

Abstract

A focused chemical optimization effort of compound 1 based on metabolite elucidation is described, resulting in 15i, a highly potent and selective mGlu5 receptor antagonist with an improved pharmacokinetic profile compared to 1. Characterization of 15iin vivo in the fear-potentiated startle (FPS) paradigm revealed a robust reduction of conditioned fear behavior. This effect nicely correlates with the rat brain pharmacokinetics.
Keywords: mGlu5, benzimidazoles, pharmacokinetics
L-Glutamate serves as the neurotransmitter at the majority of excitatory synapses in the mammalian central nervous system (CNS) and as such is involved in a variety of physiological and pathophysiological functions. The existence of neuromodulatory glutamate receptors, called metabotropic glutamate receptors (mGluRs), provides a mechanism by which glutamate can modulate cell excitability and synaptic transmission. Metabotropic glutamate receptors are G-protein-coupled receptors linked to multiple second messenger systems modulating for example ion channel functions in the neurons.1−3 Eight different types of metabotropic glutamate receptors are known (mGluR1−mGluR8), which are divided into three groups according to their sequence homology, effector coupling and pharmacology. Group I mGlu receptors (mGluR1 and mGluR5) are positively coupled to phospholipase C; group II mGlu receptors (mGluR2 and mGluR3) and group III mGlu receptors (mGluR4, mGluR6, mGluR7 and mGluR8) are negatively coupled to adenylate cyclase. mGluRs are broadly distributed throughout the CNS and are specifically localized at discrete synaptic and extrasynaptic sites.
mGlu5 receptor expression is especially high in brain regions thought to mediate and modulate emotions like fear and anxiety (amygdala, prefrontal cortex, hippocampus and basal ganglia).4−6 mGlu5 receptors are predominantly postsynaptically located and have been shown to play a role in regulating glutamatergic transmission via potentiation of NMDA receptor activity. Excessive glutamatergic transmission has been proposed to play a role in psychiatric diseases like anxiety disorders and depression.7,8 Additionally, mGlu5 receptor antagonism has been proposed as a potential target for the treatment of pain,9 obesity,10 Parkinson’s disease,11 drug abuse,12 migraine,13 gastroesophageal reflux disease (GERD)14 and fragile X syndrome (FXS).15 Therefore, the development of potent and selective mGlu5 receptor antagonists as potential therapeutic agents has been the focus of significant research in our laboratories.
We recently disclosed the piperidyl amide-based selective mGluR5 antagonist 1 (Figure 1) that shows robust anxiolytic-like activity in different animal models of fear and anxiety.16
Figure 1.

mGluR5 antagonist. ahmGluR5: Ca2+ flux using glutamate and phosphoinositol turnover using quisqualate as agonist;17 data are geometric means of n ≥ 2, IC50 (nM). bSolubility (mg/L) measured in a dissolution template potentiometric titration approach. cHLM/RLM: human and rat liver microsomal intrinsic clearance (μL/min/mg microsomal protein). dn = 3 Sprague−Dawley rats/group. eMRT = mean residence time (h).
The moderate metabolic stability of compound 1, based on in vivo plasma clearance in rat and in vitro microsomal clearance data in rat and human required further optimization.
In the present letter, the chemical derivation effort of 1 to an improved compound with a superior pharmacokinetic profile and increased potency in vivo will be discussed.
As a result of the moderate metabolic stability of 1, the biotransformation of this compound was studied in human and rat liver microsomes as well as in vivo in rat after intravenous or oral administration (Scheme 1).
Scheme 1. Proposed Chemical Structures of Metabolites of 1 Detected after Incubation with Rat and Human Liver Microsomes as Well as in Vivo in Rat Plasma.
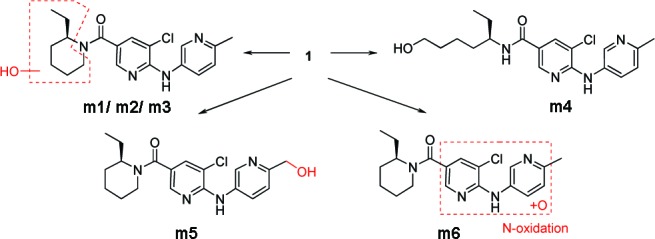
In microsomal incubations, six phase I metabolites were found and characterized by means of mass spectrometry. Metabolites m1, m2 and m3 were oxidation products of the 2-ethylpiperidin-1-yl moiety. Further oxidation and ring-opening led to metabolite m4, whereas oxidation of the methyl group in the 6-methylpyridin-3-ylamino moiety led to metabolite m5. H/D exchange indicated N-oxidation at the 5-chloro-6-(6-methylpyridin-3-ylamino)pyridin-3-yl moiety in the case of metabolite m6. In vivo, the metabolites m1−m3 were also detected in plasma besides the parent compound.
The formation of phase II metabolites was not observed in any of the in vitro or in vivo samples. The abundances of 1 and its metabolites in plasma suggested that the elimination of 1 may predominantly occur via oxidation of the 2-ethylpiperidin-1-yl moiety (m1−m3), with major metabolite being m1in vivo and m5in vitro.18
With the piperidyl moiety identified as the major site of metabolism in compound 1, the initial derivation efforts aimed at exploring bioisosteric replacements for the left-hand fragment in 1. Compound 2 was used as a reference for the optimization of this series, which contains the central and right-hand-side fragments that led to most potent derivatives described in our preceding letter, combined with the piperidyl amide left-hand-side fragment of 1.16 Several 5-ring heterocyclic bioisosteres such as oxadiazole, triazole, and isoxazole were investigated and shown to be inactive. Only the imidazole system proved to be a suitable bioisosteric replacemement for the piperidyl amide, albeit with reduced potency.
The imidazole derivatives were prepared by the methods depicted in Scheme 2, whereas the benzimidazole analogues were synthesized according to Scheme 3. The synthesis started by reacting pyridine 3 with 4-chloroaniline to afford 4, followed by boronic acid formation to give 5. Suzuki coupling with 6 provided 7a.(19) The synthesis of 7b−e commenced with a Buchwald reaction between pyridine 8 and 4-chloroaniline, leading to 9, followed by amination of the nitrile group to give the amidine 10. Condensation of 10 with α-chloroketones and subsequent N-alkylation of the imidazole ring afforded 7b−e.
Scheme 2.

Scheme 3.

The preparation of the benzimidazole derivatives started with the 2-chloronicotinic acid 11 or ester 12, which were reacted with an aromatic amine to provided 13a and 14. In the case of 14, the ester group was first hydrolyzed to 13b, and the carboxylic acid intermediates were then engaged in condensation reactions with phenylenediamine reagents to give rise to the benzimidazole compounds 15a−e, 16 and 17. For compounds 16 and 17 (W = Cl), final N-alkylation, followed by separation of the regioisomers, led to 15f−i.
The bicyclic derivative 7a, which can be seen as a cyclized version of compound 2, served as a lead for our optimization. The SAR evaluation around the imidazole moiety started with structurally simpler derivative, where the fused 6-membered ring of 7a was opened. The 1,5-dimethylimidazole derivative 7b displayed only marginal activity, whereas the 1,4-dimethyl analogue 7c exhibited micromolar potency. Exploration of the N-alkyl substituent permitted identification of the potent n-propyl derivative 7d. Further exploration of the 4- and 5-positions led to compound 7e, and subsequent bridging of those two positions into a benzimidazole moiety resulted in 15a, the most active derivatives of this subset (Table 1).
Table 1. Bioisosteric Replacement of the Piperidyl Amide Fragment: In Vitro hmGluR5 Antagonism.
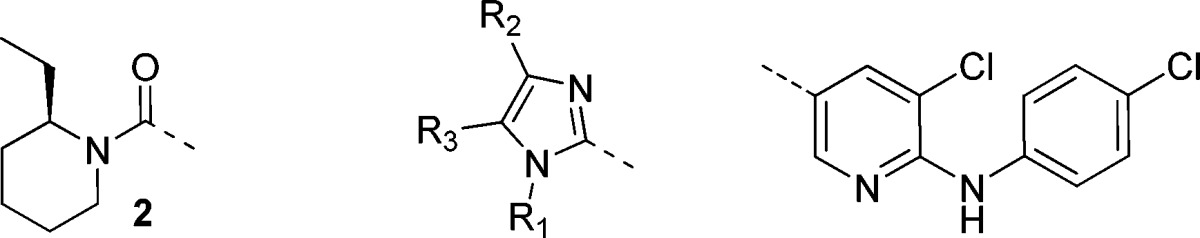
| compd | R1 | R3 | R2 | IC50 [μM]a |
|---|---|---|---|---|
| 2 | 0.064 | |||
| 7a | −(CH2)4− | H | 1.2 | |
| 7b | Me | Me | H | 48% @ 10 |
| 7c | Me | H | Me | 2.9 |
| 7d | Pr | H | Me | 0.23 |
| 7e | Pr | Me | Me | 0.25 |
| 15a | Pr | −phenyl−b | 0.18 | |
hmGluR5 Ca2+ flux7 using glutamate as agonist; data are geometric means of n ≥ 2.
Benzimidazole.
Having identified the benzimidazole moiety as a suitable bioisostere for the former piperidyl amide, the 4-chloroaniline right-hand fragment was exchanged for the less lipophilic and more soluble 3-amino-6-methylpyridyl group as in 1, leading to compound 15b.
Further exploration of the SAR around the benzimidazole moiety revealed that structural variation of the N-substituent was tolerated with minimal impact on the potency, but significant difference in terms of human microsomal stability (15c−e). Subsequently, a “chloro screen” on the benzimidazole moiety (15f−i) permitted identification of the 7-chlorobenzimidazole 15i as the most potent analogue of this series (Table 2). Due to its improved potency, 15i was selected for further profiling in vitro and in vivo despite a similarly high intrinsic clearance in liver microsomes compared to original compound 1.
Table 2. Optimization of Benzimidazole Fragment: In Vitro hmGluR5 Antagonism.
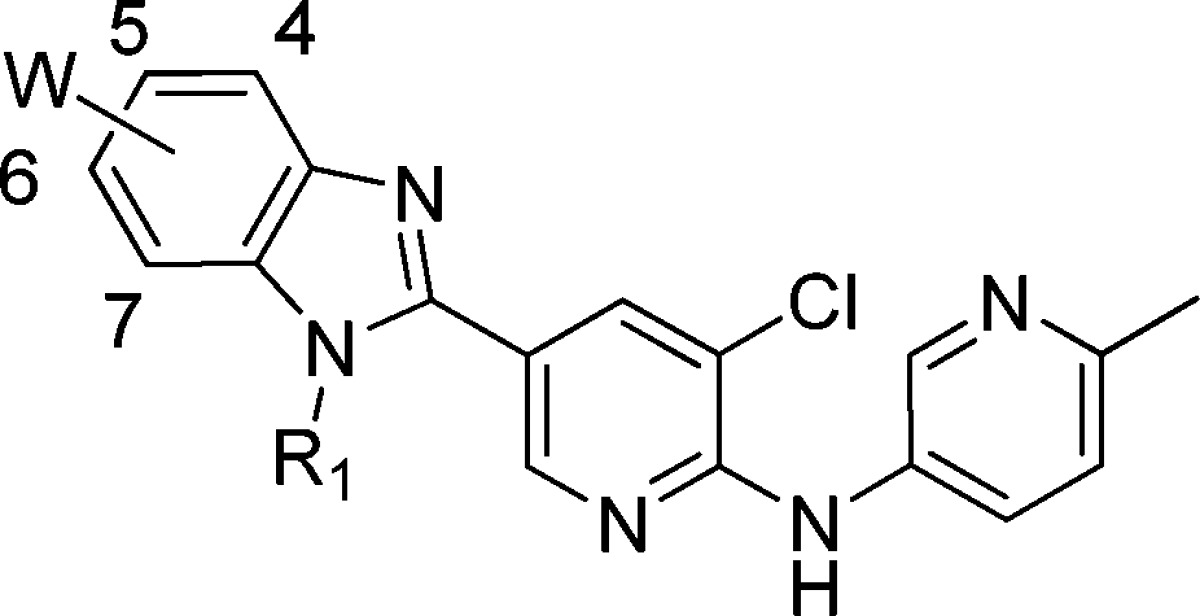
| compd | R1 | 4-W | 5-W | 6-W | 7-W | IC50 [μM]a | HLMb | RLMb |
|---|---|---|---|---|---|---|---|---|
| 15b | Pr | H | H | H | H | 0.10 | 83 | 158 |
| 15c | Et | H | H | H | H | 0.11 | 120 | 118 |
| 15d | Bu | H | H | H | H | 0.36 | 347 | 279 |
| 15e | iBu | H | H | H | H | 0.11 | 55 | 826 |
| 15f | Pr | Cl | H | H | H | 0.20 | 74 | 69 |
| 15g | Pr | H | Cl | H | H | >0.5 | 400 | 34 |
| 15h | Pr | H | H | Cl | H | 0.13 | 56 | 33 |
| 15i | Pr | H | H | H | Cl | 0.024 | 83 | 112 |
hmGluR5 Ca2+ flux7 using glutamate as agonist; data are geometric means of n ≥ 2.
HLM: human liver microsomes. RLM: rat liver microsomes. CLint (intrinsic clearance) reported in μL/min/mg protein.
In vitro radioligand displacement assays utilizing [3H]-ABP68820 showed that 15i is a high-affinity ligand at the previously characterized allosteric binding site located in the membrane-spanning region of mGlu5 receptors21 with a Ki at the human recombinant receptor of 2.4 nM.22 In addition, 15i showed a high degree of selectivity in functional assays over representatives of group I, II and III metabotropic glutamate receptor subtypes (IC50 > 10 μM for hmGluR1, -2, -7) and ionotropic glutamate receptors (IC50 > 10 μM).23
Single-dose pharmacokinetic studies of 15i in rats showed that this compound is superior to 1 in terms of lower plasma clearance, prolonged mean residence time and apparent elimination half-life. This could not be expected based on the similar intrinsic clearance of both compounds in liver microsomes, but might be explained by the higher plasma protein binding of 15i,24 resulting in a protective effect in vivo. Brain penetration and bioavailability were similar (Table 3).
Table 3. Pharmacokinetic Parametersa of 15ib and 1c.
| 15i | 1 | |
|---|---|---|
| plasmaCL (mL/min/kg) | 15 | 37 |
| MRT (h) | 2.2 | 0.9 |
| t1/2 (h) | 1.9 | 0.9 |
| Tmax (h) | 0.5 | 0.25 |
| Cmaxd (pmol/mL/μmol/kg) | 60 | 41 |
| AUCpod (pmol h/mL/μmol/kg) | 449 | 241 |
| F (%) | 41 | 54 |
| brain/plasma-AUCpo ratio | 1.67 | 1.36 |
n = 3 Sprague−Dawley rats/group.
3.6 μmol/kg po, 2.4 μmol/kg iv.
30 μmol/kg po, 10 μmol/kg iv.
Dose-normalized value.
As in a preceding study16 with 1, we assessed efficacy and potency of 15iin vivo by determining the compound’s effect on the expression of fear-potentiated startle (FPS) in rats.25,26 In the present study, rats were orally treated with 0.3, 1, and 3 mg/kg of 15i, or vehicle 60 min prior to the behavioral test (group size n = 10). The FPS response was reduced in a dose-dependent manner; the effects caused by 1 and 3 mg/kg were statistically significant vs the vehicle control (Figure 2). The minimal effective dose of 15i is thus considered to be 1 mg/kg po in this paradigm, whereas we found 3 mg/kg for 1.25,26
Figure 2.

(A) Expression of fear-potentiated and baseline startle response (0.3−3 mg/kg compound 15i). Mean startle magnitudes and SEMs of startle stimulus alone and CS-startle stimulus trials, respectively, as well as the difference. Multifactorial ANOVA: interaction treatment × trial type: F3,36 = 7.49, p < 0.001, p = 0.001; **p < 0.01 (Dunnett post hoc test vs 0 mg/kg). (B) Brain concentration (mean ± SEM) of compound 15i in the different treatment groups as well as the mean percent startle difference (±SEM). Compound 15i brain levels are negatively correlated with the expression of fear-potentiated startle. For experimental details see the Supporting Information.
The amount of FPS reduction induced by 15i was positively correlated with the compound’s plasma and brain concentrations, measured in a subset (n = 4) of the animals that underwent the FPS test (samples collected at 90 min postdose). Both plasma and brain concentrations were proportional to the applied doses (0.3 to 3 mg/kg); the brain/plasma ratio varied between 1.2 and 2.0.
In conclusion, biotransformation investigations with compound 1 identified structural liabilities that guided a focused chemical optimization effort resulting in 15i, a selective, orally bioavailable mGluR5 antagonist of similar potency and affinity in vitro and superior metabolic stability compared to 1. In vivo characterization of 15i in the FPS model revealed a robust anxiolytic-like effect with a minimal effective dose of 1 mg/kg po and positive correlation with brain and plasma exposures. In view of this promising profile, compound 15i was considered for further development.
Acknowledgments
The excellent technical assistance of Christian Boesch, Ralf Boesch, Hugo Bürki, Stefan Imobersteg, Martin Gunzenhauser, Nicole Reymann, Patrick Seitzer, Christine Stierlin, Peter Wipfli, Francis Risser, Pierrette Guntz and Valerie Cordier, Philippe Ramstein, Werner Gertsch is gratefully acknowledged.
Supporting Information Available
Experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- a Swanson C. J.; Bures M.; Johnson M. P.; Linden A.-M.; Monn J. A.; Schoepp D. D. Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat. Rev. Drug Discovery 2005, 4, 131–144. [DOI] [PubMed] [Google Scholar]
- Ritzen A.; Mathiesen J. M.; Thomsen C. Molecular Pharmacology and Therapeutic Prospects of Metabotropic Glutamate Receptor Allosteric Modulators. Basic Clin. Pharmacol. Toxicol. 2005, 97, 202–2013. [DOI] [PubMed] [Google Scholar]
- Knöpfel T.; Kuhn R.; Allgeier H. Metabotropic Glutamate Receptors: Novel Targets for Drug Development. J. Med. Chem. 1995, 38, 1417. [DOI] [PubMed] [Google Scholar]; Conn P. J.; Pin J. P. Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 205. [DOI] [PubMed] [Google Scholar]
- Shigemoto R.; Nomura S.; Ohishi H.; Sugihara H.; Nakanishi S.; Mizuno N. Immunohistochemical localization of metabotropic glutamate receptor, mGluR5, in the rat brain. Neurosci. Lett. 1993, 163, 53. [DOI] [PubMed] [Google Scholar]
- Romano C.; Sesma M. A.; McDonald C. T.; O’Malley K.; Van den Pol A. N.; Olney J. W. Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J. Comp. Neurol. 1995, 355, 455. [DOI] [PubMed] [Google Scholar]
- Rodrigues S. M.; Bauer E. P.; Farb C. R.; Schafe G. E.; LeDoux J. E. The Group I Metabotropic Glutamate Receptor mGluR5 Is Required for Fear Memory Formation and Long-Term Potentiation in the Lateral Amygdala. J. Neurosci. 2002, 22, 5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan K. L.; Fitzgerald D. A.; Cortese B. M.; Seraji-Bozorgzad N.; Tancer M. E.; Moore G. J. Anterior cingulate neurochemistry in social anxiety disorder: 1H-MRS at 4 T. NeuroReport 2005, 16, 183. [DOI] [PubMed] [Google Scholar]
- Mathew S. J.; Price R. B.; Charney D. S. Recent advances in the neurobiology of anxiety disorders: implications for novel therapeutics. Am. J. Med. Genet., Part C 2008, 148, 89. [DOI] [PubMed] [Google Scholar]
- Zhu C. Z.; Wilson S. G.; Mikusa J. P.; Wismer C. T.; Gauvin D. M.; Lynch J. L. III; Wade C. L.; Decker M. W.; Honore P. Assessing the role of metabotropic glutamate receptor 5 in multiple nociceptive modalities. Eur. J. Pharmacol. 2004, 506, 107. [DOI] [PubMed] [Google Scholar]
- Bradbury M. J.; Campbell U.; Giracello D.; Chapman D.; King C.; Tehrani L.; Cosford N. D. P.; Anderson J.; Varney M. A.; Strack A. M. Metabotropic Glutamate Receptor mGlu5 Is a Mediator of Appetite and Energy Balance in Rats and Mice. J. Pharmacol. Exp. Ther. 2005, 313, 395. [DOI] [PubMed] [Google Scholar]
- Breysse N.; Baunez C.; Spooren W.; Gasparini F.; Amalric M. Metabotropic Glutamate 5 Receptor Blockade Alleviates Akinesia by Normalizing Activity of Selective Basal-Ganglia Structures in Parkinsonian Rats. J. Neurosci. 2002, 22, 5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiamulera C.; Epping-Jordan M. P.; Zocchi A.; Marcon C.; Cottiny C.; Tacconi S.; Corsi M.; Orzi F.; Conquet F. Accruing preclinical evidence about metabotropic glutamate 5 receptor antagonists as treatments for drug dependence highlights the irreplaceable contributions of animal studies to the discovery of new medications for human disorders. Nat. Neurosci. 2001, 4, 873.11528416 [Google Scholar]
- Keywood C.; Wakefield M. ADX10059, a negative allosteric modulator of mGluR5, demonstrates proof of concept for a role in the management of migraine, in a placebo controlled phase 2A study. Neuropharmacology 2008, 55, 605. [Google Scholar]
- Lehmann A. Novel treatments of GERD: focus on the lower esophageal sphincter. Eur. Rev. Med. Pharmacol. Sci. 2008, 12, 103. [PubMed] [Google Scholar]
- Bear M. F.; Huber K. M.; Warren S. T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370. [DOI] [PubMed] [Google Scholar]
- Spanka C.; Glatthar R.; Desrayaud S.; Fendt M.; Orain D.; Troxler T.; Vranesic I. Piperidyl amides as novel, potent and orally active mGlu5 receptor antagonists with anxiolytic-like activity. Bioorg. Med. Chem. Lett. 2010, 20, 184. [DOI] [PubMed] [Google Scholar]
- Knöpfel T.; Sakaki J.; Flor P. J.; Baumann P.; Sacaan A. I.; Velicelebi G.; Kuhn R.; Allgeier H. Profiling of trans-azetidine-2,4-dicarboxylic acid at the human metabotropic glutamate receptors mGlu1b, -2, -4a and -5a. Eur. J. Pharmacol. 1994, 288, 389. [DOI] [PubMed] [Google Scholar]
- Abundances of components are not corrected for different ionization efficiencies in electrospray
- Compound 6 was prepared by bromination of commercially available 5,6,7,8-tetrahydroimidazo[1,5-a]pyridine. See Supporting Information for experimental details.
- Hintermann S.; Vranesic I.; Allgeier H.; Bruelisauer A.; Hoyer D.; Lemaire M.; Moenius T.; Urwyler S.; Whitebread S.; Gasparini F.; Auberson Y. P. ABP688, a novel selective and high affinity ligand for the labeling of mGlu5 receptors: identification, in vitro pharmacology, pharmacokinetic and biodistribution studies. Bioorg. Med. Chem. 2007, 15, 903. [DOI] [PubMed] [Google Scholar]
- Pagano A.; Rüegg D.; Litschig S.; Stoehr N.; Stierlin C.; Heinrich M.; Floersheim P.; Prezeau L.; Carroll F.; Pin J. P.; Cambria A.; Vranesic. I.; Flor P. J.; Gasparini F.; Kuhn R. The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J. Biol. Chem. 2000, 27533750. [DOI] [PubMed] [Google Scholar]
- A similar binding affinity (Ki: 30 nM) was observed in membranes prepared from cells expressing rat brain tissue (cortex/hippocampus).
- Selectivity to ionotropic glutamate receptors was assessed with radioligand displacement assays in rat brain membranes at the AMPA (glutamate binding site), kainate (glutamate binding site) and NMDA (glutamate and glycine binding sites) subtypes.
- Rat plasma protein binding of compounds 15i and 1 were assessed by equilibrium dialysis. 15i showed a free fraction of 1.7% whereas 1 showed a free fraction of 12.7%.
- Davis M.; Falls W. A.; Campeau S.; Kim M. Fear-potentiated startle: a neural and pharmacological analysis. Behav. Brain Res. 1993, 58, 175. [DOI] [PubMed] [Google Scholar]
- Fendt M.; Fanselow M. S. The neuroanato- mical and neurochemical basis of conditioned fear. Neurosci. Biobehav. Rev. 1999, 23, 743. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.