pharmacokinetic profiles of the coadministered drug.
Acknowledgments
We gratefully thank Dr. Micheline Piquette-Miller for her supply of the A2780 adriamycin-resistant cell line.
Supporting Information Available
Experimental details and supporting data. This material is available free of charge via the Internet at http://pubs.acs.org.
We thank the Canadian Foundation for Innovation (S.O.K.) and NSERC Canada Graduate Scholarship (S.B.F.) for funding of this project.
Supplementary Material
References
- Higgins C. F.; Linton K. J. Nat. Struct. Mol. Biol. 2004, 11, 918–926. [DOI] [PubMed] [Google Scholar]
- Sauna Z. E.; Ambudkar S. V. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 2515–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M.; Hamon Y.; Chimini G. J. Lipid. Res. 2001, 42, 1007–1017. [PubMed] [Google Scholar]
- Thiebaut F.; Tsuruo T.; Hamada H.; Gottesman M. M.; Pastan I.; Willingham M. C. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 7735–7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckford P. D.; Sharom F. J. Chem. Rev. 2009, 109, 2989–3011. [DOI] [PubMed] [Google Scholar]
- Ambudkar S. V.; Dey S.; Hrycyna C. A.; Ramachandra M.; Pastan I.; Gottesman M. M. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [DOI] [PubMed] [Google Scholar]
- Gottesman M. M.; Fojo T.; Bates S. E. Nat. Rev. Cancer 2002, 2, 48–58. [DOI] [PubMed] [Google Scholar]
- Zamora J. M.; Pearce H. L.; Beck W. T. Mol. Pharmacol. 1988, 33, 454–462. [PubMed] [Google Scholar]
- Prados J.; Melguizo C.; Marchal J. A.; Velez C.; Alvarez L.; Aranega A. Int. J. Cancer 1998, 75, 379–383. [DOI] [PubMed] [Google Scholar]
- Qadir M.; O'Loughlin K. L.; Fricke S. M.; Williamson N. A.; Greco W. R.; Minderman H.; Baer M. R. Clin. Cancer Res. 2005, 11, 2320–2326. [DOI] [PubMed] [Google Scholar]
- Szakacs G.; Paterson J. K.; Ludwig J. A.; Booth-Genthe C.; Gottesman M. M. Nat. Rev. Drug Discovery 2006, 5, 219–234. [DOI] [PubMed] [Google Scholar]
- Glavinas H.; Krajcsi P.; Cserepes J.; Sarkadi B. Curr. Drug Delivery 2004, 1, 27–42. [DOI] [PubMed] [Google Scholar]
- Marques-Santos L. F.; Bernardo R. R.; de Paula E. F.; Rumjanek V. M. Pharmacol. Toxicol. 1999, 84, 125–129. [DOI] [PubMed] [Google Scholar]
- Horton K. L.; Stewart K. M.; Fonseca S. B.; Guo Q.; Kelley S. O. Chem. Biol. 2008, 15, 375–382. [DOI] [PubMed] [Google Scholar]
- Yousif L. F.; Stewart K. M.; Horton K. L.; Kelley S. O. Chembiochem 2009, 10, 2081–2088. [DOI] [PubMed] [Google Scholar]
- Fonseca S. B.; Pereira M. P.; Gronda M.; Horton K. L.; Hurren R.; Minden M. D.; Schimmer A. D.; Kelley S. O.. Chem. Biol. 2011, in press. [DOI] [PubMed]
- Li P. Y.; Lin C. Yao Xue Xue Bao 1995, 30, 258–262. [PubMed] [Google Scholar]
- Jekerle V.; Klinkhammer W.; Scollard D. A.; Breitbach K.; Reilly R. M.; Piquette-Miller M.; Wiese M. Int. J. Cancer 2006, 119, 414–422. [DOI] [PubMed] [Google Scholar]
- Asin-Cayuela J.; Manas A. R.; James A. M.; Smith R. A.; Murphy M. P. FEBS Lett. 2004, 571, 9–16. [DOI] [PubMed] [Google Scholar]
- Rosania G. R. Curr. Top. Med. Chem. 2003, 3, 659–685. [DOI] [PubMed] [Google Scholar]
- Zhou S. F.; Wang L. L.; Di Y. M.; Xue C. C.; Duan W.; Li C. G.; Li Y. Curr. Med. Chem. 2008, 15, 1981–2039. [DOI] [PubMed] [Google Scholar]
- Gatlik-Landwojtowicz E.; Aanismaa P.; Seelig A. Biochemistry 2006, 45, 3020–3032. [DOI] [PubMed] [Google Scholar]
- Sauna Z. E.; Andrus M. B.; Turner T. M.; Ambudkar S. V. Biochemistry 2004, 43, 2262–2271. [DOI] [PubMed] [Google Scholar]
- Namanja H. A.; Emmert D.; Pires M. M.; Hrycyna C. A.; Chmielewski J. Biochem. Biophys. Res. Commun. 2009, 388, 672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart K. M.; Horton K. L.; Kelley S. O. Org. Biomol. Chem. 2008, 6, 2242–2255. [DOI] [PubMed] [Google Scholar]
- Ross M. F.; Filipovska A.; Smith R. A.; Gait M. J.; Murphy M. P. Biochem. J. 2004, 383, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.