

Abstract

The iminothiazolidinone BMS-858 (2) was identified as a specific inhibitor of HCV replication in a genotype 1b replicon assay via a high-throughput screening campaign. A more potent analogue, BMS-824 (18), was used in resistance mapping studies, which revealed that inhibitory activity was related to disrupting the function of the HCV nonstructural protein 5A. Despite the development of coherent and interpretable SAR, it was subsequently discovered that in DMSO 18 underwent an oxidation and structural rearrangement to afford the thiohydantoin 47, a compound with reduced HCV inhibitory activity. However, HPLC bioassay fractionation studies performed after incubation of 18 in assay media led to the identification of fractions containing a dimeric species 48 that exhibited potent antiviral activity. Excision of the key elements hypothesized to be responsible for antiviral activity based on SAR observations reduced 48 to a simplified, symmetrical, pharmacophore realized most effectively with the stilbene 55, a compound that demonstrated potent inhibition of HCV in a genotype 1b replicon with an EC50 = 86 pM.
Keywords: HCV NS5A inhibitor, iminothiazolidinone, stilbene
Almost 200 million individuals worldwide are infected with hepatitis C virus (HCV), a disease for which the current, optimal therapy consists of weekly subcutaneous injections of pegylated interferon-α (PEG-IFN) in combination with twice-daily oral doses of ribavirin (RBV).1−3 The inadequacies of this treatment regimen are evident in the low response rates for genotype 1-infected patients and the significant incidence of side effects. The development of direct acting antiviral agents (DAAs) to treat HCV has focused primarily on inhibitors of NS3 protease and the NS5B RNA-dependent RNA polymerase, and several compounds are presently in clinical trials as add-on to PEG-IFN/RBV therapy.4 We have recently reported the discovery of the HCV NS5A inhibitor BMS-790052 (1), a compound that demonstrates a potent antiviral effect in HCV genotype 1-infected subjects following single oral doses.5 In this article we describe the preliminary structure−activity relationships (SAR) developed for the HTS screening hit 2 (BMS-858) and delineate a chemical reactivity inherent to this class of compound that led to the elucidation of a structurally complex dimeric NS5A inhibitor from which a simplified, symmetrical pharmacophore was excised as a prelude to the design of BMS-790052 (1).

Using a dual HCV genotype 1b/bovine viral diarrhea virus replicon screen in high throughput mode, the thiazolidinone 2 was identified as a potent HCV inhibitor, EC50 = 0.58 μM, that demonstrated a good therapeutic window with respect to cytotoxicity (CC50 = >100 μM) and showed no significant inhibitory activity in counterscreens.6,7 This compound had been prepared as part of a prospective library according to the synthetic route depicted in Scheme 1, where regiocontrol during ring closure occurred as anticipated based on literature precedent.8−10 Assignment of the 2-arylimino Z-configuration is also based on prior studies, a function of minimizing unfavorable steric interactions,11,12 while the C-5 position is configurationally unstable and cannot be defined.13 Screening results for additional library members were available (see compounds 3−7) and gave indication that antiviral activity was closely associated with the embedded amino acid moiety (Table 1).7 To expand upon this observation, a survey of amino acid analogues was conducted that confirmed that the l-configuration was essential for antiviral activity since the unnatural, d-configured 8 was over 150-fold weaker, an observation reproduced in the proline derivatives 9 and 10. Lastly, the data obtained for compounds 12−15 communicate a clear preference for smaller Cα substituents.14
Scheme 1. Synthetic Approach to Iminothiazolidinones Using Parallel Solution Phase Chemistry.

Table 1. Structure−Activity Relationships Associated with Thiazolidinones as Inhibitors of HCV 1b Replicon Activity.

 |
EC50 values are averages of at least two independent determinations.
Based on the results obtained for 2 and 9 in Table 1, l-alanine and l-proline were selected as substrates to probe the SAR at the carboxybenyl (Cbz) terminus (R3), the 3-fluorophenyl ring (R2), and furan heterocycle (R1), as collated in Tables 2 and 3. Exchange of the Cbz for either benzoyl (16 and 17) or the isosteric phenylpropionoyl (20 and 21) moieties resulted in reduced potency, whereas a phenacetyl proved to be optimal in both the alanine 18 and proline 19 analogues, enhancing potency by ∼100-fold compared to the cases of 2 and 9, respectively. The high level of potency observed for 18 (BMS-824), EC50 = 0.006 μM, facilitated its use in resistance mapping studies designed to illuminate aspects of the mode of antiviral activity. These studies implicated the NS5A protein as the target of this class of inhibitor based on the identification of a Y93H change that, when introduced into the replicon, significantly compromised the potency of 18, EC50 > 5 μM.5,15
Table 2. Comparison of the Effect of Structural Variation of the Amino Acid N Substituent on Replicon Potency in the l-Alanine and l-Proline Series.
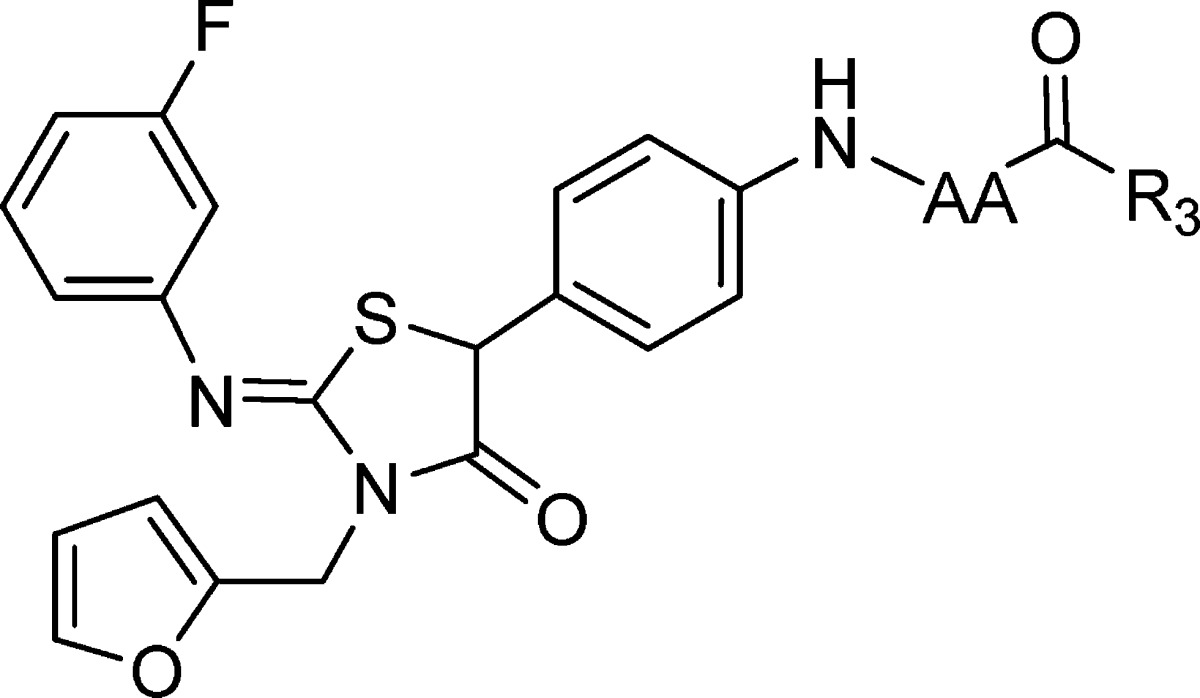
| R3 | cmpd | AA = l-Alanine EC50 (μM)a | cmpd | AA = l-Proline EC50 (μM)a |
|---|---|---|---|---|
| Ph | 16 | 1.9 | 17 | 7.9 |
| CH2Ph | 18 (BMS-824) | 0.006 | 19 | 0.023 |
| CH2CH2Ph | 20 | 3.1 | 21 | 5.1 |
EC50 values are averages of at least two independent determinations.
Table 3. Effect of Variation of the 2-Furyl and 3-Fluorophenyl Moieties in Both the l-Alanine and l-Proline Series on Antiviral Activity in a HCV Genotype 1b Replicon.
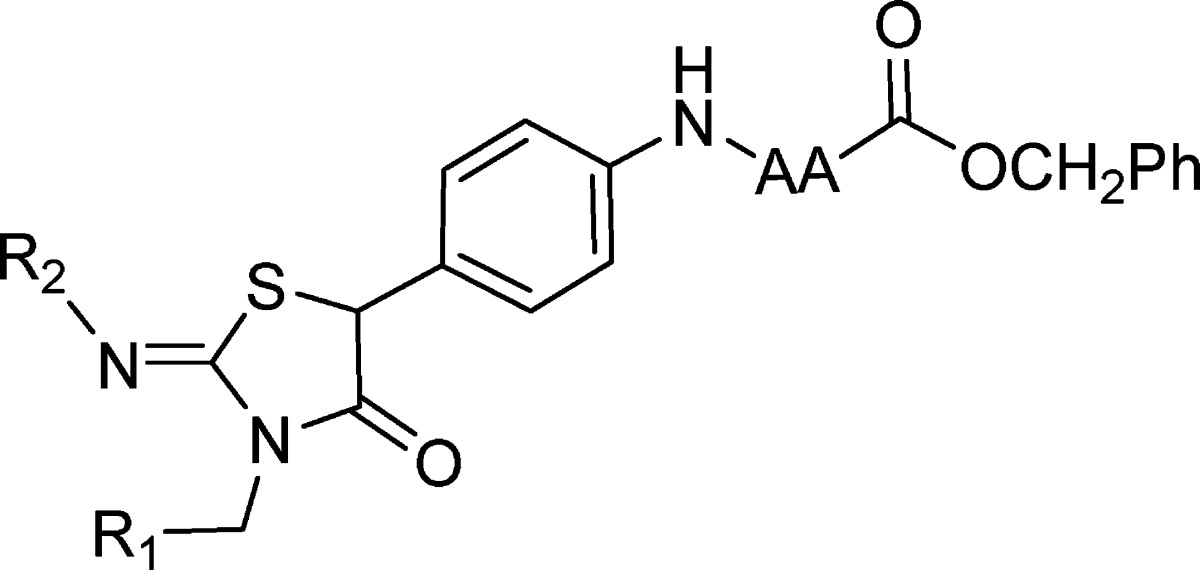
| R1 | R2 | cmpd | AA = l-alanine EC50 (μM)a | cmpd | AA = l-proline EC50 (μM)a |
|---|---|---|---|---|---|
| 2-tetrahydrofuranyl | 3-F-C6H4 | 22 | 0.65 | ||
| 2-pyrazinyl | 3-F-C6H4 | 23 | 0.025 | 24 | 0.038 |
| 2-pyridinyl | 3-F-C6H4 | 25 | 0.035 | 26 | 0.002 |
| 3-pyridinyl | 3-F-C6H4 | 27 | 0.026 | ||
| 2,3,4-F3-C6H2 | 3-F-C6H4 | 28 | 0.55 | 29 | 0.6 |
| 2-furyl | 4-CF3-C6H4 | 30 | 2.2 | 31 | 0.98 |
| 2-furyl | 3-F-4-CF3-C6H3 | 32 | 2.5 | 33 | 2.2 |
| 2-furyl | 4−Cl-C6H4 | 34 | 0.51 | 35 | 1.17 |
| 2-furyl | 4-Et-C6H4 | 36 | 1.5 | 37 | 1.8 |
| 2-furyl | 4-CN-C6H4 | 38 | 0.037 | 39 | 0.086 |
| 2-furyl |  |
40 | 0.01 | 41 | 0.009 |
| 2-furyl | 5-quinolinyl | 42 | 0.02 | 43 | 0.01 |
| 2-furyl | 4-morpholinylphenyl | 44 | 0.015 | 45 | 0.02 |
EC50 values are averages of at least two independent determinations.
The parallel SAR observed between the two amino acid series extended to variation of substituents attached to the iminothiazolidinone ring, as shown in Table 3. For example, 2-pyridyl and 3-pyridyl heterocycles in place of the furan enhanced potency by >15-fold compared to the case of 2 in both the alanine (23 and 25) and proline (24 and 26) series, whereas the more lipophilic 2,3,4-trifluorophenyl ring merely preserved potency (28 and 29). Somewhat analogously, lipophilic substituents appended to the 3-phenyl ring (compounds 30−37) generally resulted in reduced antiviral activity compared to the case of 2, while more polar substituents were beneficial in improving potency, as illustrated by compounds 38−45.
Although the data reported in Tables 1−3 provide a coherent and interpretable SAR, we subsequently discovered C-5 phenyl-iminothiazolidinones 2 and 18 were consumed during the course of the 3 day replicon assay. The first indication of an inherent chemical reactivity emerged when it was observed that, on standing in DMSO, compound 2 underwent an oxidative rearrangement to generate the thiohydantoin 46 (Scheme 2).16,17 The structure of 46 was elucidated by correlation spectroscopy, which established the proximity of the furanyl-methylene protons (δ5.2, dd, 2H) to the amide carbonyl carbon (δ 171.4 ppm) and the thiourea carbon (δ 182.5 ppm) atoms. In addition, nuclear Overhauser effects (nOe) were observed between the ortho protons on the two phenyl rings at δ 9.09 (d, 1H) and δ 7.30 (d, 2H), as depicted in structure 46.16
Scheme 2. Oxidative Rearrangement of the Iminothiazolidinones 2 and 18.

Thiohydantoin 46, however, exhibited poor activity in the replicon assay, with an EC50 = 13 μM, suggesting that other species in the cell culture media may be responsible for the observed inhibitory activity of 2, and this prompted a more detailed analysis of compound 18.17 While stable indefinitely in solid form and quite stable in solvents such as MeOH and CH3CN, the iminothiazolidinone 18 underwent the same oxidative transformation in DMSO as 2, to form 47 (EC50 > 20 μM).16 Moreover, preincubation of 18 in assay media until completely consumed, as determined by HPLC, followed by analysis of the replicon inhibitory activity of the solution, produced an equivalent antiviral effect to that recorded for parent compound 18 under standard assay conditions. HPLC analysis of the preincubated media detected the thiohydantoin 47 as the predominant product; however, it was observed to undergo degradation over time to the thiourea starting material depicted in Scheme 1, presumably via hydrolysis of intermediate C (Scheme 2). As a result of these findings, HPLC biogram fractionation experiments were performed, an exercise that traced potent HCV inhibitory activity to two minor components in the media identified as isomers of the dimeric species 48 (Figure 1).17 The connectivity of the dimer was established by 1H and 13C NMR to be at the C-5 methine carbon, with the less mobile isomer on reversed phase LC analysis being the more potent compound, EC50 = 0.6 nM, and which converted to the more mobile but less potent (EC50 = 43 nM) isomer upon heating at 55 °C.17
Figure 1.

Dimeric species identified in replicon assay media and pharmacophore hypothesis.
In considering how compounds 46−48 might be generated, it was recognized that hydrogen abstraction at the C-5 methine carbon of 18 would form a classic captodative radical stabilized by the electron donating sulfur atom and the electron withdrawing carbonyl group.18,19 A radical mechanism is consistent with the formation of 47 by a process involving combination of a C-5 radical with oxygen that would give hydroperoxide A (Scheme 2). Precedent for peroxide formation and reductive cleavage in/by DMSO, which in this case affords B, is available in the literature for other systems.20,21 Unique to B, however, is the potential for ring-opening to the α-ketoamide C, which can reclose in a complementary fashion to form the 2-thiohydantoin 47, with experimental support for this sequence of events obtained as follows. Snider has reported that radicals generated in the presence of Mn(OAc)3/Cu(OAc)2 can undergo further oxidation in which the resulting cation is trapped by acetic acid.22,23 The phenyl acetamide 49 was exposed to these conditions (Scheme 3) and gave 50, an isolable derivative of B, in 59% isolated yield together with some of the rearranged thiohydantoin 51 in 16% yield. The structure of compound 50 was established by N−H long-range NMR correlation in order to rule out the acetoxy derivative of 51, which may result simply from exchange of the hydroxyl group with acetic acid.24
Scheme 3. Radical Generation, Oxidation, and Trapping with Acetic Acid.

Exposure of 2 and 18 to the same reaction conditions gave the acetoxy derivatives 52 (48%) and 53 (78%), respectively, with no evidence of thiohydantoin formation. In the latter case, a small amount of dimeric product 48 was obtained (12 mg, 2.3%), which was shown by 1H NMR and LC to be identical with the more mobile dimeric isomer isolated from media.25,26 In a further experiment, acetates 52 and 53 were subjected to basic methanol (K2CO3/MeOH), which effected rapid conversion to thiohydantoins 46 and 47.27
Taken together, these results are consistent with a mechanism involving hydrogen atom abstraction from C-5 of the thiazolidinone ring, presumably mediated by oxygen, to afford a stabilized radical that can either react with oxygen to produce hydroperoxide A or combine to produce dimer 48. The absolute requirement for an l-configured amino acid coupled with the high sensitivity of potency to changes in the amine capping element suggested that this region of the molecule made an important contribution to the antiviral activity observed with dimer 48. This appreciation led to the hypothesis that excision of the iminothiazolidinone heterocycles would simplify the NS5A-inhibiting pharmacophore in 48 to that of the palindromic bibenzyl derivative 54, as depicted in Figure 1. Synthetic accessibility facilitated the testing of this hypothesis, and 54, a compound quite stable to replicon assay conditions, demonstrated potent inhibition, EC50 = 30 nM.28 Furthermore, the more rigidly disposed stilbene 55 proved to be an even more potent inhibitor of the 1b HCV replicon with an EC50 = 0.086 ± 10 nM (n = 4).28,29 As is evident from Table 4, SAR for this new class of stilbene inhibitors closely resembled that of the iminothiazolidinones. Replacing the phenylacetic acid amine cap with Cbz (compound 57) effected a 100-fold loss in potency, similar in magnitude to that observed between 18 and 2, while the l-configuration was again essential for potency based on the comparison of 56 and 58 with the natural proline derivatives 55 and 57. Additional profiling of stilbene 55 in the Y93H replicon revealed reduced potency, EC50 = 4 μM, confirming its association of inhibition with the NS5A protein. In a genotype 1a replicon, stilbene 55 was found to be inactive, EC50 > 10 μM, providing a clear objective for the next phase of optimization.
Table 4. SAR Associated with Stilbene-Based HCV NS5A Inhibitors.
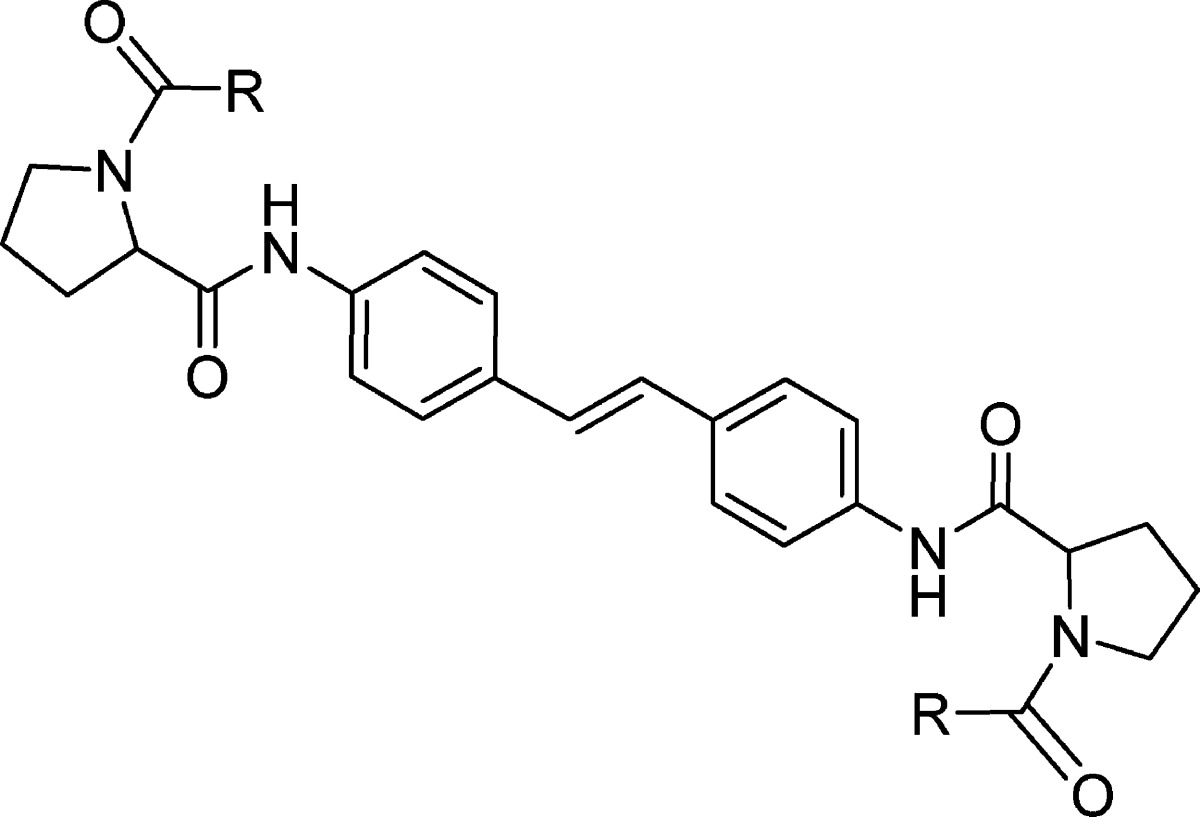
 |
EC50 values are averages of at least two independent determinations.
BMS-790052 (1) is a first-in-class inhibitor for HCV NS5A with high selectivity, picomolar potency, and broad genotype coverage in vitro that likely acts by disrupting a replication−protein assembly process critical to viral replication.5 The synthesis of stilbene 55 was a critical step in the design of BMS-790052 (1), a compound that has established the clinical relevance of interfering with the function of NS5A as an approach to the treatment of HCV infection.5 Interestingly, the discovery of symmetry within HCV NS5A inhibitors coincides well with X-ray crystallographic data subsequently published for the amino terminus of the NS5A protein.30,31 In one report, NS5A residues 36−198 crystallized as a dimer forming a U-shaped structure with the inner surface lined with a preponderance of basic amino acids proposed to form a binding site for viral RNA.30 The residues conferring resistance to inhibitor 55 are proximal to the amino terminus of NS5A and are located between the protein and the membrane, suggesting that the symmetrical compounds bind across the dimer interface.5 Although symmetry is not essential for inhibition of HCV replication in genotype 1b replicon, as reported elsewhere, genotype 1a inhibition is most optimally achieved in symmetrically disposed compounds.32
In summary, we describe here fundamental aspects associated with the HCV inhibitory activity of a series of C-5-phenylated iminothiazolidinones discovered in a high throughput replicon screen, compounds that target the NS5A protein. The SAR observed for this chemotype was highly sensitive to the absolute configuration of the amino acid and to structural changes in the capping element at the amino terminus, and it was consistent between the alanine and proline series. Chemical reactivity at the C-5 position of iminothiazolidinones in DMSO was based on a propensity to form radicals at this site, resulting in oxidation via reaction with oxygen or the formation of dimeric species upon radical combination. The precise SAR associated with the amino acid moiety suggested that this element was a critical determinant of biological activity and lead to a hypothesis that the pharmacophore inherent in 48 could be simplified to that of a symmetrical bibenzyl derivative, an idea initially vindicated upon synthesis and evaluation of 54. The potent inhibition of HCV replication in the replicon assay by symmetrically disposed stilbene 55 ultimately lead to the design of BMS-790052 (1), the compound that established proof-of-concept for HCV NS5A inhibition as a treatment of HCV infection.
Acknowledgments
We wish to express our gratitude to the Bristol-Myers Squibb Research and Development, Department of Analytical Services, in general, and the mass spectroscopy group, in particular.
Author Present Address
⊥ Novartis Institute for BioMedical Research, Cambridge, MA 02139.
Author Present Address
# Achillion Pharmaceuticals, New Haven, CT 06511.
Supporting Information Available
Experimental details for synthetic procedures and associated chemical data for compounds 2−58 and the experimental protocol used to evaluate inhibitors in the replicon assay. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Kim W. R. Global Epidemiology and Burden of Hepatitis C. Microbes Infection 2002, 4, 1219–1225. [DOI] [PubMed] [Google Scholar]
- Hoofnagle J. H.; Seeff L. B. Peginterferon and Ribavirin for Chronic Hepatitis C. N. Engl. J. Med. 2006, 355, 2444–2451. [DOI] [PubMed] [Google Scholar]
- Pearlman B. L. Extended-Therapy Duration for Chronic Hepatitis C, Genotype 1: The Long and the Short of It. World J. Gastroenterol. 2008, 14, 3621–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Francesco R.; Migliaccio G. Challenges and Successes in Developing New Therapies for Hepatitis C. Nature 2005, 436, 953–960. [DOI] [PubMed] [Google Scholar]
- Gao M.; Nettles R. E.; Belema M.; Snyder L. B.; Nguyen V. N.; Fridell R. A.; Serrano-Wu M. H.; Langley D. R.; Sun J.-H.; O’Boyle D. R.; Lemm J. A.; Wang C.; Knipe J. O.; Chien C.; Colonno R. J.; Grasela D. M.; Meanwell N. A.; Hamann L. G. Chemical Genetics Strategy Identifies an HCV NS5A Inhibitor with a Potent Clinical Effect. Nature 2010, 465, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle D. R.; Nower P. T.; Lemm J. A.; Valera L.; Sun J.-H.; Rigat K.; Colonno R.; Gao M. Development of a Cell-Based High-Throughput Specificity Screen Using a Hepatitis C Virus-Bovine Viral Diarrhea Virus Dual Replicon Assay. Antimicrob. Agents Chemother. 2005, 49, 1346–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romine J. L.; Martin S. W.; Snyder L. B.; Serrano-Wu M.; Desphande M. S.; Whitehouse D. PCT WO 2004/014852 A2; 2007, US Patent 7,183,302 B2.
- Erol S.; Dogan I. Axially Chiral 2-Arylimino-3-aryl-thiazolidine-4-one Derivatives: Enantiomeric Separation and Determination of Racemization Barriers by Chiral HPLC. J. Org. Chem. 2007, 72, 2494–2500. [DOI] [PubMed] [Google Scholar]
- Garnaik M. B. K.; Behera R.. Influence of Substituents on the Synthesis of Thiazolidinones. Indian J. Chem. 1987, 26B, 779−781. [Google Scholar]
- St. Laurent D. R.; Gao Q.; Wu D.; Serrano-Wu M. H. Regioselective Synthesis of 3-(Heteroaryl)-iminothiazolindin-4-ones. Tetrahedron Lett. 2004, 45, 1907–1910. [Google Scholar]
- Bolli M. H.; Abele S.; Binkert C.; Bravo R.; Buchmann S.; Bur D.; Gatfield J.; Hess P.; Kohl C.; Mangold C.; Mathys B.; Menyhart K.; M€uller C.; Nayler O.; Scherz M.; Schmidt G.; Sippel V.; Steiner B.; Strasser D.; Treiber A.; Weller T. 2-Imino-thiazolidin-4-one Derivatives as Potent, Orally Active S1P1 Receptor Agonists. J. Med. Chem. 2010, 53, 4198–4211. [DOI] [PubMed] [Google Scholar]
- Kumar G.; Parasuraman P.; Sharma S. K.; Banerjee T.; Karmodiya K.; Surolia N.; Surolia A. Discovery of a Rhodanine Class of Compounds as Inhibitors of Plasmodium falciparum Enoyl-Acyl Carrier Protein Reductase. J. Med. Chem. 2007, 50, 2665–2675. [DOI] [PubMed] [Google Scholar]
- The diastereomers at the C-5 position of 2 were separated by chiral prep HPLC but observed to rapidly equilibrate due to facile keto−enol tautomerism (see Supporting Information for LC trace).
- Compounds 2−15 were prepared in library format using parallel synthesis methodology and subject to minimal purification. All compounds were of at least 70% purity, and MWs were confirmed by LCMS.
- Lemm J. A.; O’Boyle D. R.; Liu M.; Nower P. T.; Colonno R.; Deshpande M. S.; Snyder L. B.; Martin S. W.; St. Laurent D. R.; Serrano-Wu M. H.; Romine J. L.; Meanwell N. A.; Gao M. Identification of Hepatitis C Virus NS5A Inhibitors. J. Virol. 2010, 84, 482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Supporting Information for HPLC stability studies and experimental details. In DMSO, compounds 2 and 18 underwent complete conversion to 46 and 47, respectively. All compounds were dissolved and diluted in DMSO prior to evaluation in replicon and preincubation in media.
- Lemm J. A.; Leet J. E.; O’Boyle D. R. II; Romine J. L.; Huang X.; Schroeder D. R.; Alberts J.; Cantone J. L.; Sun J.-H.; Nower P. T.; Martin S. W.; Serrano-Wu M. H.; Meanwell N. A.; Snyder L. B.; Gao M. Discovery, Isolation and Synthesis of Potent Hepatitis C Virus NS5A Inhibitors with Dimeric Structures. Manuscript submitted.
- Giese B.Radicals in Organic Synthesis: Formation of Carbon-Carbon Bonds; Pergamon Press: New York, 1986. [Google Scholar]
- Viehe H. G.; Janousek Z.; Merenyi R.; Stella L. The Captodative Effect. Acc. Chem. Res. 1985, 18, 148–154. [Google Scholar]
- Schmidt V. A.; Alexanian E. J. Metal-Free, Aerobic Dioxygenation of Alkenes Using Hydroxamic Acids. Angew. Chem., Int. Ed. 2010, 49, 4491–4494. [DOI] [PubMed] [Google Scholar]
- Rowlands G. J. Radicals in Organic Synthesis. Part 1. Terahedron 2009, 65, 8603–8655. [Google Scholar]
- Kates S. A.; Dombroski M. A.; Snider B. B. Manganese(III)-Based Oxidative Free-Radical Cyclization of Unsaturated β-Keto Esters, 1,3-Diketones, and Malonate Diesters. J. Org. Chem. 1990, 55, 2427–2436. [Google Scholar]
- Snider B. B. Manganese(III)-Based Oxidative Free-Radical Cyclizations. Chem. Rev. 1996, 96, 339–363. [DOI] [PubMed] [Google Scholar]
- Chemical shifts were consistent with the presence of an imine bond (not present in 51), while H−N long-range correlation indicated an amide nitrogen (see Supporting Information).
- Dimers derived from 2 and 49 were detectable by LCMS in low amounts, <1%. Dimer isolated from media was obtained from 1 L scale incubation of 18 at a concentration of 100 μM and yielded 1.1 mg each of the two isomers (see ref (17) for a more detailed account). The synthetic material was identical with the more mobile isomer and demonstrated an EC50 = 2 nM. Dimer and acetates 52 and 53 were formed as single compounds with no evidence of diastereomers being formed (see Supporting Information).
- For the radical dimerization process, see:Obata N.; Niimura K. Oxidative Dehydrodimerization of N-Acyl α-Amino-acids: Synthesis of Novel Di-αα′-amino-acid Derivatives. Chem. Commun. 1977, 238–239. [Google Scholar]
- Thiohydantoins hydrolyze under these conditions to the thioureas if not isolated promptly.
- Serrano-Wu M.; Belema M.; Snyder L. B.; Meanwell N. A.; St. Laurent D. R.; Kakarl R.; Nguyen V. N.; Qui Y.; Yang X.; Leet J. E.; Gao M.; O’Boyle D. R. II; Lemm J. A.; Yang F. US 2006/0276511 A1.
- The alanine analogue of stilbene 55 proved to be too insoluble in DMSO to evaluate in the replicon assay.
- Tellinghuisen T. L.; Marcotrigiano J.; Rice C. M. Structure of the Zinc-Binding Domain of an Essential Component of the Hepatitis C Virus Replicase. Nature 2005, 435, 374–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love R. A.; Brodsky O.; Hickey M. J.; Wells P. A.; Cronin C. N. Crystal Structure of a Novel Dimeric Form of NS5A Domain I Protein from Hepatitis C Virus. J. Virol. 2009, 83, 4395–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz U.; Tan S.-L. NS5A—From Obscurity to New Target for HCV Therapy. Recent Pat. Anti-Infect. Drug Discov. 2008, 3, 77–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.