

Abstract
The synthesis of novel, selective, orally active 2,5-disubstituted 6H-pyrimido[1,6-b]pyridazin-6-one p38α inhibitors is described. Application of structural information from enzyme–ligand complexes guided the selection of screening compounds, leading to the identification of a novel class of p38α inhibitors containing a previously unreported bicyclic heterocycle core. Advancing the SAR of this series led to the eventual discovery of 5-(2,6-dichlorophenyl)-2-(2,4-difluorophenylthio)-6H-pyrimido[1,6-b]pyridazin-6-one (VX-745). VX-745 displays excellent enzyme activity and selectivity, has a favorable pharmacokinetic profile, and demonstrates good in vivo activity in models of inflammation.
Keywords: p38 inhibitor, VX-745, inflammatory diseases
Mammalian mitogen-activated protein (MAP) kinases are serine/threonine kinases that mediate pro-inflammatory cytokine biosynthesis at the transcriptional and translational level.1−3 Tumor necrosis factor α (TNFα) and interleukin-1β (IL-1β) are cytokines involved in various cellular functions and, when present in elevated levels, are associated with the pathogenesis of a variety of inflammatory diseases, including rheumatoid arthritis (RA).4 It is known that small molecule inhibitors of p38α MAP kinase (i.e., inhibitors of the p38α and p38β isoforms) suppress the production of TNFα and IL-1β in vitro and in animal models,5−8 suggesting that the stress-activated signal transduction pathway leading to these cytokines is at least partially regulated by p38. The clinical success in the treatment of RA patients by targeting TNFα mediated inflammatory pathways using, for example, the soluble TNFα receptor fusion protein etanercept (Enbrel) and the monoclonal anti-TNFα products infliximab (Remicade) and adalimumab (Humira) provides the rationale of targeting these cytokines for therapeutic effect.9−14 Thus, novel orally administered small molecules that selectively inhibit p38 would be desirable to provide therapeutic intervention for inflammatory disorders and to more clearly define the physiological role of this protein kinase-mediated pathway.
First disclosed in 1994, pyridinylimidazole compounds, exemplified by SB 203580 (1) and VRT-19911 (2) (see Figure 1), block the production of TNFα and IL-1β from monocytes stimulated by bacterial lipopolysaccharides (LPS) through the inhibition of p38 MAP kinase.2 Compound 1 has been shown to be effective in animal models of arthritis, bone resorption, and endotoxin shock.15 Compound 1 is a selective inhibitor of p38α and p38β (IC50 = 0.1 and 0.43 μM respectively, but does not inhibit the closely related p38γ or p38δ kinases or other MAP kinase family members such as the extracellular-signal regulated kinases (ERK) and most isoforms of the c-Jun N-terminal kinases (JNKs).16−18
Figure 1.

Inhibitors of p38 MAP kinase.
Compound 2 has been shown to bind in the ATP binding site of p38α and is a selective and potent inhibitor with a Ki of 60 nM.19 However, it has been previously reported that the presence of the pyridyl moiety in 1 and 2 leads to significant inhibition of hepatic cytochrome p450 isozymes in vitro,20 rendering these compounds unsuitable for development in the treatment of chronic disease.
Analysis of our previously reported crystal structure of unphosphorylated p38α MAP kinase bound to 2(21) facilitated the early design and eventual discovery of novel p38 inhibitors. We utilized virtual screening and a diverse selection of shape similarity methods to search commercially available compound databases for molecules with similar configuration, yet different chemical connectivity when compared to 2. The selected compounds were screened for p38 inhibition, and based on this approach, we identified several inhibitors of p38α in the range of IC50 = 1–30 μM. We report here the optimization of one pyridazine-containing class of inhibitors, leading to the identification of VX-745 (3), a first-generation p38 inhibitor clinical candidate.
Structure 4 (see Figure 2), a commercially procured sample, emerged as a p38α enzyme inhibitor screening hit22 selected for further evaluation. In an attempt to resynthesize 4, we performed the synthesis outlined in Scheme 1.23 2,4-Dichlorophenylacetonitrile (5) was treated with sodium tert-butoxide followed by 3,6-dichloropyridizine to give the corresponding pyridazinyl-phenylacetonitrile (6). Displacement of the second chloro group with phenylthiosodium gave the 3,6-disubstituted pyridazine (7). Compound 7 was subsequently hydrated to amide 8 using concentrated sulfuric acid. The attempted synthesis of 4 from 8 using DMF–DMA in toluene at 100 °C did not result in the desired product but afforded a product in which the N,N-dimethyl group was absent from the 1H NMR spectrum. This was confirmed by mass spectrometry, which showed m/z = 400 (M + H), not 445 as might be expected, indicating the loss of dimethylamine. We hypothesized the in situ formation of compound 4, and tautomerization of the α-pyridazine ring system to compound 9 allowed for nucleophilic collapse of the pyridazine ring nitrogen on the ethanamide carbon to afford compound 10.
Figure 2.

Structure of screening hit, as reported by vendor.
Scheme 1.

Small molecule X-ray crystallography24 of a closely related analogue (23), also prepared via Scheme 1, confirmed the structure we pursued was not 4, as assigned by the vendor, but 5-(2,4-dichlorophenyl)-2-(phenylthio)-6H-pyrimido[1,6-b]pyridazin-6-one (10). The core bicyclic system, 6H-pyrimido[1,6-b]pyridazin-6-one, was, at this time, unreported in the literature.
Having developed a practical synthetic route, we initiated exploration of the SAR of the two pendant aryl rings. Initial studies focused on the substitutions of the directly attached phenyl ring (5-position substituent of pyrimido[1,6-b]pyridazin-6-one: Table 1), prepared from commercially available phenylacetonitriles, by the same methods shown in Scheme 1. Evaluation of the products 10–21 (Table 1) indicated that the unsubstituted phenyl (11) and the 4-substituted systems 16, 17, and 18 showed poor enzyme inhibitory activity. Introduction of a 2-substituent (12–15) showed modest potency, similar to the screening hit 10, suggesting that, of the two substituents, the 2-chloro is the key to enzyme activity. The 2,6-dichloro substitution emerged as optimal, as indicated by compound 21, which also demonstrated corresponding inhibition of IL-1β and TNFα release from lipopolysaccharide (LPS) treated human peripheral blood mononuclear cells (PBMCs). For other 2,6-disubstituted systems examined (19, 20), potency was approximately 10-fold weaker than that of 21.
Table 1. Inhibition of p38α and PBMC Cytokine Release by 5-Aryl-2-(phenylthio)-6H-pyrimido[1,6-b]pyridazin-6-ones.
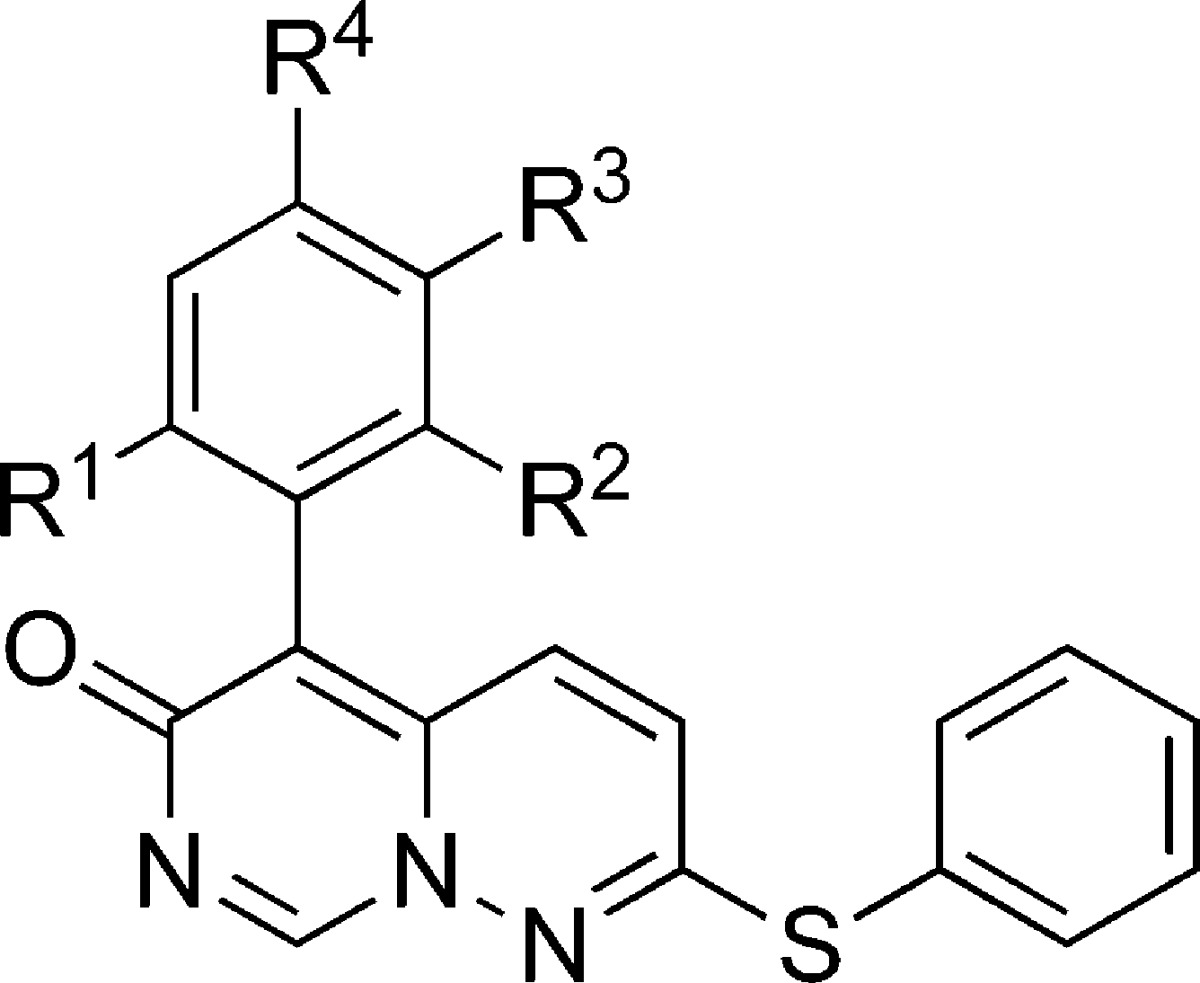
| compd | R1 | R2 | R3 | R4 | p38α IC50 (μM)a | PBMC IL-1β IC50b (μM) | PBMC TNFα IC50b (μM) |
|---|---|---|---|---|---|---|---|
| 10 | Cl | H | H | Cl | 5 | 20 | 20 |
| 11 | H | H | H | H | >20 | NDc | ND |
| 12 | Me | H | H | H | 4.4 | 3.5 | 9.7 |
| 13 | OMe | H | H | H | 12 | 2.8 | >20 |
| 14 | CF3 | H | H | H | 11 | 2.1 | >20 |
| 15 | Cl | H | H | H | 4.1 | 5.0 | 19.9 |
| 16 | H | H | H | F | >20 | 15 | >20 |
| 17 | H | H | H | OMe | >20 | 20 | >20 |
| 18 | H | H | Cl | Cl | >20 | >20 | >20 |
| 19 | CF3 | F | H | H | 1.7 | 1.0 | 3.1 |
| 20 | F | F | H | H | 3.5 | 3.7 | 15.3 |
| 21 | Cl | Cl | H | H | 0.25 | 0.40 | 0.46 |
Assayed according to ref (22).
See Supporting Information for assay details.
ND = not determined.
With 21 representing optimal substitution of the 5-aryl ring, we focused on the S-linked phenyl ring at the pyrimido[1,6-b]pyridazin-6-one 2-position (Table 2) using commercially available thiophenols. Addition of a 4-fluoro group, exemplified by 23, resulted in a slight improvement in enzyme and cellular activity versus the corresponding unsubstituted thiophenyl analogue 21 (Table 1). Small alkyl groups or halogens at the meta position resulted in a reduction of affinity, as in 24, 25, 26, 30, 31, and 32 without regard to the nature of the substituent at the para position. Small polar functionalities at the ortho position were tolerated, as in 29 and 33. However, the most significant improvement was observed with a 2,4-dihalo substitution of the thiophenol. The 2-chloro-4-fluoro compound 34 showed excellent potency, while the 2,4-difluoro exhibited the best potency, resulting in 3. As before, cellular data tracked enzyme inhibition. Compound 3 exhibits PBMC IL-1β and TNFα IC50 values of 45 and 51 nM, respectively. Compound 3 is also effective in whole blood, blocking IL-1β and TNFα release with IC50 values of 150 and 180 nM, respectively. Conversely, compound 3 does not affect proliferation of phytohemaglutinin-stimulated PBMCs up to 20 μM, indicating cellular selectivity for p38 versus other kinases.
Table 2. Inhibition of p38α and PBMC Cytokine Release by 5-(2,6-Dichlorophenyl)-2-(arylthio)-6H-pyrimido[1,6-b]pyridazin-6-ones.
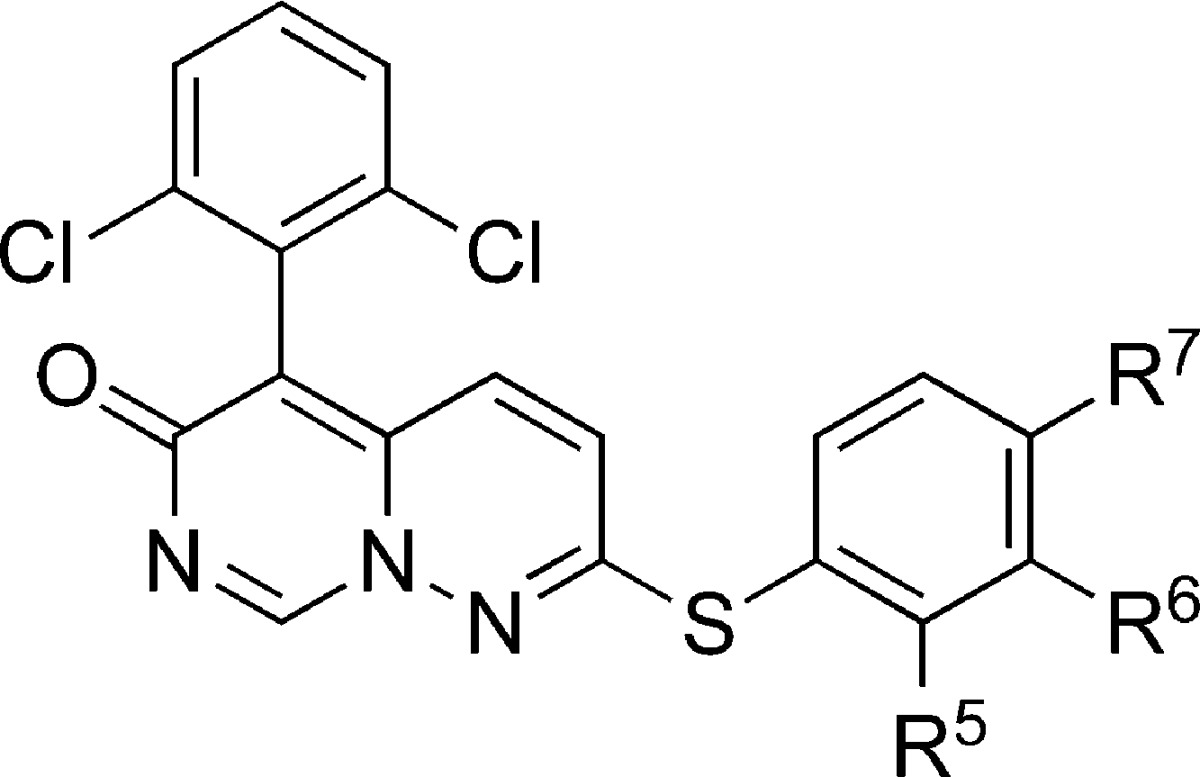
| compd | R5 | R6 | R7 | p38α IC50 (μM) | PBMC IL-1β IC50 (μM) | PBMC TNFα IC50 (μM) |
|---|---|---|---|---|---|---|
| 21 | H | H | H | 0.25 | 0.40 | 0.45 |
| 22 | H | H | Me | 1.4 | 0.37 | 0.87 |
| 23 | H | H | F | 0.123 | 0.099 | 0.293 |
| 24 | H | F | H | 0.45 | 0.44 | 0.72 |
| 25 | H | Cl | H | 0.28 | 0.35 | 0.90 |
| 26 | H | Me | H | 1.4 | 0.21 | 0.28 |
| 27 | Et | H | H | 0.75 | 0.98 | 2.8 |
| 28 | Me | H | H | 0.20 | 0.33 | 3.7 |
| 29 | OH | H | H | 0.36 | 0.14 | 0.49 |
| 30 | H | Cl | Cl | 0.8 | 0.97 | 1.2 |
| 31 | H | Me | Me | 1.9 | 2.4 | 3.5 |
| 32 | H | Cl | F | 0.5 | 0.18 | 0.48 |
| 33 | NH2 | H | F | 0.22 | 0.28 | 1.2 |
| 34 | Cl | H | F | 0.036 | 0.053 | 0.275 |
| 3 | F | H | F | 0.009 | 0.045 | 0.051 |
| 35 | Me | H | Cl | 0.18 | 0.18 | 0.16 |
| 36 | Me | H | Me | 0.26 | 0.69 | 0.32 |
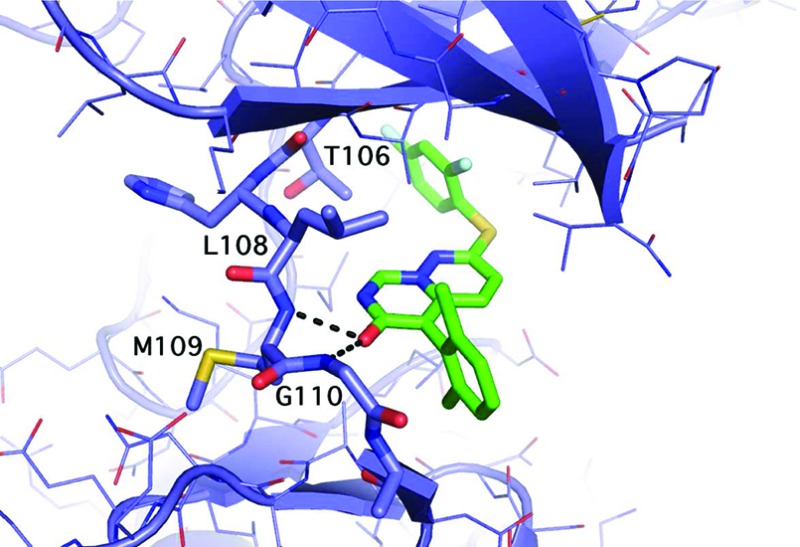
The X-ray cocomplex of 3 and p38α at 2.4 Å resolution (Figure 3) shows favorable van der Waals interactions that stabilize the interaction of the 2,6-dichloro phenyl ring with p38.25 Each chlorine atom occupies a hydrophobic pocket with contact to residues V30, L108, A157, and L167. The phenyl ring itself makes favorable van der Waals interactions with backbone residue atoms G110, A111, and D112. The para position of the ring faces bulk solvent. The 2,4-difluorophenyl ring of 3 occupies a hydrophobic pocket at the gatekeeper (T106) residue and makes extensive van der Waals contacts. In addition, the enzyme facilitates the binding of 3 by flipping the G110 backbone.26 This flip is possible because glycine lacks an α-carbon substitution. This unique enzyme conformation allows the carbonyl oxygen of 3 to accept an additional H-bond from the p38 backbone. This combination of interactions is a key determinant of enzyme specificity.19,21
Figure 3.
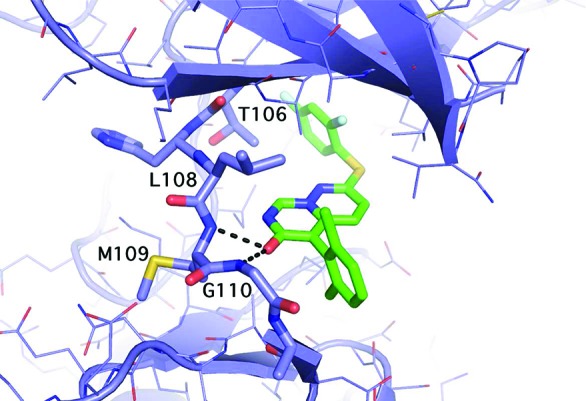
Structure of 3 bound to p38.
The lead compound, 3, was tested against the different p38 isozymes and closely related MAP kinases (ERK, JNK) as well as a panel of 50 other kinases to examine its selectivity profile. Compound 3 showed a promising selectivity profile, with 20-fold selectivity for p38α over p38β (Ki = 220 nM),27 and no significant inhibition of other MAP kinases (with the exception of MKK6, itself an activator of p38)28 or any of the other 50 kinases tested at 2 μM concentration (see Supporting Information). Similar selectivity results have been reported elsewhere.29
The pharmacokinetic parameters obtained for 3 in three species are summarized in Table 3. Systemic clearance was slightly higher than hepatic blood flow for rat and dog, and 3 appears to have significant extravascular distribution in all three species studied, as indicated by the volume of distribution at steady state. The oral pharmacokinetic profile of 3 in TPGS/PEG-400/water (2:7:1) was characterized in male BALB/c mice, Sprague–Dawley rats, and beagle dogs. The results of these single dose studies showed that 3 has excellent bioavailability in all three species (87%, 56%, and 69%, respectively). Compound 3 demonstrated a longer half-life following oral administration as compared to IV administration, suggesting an absorption-rate limited elimination process in the three species. Compound 3 is neither a significant inhibitor nor an inducer of human hepatic cytochrome p450 isozymes. The IC50 values for the inhibition of CYPs 3A4, 2D6, 2C19, 2C9, and 1A2 were determined to be >40 μM. The fraction of 3 bound to plasma protein was 98% and 92% in the rat and the dog, respectively.
Table 3. Pharmacokinetic Properties of 3.
| parameter | rat | mouse | dog |
|---|---|---|---|
| IV dose (mg/kg) | 4.8 | 2 | 13.8 |
| n | 3 | 3 | 4 |
| T1/2 (h) | 1.9 | 2.6 | 1.5 |
| Vss (L/kg) | 3.9 | 2.2 | 2.3 |
| AUC (μg·h/mL) | 1.26 | 1.54 | 6.12 |
| Cl (mL/min/kg) | 63 | 21 | 39 |
| PO dose (mg/kg) | 43 | 40 | 33 |
| n | 3 | 3 | 4 |
| T1/2 (h) | 4.5 | 4.5 | 4.5 |
| Cmax (μg/mL) | 0.89 | 7.85 | 1.37 |
| AUC (μg·h/mL) | 6.3 | 26.96 | 4.34 |
| F (%) | 56 | 87 | 69 |
To evaluate the in vivo therapeutic potential of compound 3, we employed the murine collagen-induced arthritis model of human rheumatoid arthritis, in which DBA/1J mice were immunized twice with chick type II collagen. Treatment was initiated once an inflammation score of 2 in each paw had been reached, corresponding to focal swelling of the wrist joint. Results showed that mice treated with 2.5, 5, and 10 mg/kg of compound 3 twice daily for 20 days had 27%, 31%, and 44% improvement in the inflammatory scores, respectively, when compared to vehicle-treated mice at the end of treatment (Table 4; see Supporting Information for details). In addition, histological scores showed a 32–39% protection of bone and cartilage erosion.
Table 4. Therapeutic Effect of 3 in Type II Murine Collagen-Induced Arthritis (CIA) Model.
| clinical score on paw inflammationa |
histology scoreb |
|||||
|---|---|---|---|---|---|---|
| dose | meana | SEM | % inh | meanb | SEM | % inh |
| vehicle (100% polyethylene glycol) | 3.81 | 0.13 | NA | 2.04 | 0.09 | NA |
| compound 3, 2.5 mg/kg | 2.77 | 0.17 | 27 | 1.33 | 0.09 | 35 |
| compound 3, 5 mg/kg | 2.65 | 0.28 | 31 | 1.38 | 0.11 | 32 |
| compound 3, 10 mg/kg | 2.13 | 0.13 | 44 | 1.25 | 0.09 | 39 |
Clinical scores measured on final day 20. Clinical scores are defined in the Supporting Information.
Histology scores are defined in the Supporting Information.
Compound 3 (VX-745) and other analogues described in this study represent the first known examples of 5-phenyl-2-(phenylthio)-6H-pyrimido[1,6-b]pyridazin-6-ones as p38 inhibitors. These compounds show potent inhibition of the key inflammatory mediators, IL-1β and TNFα, production in vitro in isolated PBMCs and whole blood. Compound 3 has also been shown to demonstrate anti-inflammatory efficacy in an animal model of rheumatoid arthritis and was designated for further development.30
Acknowledgments
We thank Dr. Mariusz Krawiec for X-ray crystallographic support, Dr. Nigel Ewing for high resolution mass spectral support, Dr. Patricia McCaffrey and Dr. Karyn Cepek for cellular assays support, and Dr. George Ku for in vivo pharmacology support.
Supporting Information Available
Full experimental details for representative compounds synthesized, HRMS results, description of assays, animal efficacy studies, and X-ray crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
‡ Novartis, 250 Massachusetts Avenue, Cambridge, MA 02139.
Author Present Address
§ Agios Pharmaceuticals, 38 Sidney Street, Cambridge, MA 02139.
Author Present Address
∥ GlaxoSmithKline, 830 Winter Street, Waltham, MA 02451.
Author Present Address
⊥ AstraZeneca, 35 Gatehouse Drive, Waltham, MA 02451.
Author Present Address
# Takeda, 10410 Science Center Drive, San Diego, CA 92121.
Author Present Address
∇ Constellation Pharmaceuticals, 215 First Street, Suite 200, Cambridge, MA 02142.
Author Present Address
○ Amgen, 1 Amgen Center Drive, Thousand Oaks, CA 91320.
Supplementary Material
References
- Cuenda A.; Rousseau S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [DOI] [PubMed] [Google Scholar]
- Lee J. C.; Laydon J. T.; McDonnell P. C.; Gallagher T. F.; Kumar S.; Green D.; McNulty D.; Blumenthal M. J.; Heys J. R.; Landvatter S. W.; Strickler J. E.; McLaughlin M. M.; Siemens I. R.; Fisher S. M.; Livi G. P.; White J. R.; Adams J. L.; Young P. R. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372, 739–746. [DOI] [PubMed] [Google Scholar]
- Schieven G. L. The Biology of p38 Kinase: A Central Role in Inflammation. Curr. Top. Med. Chem. 2005, 5, 921–928. [DOI] [PubMed] [Google Scholar]
- Feldmann M. L.; Brennan F. M.; Maini R. N. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 1996, 14, 397–440. [DOI] [PubMed] [Google Scholar]
- Wagner G.; Laufer S. Small molecular anti-cykotine agents. Med. Res. Rev. 2006, 26, 1–62. [DOI] [PubMed] [Google Scholar]
- Adams J. L.; Badger A. M.; Kumar S.; Lee J. C. p38 MAP kinase: molecular target for the inhibition of pro-inflammatory cytokines. Prog. Med. Chem. 2001, 38, 1–60. [DOI] [PubMed] [Google Scholar]
- Liverton N. J.; Butcher J. W.; Claiborne C. F.; Claremon D. A.; Libby B. E.; Nguyen K. T.; Pitzenberger S. M.; Selnick H. G.; Smith G. R.; Tebben A.; Vacca J. P.; Varga S. L.; Agarwal L.; Dancheck K.; Forsyth A. J.; Fletcher D. S.; Frantz B.; Hanlon W. A.; Harper C. F.; Hofsess S. J.; Kostura M.; Lin J.; Luell S.; O’Neill E. A.; Orevillo C. J.; Pang M.; Parsons J.; Rolando A.; Sahly Y.; Visco D. M.; O’Keefe S. J. Design and synthesis of potent, selective, and orally bioavailable tetrasubstituted imidazole inhibitors of p38 mitogen-activated protein kinase. J. Med. Chem. 1999, 42, 2180–2190. [DOI] [PubMed] [Google Scholar]
- Gallagher T. F.; Seibel G. L.; Kassis S.; Laydon J. T.; Blumenthal M. J.; Lee J. C.; Lee D.; Boehm J. C.; Fier-Thompson S. M.; Abt J. W.; Soreson M. E.; Smietana J. M.; Hall R. F.; Garigipati R. S.; Bender P. E.; Erhard K. F.; Krog A. J.; Hofmann G. A.; Sheldrake P. L.; McDonnell P. C.; Kumar S.; Young P. R.; Adams J. L. Regulation of stress-induced cytokine production by pyridinylimidazoles; inhibition of CSBP kinase. Bioorg. Med. Chem. 1997, 5, 49–64. [DOI] [PubMed] [Google Scholar]
- Pugsley M. K. Etanercept: Immunex. Curr. Opin. Invest. Drugs 2001, 2, 1725–1731. [PubMed] [Google Scholar]
- Bondeson J.; Maini R. N. Tumor necrosis factor as a therapeutic target in rheumatoid arthritis and other chronic inflammatory diseases: The clinical experience with infliximab (Remicade). Int. J. Clin. Pract. 2001, 55, 211–216. [PubMed] [Google Scholar]
- Saleem B.; Mackie S.; Emery P. Infliximab for rheumatoid arthritis. Expert Rev. Clin. Immunol. 2006, 2, 193–207. [DOI] [PubMed] [Google Scholar]
- Cottone M.; Mocciaro F.; Modesto I. Infliximab and ulcerative colitis. Expert Opin. Biol. Ther. 2006, 6, 401–408. [DOI] [PubMed] [Google Scholar]
- Kavanaugh A. Anakinra (interleukin-1 receptor antagonist) has positive effects on function and quality of life in patients with rheumatoid arthritis. Adv. Ther. 2006, 23, 208–217. [DOI] [PubMed] [Google Scholar]
- Fleischmann R. Anakinra in the treatment of rheumatic disease. Expert Rev. Clin. Immunol. 2006, 2, 331–341. [DOI] [PubMed] [Google Scholar]
- Badger A. M.; Bradbeer J. N.; Votta B.; Lee J. C.; Adams J. L.; Griswold D. E. Pharmacological Profile of SB 203580, a Selective Inhibitor of Cytokine Suppressive Binding Protein/p38 Kinase, in Animal Models of Arthritis, Bone Resorption, Endotoxin Shock and Immune Function. J. Pharmacol. Exp. Ther. 1996, 279, 1453–1461. [PubMed] [Google Scholar]
- Cuenda A.; Rouse J.; Doza Y. N.; Meier R.; Cohen P.; Gallagher T. F.; Young P. R.; Lee J. C. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995, 364, 229–233. [DOI] [PubMed] [Google Scholar]
- Davies S. P.; Reddy H.; Caivano M.; Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y.; Gram H.; Zhao M.; New L.; Gu J.; Feng L.; DiPadova F.; Ulevitch R. J.; Han J. Characterization of the Structure and Function of the Fourth Member of p38 Group Mitogen-activated Protein Kinases, p38δ. J. Biol. Chem. 1997, 272, 30122–30128. [DOI] [PubMed] [Google Scholar]
- Salituro F. G.; Germann U. A.; Wilson K. P.; Bemis G. W.; Fox T.; Su M. S. Inhibitors of p38 MAP Kinase: Therapeutic Intervention in Cytokine-Mediated Diseases. Curr. Med. Chem. 1999, 6, 807–823. [PubMed] [Google Scholar]
- Adams J. L.; Boehm J. C.; Kassis S.; Gorycki P. D.; Webb E. F.; Hall R.; Sorenson M.; Lee J. C.; Ayrton A.; Griswold D. E.; Gallagher T. F. Pyrimidinylimidazole Inhibitors Of CSBP/P38 Kinase Demonstrating Decreased Inhibition Of Hepatic Cytochrome P450 Enzymes. Bioorg. Med. Chem. Lett. 1998, 8, 3111–3116. [DOI] [PubMed] [Google Scholar]
- Wilson K. P.; McCaffrey P. G.; Hsiao K.; Pazhanisamy S.; Galullo V.; Bemis G. W.; Fitzgibbon M. J.; Caron P. R.; Murcko M. A.; Su M. S.-S. The structural basis for the specificity of pyridinylimidazole inhibitors of p38 MAP kinase. Chem. Biol. 1997, 4, 423–431. [DOI] [PubMed] [Google Scholar]
- Compounds were assayed by the coupled assay method described in:Fox T.; Coll J. T.; Xie X.; Ford P. J.; Germann U. A.; Porter M. D.; Pazhanisamy S.; Fleming M. A.; Galullo V.; Su M. S.; Wilson K. P. Protein Sci. 1998, 7, 2249.Reactions were carried out in 100 mM HEPES pH 7.6, 10 mM MgCl2, 25 mM NaCl, 1 mM DTT, 20 μg/mL BSA, and 1.5% DMSO. Final substrate concentrations in the assay were 100 μM ATP and 200 μM peptide (KRELVEPLTPSGEAPNQALLR). Assays were carried out at 30 °C using 15 nM p38. The final concentrations of the components of the coupled enzyme system were 2.5 mM phosphoenolpyruvate, 200 μM NADH, 140 μg/mL pyruvate kinase, and 10 μg/mL lactate dehydrogenase.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemis G. W.; Salituro F. G.; Duffy J. P.; Cochran J. E.; Harrington E. M.; Murcko M.; Wilson K. P.; Su M.; Galullo V. P. Substituted nitrogen containing heterocycle as inhibitors of p38 protein kinase. WO9827098 Publ 6/25/1998.
- See Supporting Information for X-ray crystallographic data.
- PDB accession code 3fc1.
- Fitzgerald C. E.; Patel S. B.; Becker J. W.; Cameron P. M.; Zaller D.; Pikounis V. B.; O’Keefe S. J.; Scapin G. Structural basis for p38αMAP kinase quinazolinone and pyridol-pyrimidine inhibitor specificity. Nat. Struct. Biol. 2003, 10, 764–769. [DOI] [PubMed] [Google Scholar]
- Although p38β is homologous to p38α in both primary sequence and tertiary structure, conformational differences in the ATP binding site lead to different selectivities of small molecule inhibitors.Patel S. B.; Cameron P. M.; O’Keefe S. J.; Frantz-Wattley B.; Thompson J.; O’Neill E. A.; Tennis T.; Liu L.; Becker J. W.; Scapin G. The three-dimensional structure of MAP kinase p38β: different features of the ATP-binding site in p38β compared with p38α. Acta Crystallogr. 2009, D65, 777–785. [DOI] [PubMed] [Google Scholar]
- Remy G.; Risco A. M.; Iñesta-Vaquera F. A.; González-Terán B.; Sabio G.; Davis R. J.; Cuenda A. Differential activation of p38 MAPK isoforms by MKK6 and MKK3. Cell. Signalling 2010, 22, 660–667. [DOI] [PubMed] [Google Scholar]
- Fabian M. A.; Biggs W. H.; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lélias J.-M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J. A small molecule–kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [DOI] [PubMed] [Google Scholar]
- Compound 3 (VX-745) attained clinical proof-of-principle correlating inhibition of p38 with anti-inflammatory effect, but clinical development was suspended following adverse effects findings in non-clinical studies. http://www.prnewswire.com/news-releases/vertex-moves-to-re-allocate-resources-from-vx-745-in-p38-map-kinase-program-to-accelerate-development-of-second-generation-drug-candidates-vx-702-and-vx-850-72104487.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.